
Abstract
A simple, inexpensive and robust high-performance liquid chromatography diode array detector (HPC-DAD) procedures are proposed to analyse food dyes in beverages, hard candy and fish roe samples. An ether-linked phenyl stationary phase provides sufficient selectivity and chromatographic performance for separation of 11 sulfonated azo dyes. Beverage samples were only diluted (and degassed when needed) before analysis. Solid-phase extraction (SPE) or matrix solid-phase dispersion (MSPD) procedures are proposed for efficient extraction of the analytes from candies or fish roe samples, respectively. Limits of detection (LODs) were from 0.005 to 0.013 μg mL−1 and limits of quantification (LOQs) between 0.014 and 0.038 μg mL−1. HPLC-DAD method was validated in terms of intra- and inter-day accuracy and precision at three concentration levels 2, 1, and 0.1 μg mL−1. Validation was also performed for SPE and MSPD extraction procedures including intra- and inter-day accuracy (Recovery %) and precision (RSD%), as well as intra-laboratory reproducibility. Application to analysis of beverages and food samples available to consumers proved that described methods are suitable for the routine analysis of dyes in food products.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Synthetic dyes are widely used as food additives, which are added to foodstuffs in order to compensate for the loss of natural colours destroyed during processing and/or storage, to enhance natural colour or add colour to foods that would otherwise be colourless or coloured differently (Jia et al. 2014). There are evidences indicating that dyes and their metabolites pose potential health risk to human, including allergy and asthmatic reaction, DNA damage, hyperactivity and carcinogenesis (Zou et al. 2013; Rovina et al. 2016). In order to ensure food safety and control international trade, different legislative efforts were paid to the food colourants regulation (Burrows 2009). Many countries have their own regulations about the food dyes permitted to be used in foods. Both, maximum level of dyes used in different foodstuffs as well as acceptable daily intake (ADI), were established by the respective institutions. According to the Regulation (EC) No 1333/2008 (2008), all food additives authorized for use in the EU before 20 January 2009 should be subjected to a new risk assessment by the European Food Safety Authority (EFSA). In EU, Tartrazine (E 102); Quinoline yellow (E 104); Sunset yellow (E 110); Azorubin (E 122); Ponceau 4R (E 124) and Allura red (E 129) should be labelled with additional information: ‘name or E number of the colour(s): may have an adverse effect on activity and attention in children’. Some of these colourants are banned in other countries. Considering wide availability, low cost and chemical stability of the synthetic dyes, there is a risk of their illegal usage by unfair manufacturers. Food producers may exceed the maximum levels of approved dyes or use banned substances in order to increase the attractiveness of food products. There are also possible misstatements by adding additional dyes without their proper labelling. As a consequence, analysis of synthetic dyes in food products plays an important role in assessing food quality and evaluating food safety, including possible risks to human. Therefore, reliable analytical methods are needed to monitor dyes in processed products brought to the consumer.
Up to date, several review papers concerning analytical approaches for extraction and reliable identification and quantification of food dyes have been published in the literature. Ahlström et al. (2005) have reviewed the development of analytical procedures for determination of banned azo dyes in consumer goods. Kucharska and Grabka (2010) reviewed chromatographic methods for the determination of synthetic dyes. Rebane et al. (2010) reviewed methods utilizing liquid chromatography (LC) coupled with UV–VIS detection and LC coupled with mass spectrometry (MS) for analysis of Sudan I–IV dyes along with their extraction procedures in various food matrices. Kaur and Gupta (2012) have reviewed the determination of water-soluble and water-insoluble food dyes by spectrophotometry. Yamjala et al. (2016) comprehensively described methods for the analysis of azo dyes employed in food industry. Liquid chromatography (LC) is preferred analytical technique for artificial colourants analysis. Synthetic food dyes are complex molecules, which may cause some difficulties during chromatographic analysis. These compounds are generally highly polar and elute very fast near the dead volume. Significant influence on chromatographic behaviour have sulfonyl acidic groups in their structure. The presence of these ionisable groups leads to different interactions of neutral and ionic forms with the stationary phase resulting in peak tailing, low system efficiency and poor reproducibility of retention data. Dyes in their anionic forms are poorly retained in RP-LC systems, which may be explained by electrostatic repulsion between anions and the deprotonated residual silanol groups present on the silica-based stationary phase. Separation of different dyes is most often performed in reversed phase (RP) or ion pair (IP) systems, while quantitative analysis is usually based on measurements via UV–VIS detection, especially using diode array detector (DAD) (Fuh and Chia 2002; Kiseleva et al. 2003, Tuzimski and Woźniak 2008; Culzoni et al. 2009; Tuzimski 2011; Yan et al. 2012; Petigara Harp et al. 2013; Zou et al. 2013; De Andrade et al. 2014; Li et al. 2015; Qi et al. 2015; Šuleková et al. 2016), or employing MS (Fuh and Chia 2002; Sun et al. 2007; Pardo et al. 2009; Feng et al. 2011; Liu et al. 2011; Chen et al. 2013; Li et al. 2013; Zou et al. 2013; Li et al. 2014; Jia et al. 2014; Li et al. 2015; Tsai et al. 2015; Qi et al. 2015). There are also some literature reports devoted to utilization of gas chromatography coupled with MS (GC-MS) (Otero et al. 2016) or capillary electrophoresis (CE) (Del Giovine and Bocca 2003; Huang et al. 2005; Prado et al. 2006) for food colours analysis.
The presence of chromophore groups in synthetic dyes allowed their determination and quantification at sufficiently low concentration levels by measuring their absorbance in visible wavelengths range. Therefore, matrix effect that can be observed in food analysis is practically limited. Only natural pigments (e.g. anthocyanins) present in food samples may affect proper quantification of analytes. However, these natural pigments often differs significantly in terms of chemical structure and properties from synthetic colourants and can be removed during sample preparation and/or chromatographically separated from peaks of interest. In case of LC-MS/MS, any of co-eluting compounds from a food extract (if present in the sample) can cause significant differences in signals in comparison to solvent-only samples. The co-eluting compounds can either suppress or enhance ionization of the analyte in the electrospray ion source (ESI). These difficulties are partially overcome by plotting of matrix-matched calibration and carful matrix effect assessment. Nevertheless, some differences observed from sample to sample may still occur and affect proper quantification of the analytes. Considering all of the above, HPLC-DAD can be simple, cost-effective, reliable and sufficiently sensitive analytical platform for dyes analysis in beverages and foodstuffs. As a result, it may successfully serve as alternative to expensive and more sophisticated LC-MS/MS instrumentation.
First stage of dyes analysis is extracting them from food sample, which may be, in many cases, more complicated then instrumental analysis itself (Kucharska and Grabka 2010). Selection of proper extraction technique depends on the kind of sample matrix and is also strictly connected with analytical technique that will be used to determinate the food dyes (Kucharska and Grabka 2010). Due to its advantages, like simplicity and rapidity, solid-phase extraction (SPE) is the most commonly used technique to concentrate and purify these target compounds (Tuzimski and Woźniak 2008; Baggiani et al. 2009; Tuzimski 2011; Yan et al. 2012; Chen et al. 2013; Li et al. 2014; Qi et al. 2015; Yamjala et al. 2016). However, in case of solid food samples, dyes ought to be extracted prior to SPE into appropriate solvent-media. For this purpose, solvent extraction is preferably selected (Zou et al. 2013; Tsai et al. 2015; Otero et al. 2016). Solvent extraction could be also assisted by ultrasounds (UAE) (Bonan et al. 2013; Li et al. 2013; Khalikova et al. 2015) or microwaves (MAE) (Sun et al. 2013). There are also literature reports on applying of quick, easy, cheap, effective, rugged and safe (QuEChERS) approach for dyes extraction (Jia et al. 2014; Zhu et al. 2014; Rejczak and Tuzimski 2015).
In this study, authors developed an easy and cost-effective HPLC-DAD method for analysis of 11 synthetic dyes. Extraction of the analytes was carried out by means of anion exchange SPE or matrix solid-phase dispersion (MSPD). The procedures were preliminary validated and applied to analysis of different beverages, as well as solid food samples, such as hard candies and fish roe.
Experimental
Chemicals and Reagents
Solvents and Mobile-Phase Solutions
Acetonitrile (MeCN) and methanol (MeOH) were pro chromatography grade and were obtained from E. Merck. Deionized water (0.07–0.09 μS cm−1) was obtained by means of Hydrolab System (Gdansk, Poland) in our laboratory. Ammonium acetate and aqueous ammonia solution were obtained from POCH (Gliwice, Poland).
Dyes Standards
Standards for the 11 synthetic colourants under investigation, such as Allura Red AC (E129), Amaranth (E123), Azorubin (E122), Brilliant Black PN (E151), Brilliant Blue FCF (E133), Brilliant Green BS (E142), Patent Blue V (E131), Ponceau 4R (E124), Red 2G (E128), Sunset Yellow FCF (E110) and Tartrazine (E102), were obtained from Institute for Engineering of Polymer Materials and Dyes (Zgierz, Poland).
Individual stock standard solutions (200 mg L−1) were prepared in methanol and were stored at 6 ± 2 °C. The working standard solutions and their mixtures were prepared by combining suitable aliquots of each individual standard stock solution and diluting them with 50 mM ammonium acetate.
Solid-Phase Extraction Materials
Strata X-AW cartridges, containing weak anion exchange functionalized polymeric sorbent, were obtained from Phenomenex Inc. (Torrance, CA, USA).
Sample Preparation
Beverages Procedure
Several different types of beverages were selected for the analysis including isotonic drinks (OSHEE Multifruit, OSHEE Red, OSHEE Pink, OSHEE Orange for Runners and 4Move Lime&Mint Flavour), carbonated alcoholic beverages (Sobieski Impress Cranbery and Sobieski Impress Kamikaze), flavoured vodka (Lubelska Grapefruit) and syrups (Victoria’s Blue Curacao and Bols Grenadine). Simple dilute and shoot method was applied for beverage analysis. Before injection, samples were prepared by combining suitable aliquots of each beverage and diluting them with 50 mM ammonium acetate up to 5 mL in volumetric flasks. Isotonic drinks and flavoured vodka were diluted fiftyfold, when syrup samples were diluted hundredfold before analysis. In the case of carbonated alcoholic drinks, gas in beverages was removed by placing them into an ultrasonic bath at room temperature for 20 min. Afterwards, fiftyfold dilution was used to prepare final samples. Each beverage sample was prepared in triplicates (n = 3).
Hard Candy Procedure
Hard candies were thoroughly grinded down in a ceramic mortar with a pestle. Then, 100 mg of the crumbled candy was weighted into a 12 mL polypropylene (PP) tube and 10 mL of deionized water was added. Closed tube was shaken by hand for 5 min in order to dissolve the sample. Afterwards, SPE procedure using Strata X-AW cartridges was conducted.
Initially, a SPE cartridge was conditioned with 5 mL of methanol and 5 mL of deionized water. Next, sample was loaded onto the cartridge (negative pressure 300–400 mbar on the pump connected to the SPE vacuum manifold). Afterwards, cartridge was washed with 2 mL of 25 mM ammonium acetate and 3 mL of methanol and dried for 1 min (negative pressure about 750 mbar). Finally, dyes were eluted with 5% (v/v) solution of aqueous ammonia in methanol up to 5 mL in volumetric flask. The eluate was transferred into an evaporating dish and evaporated to dryness under a fume hood with air intake switched on. Dry residues were reconstituted in 1 mL of 50 mM ammonium acetate producing final sample for HPLC-DAD analysis.
Caviar/Fish Roe Procedure
Matrix solid-phase dispersion (MSPD) procedure was developed for caviar/fish roe samples. First, 250 mg of the sample was weighted into ceramic mortar and 200 mg of Strata X-AW sorbent was added. The whole content of the mortar was blended using a pestle for approximately 2 min until a visually homogeneous mixture was obtained. Then, 1 mL of deionized water was added and the mixture was blended for additional 1 min in order to enable/improve retention of dyes by anion exchange mechanism. The mixture was transferred into a 6 mL empty SPE syringe with a frit disk packed at the bottom. Water was pulled away from the sample in the SPE vacuum manifold and discarded. Next, 1 mL 5% (v/v) solution of aqueous ammonia in acetonitrile was used to wash the mortar carefully and transferred to the syringe with packed sample. Finally, dyes were eluted with another portion of 5% (v/v) aqueous ammonia solution in acetonitrile, together up to 5 mL in volumetric flask. The eluate was transferred into 12 mL PP centrifuge tube and stored in a freezer compartment at temperature about −20 °C for 15 min in order to improve protein/peptide precipitation. Afterwards, the sample was centrifuged for 5 min at 6000 rmp (3480 rcf) using a laboratory centrifuge (MPW-223e, Warsaw, Poland). After centrifugation, 200 μL of extract was transferred into an evaporating dish and evaporated to dryness under a fume hood with air intake switched on. Dry residues were reconstituted in 1 mL of 50 mM ammonium acetate producing final sample for HPLC-DAD analysis.
HPLC-DAD Procedure
Agilent Technologies 1200 HPLC system with a quaternary pump was used for the LC analysis. Analytes were separated using a Synergi Polar RP 150 mm × 4.6 mm column, with 4-μm particle size (Phenomenex, Torrance, CA, USA). The column was thermostated at 22 °C. Mobile phase consisted of 50 mM CH3COONH4 in water (component A) and acetonitrile (component B). Linear gradient elution programme was used (5% B at start; linear to 40% B in 20 min). Mobile phase flow was 1 mL min−1. Final samples were injected onto the column using a Rheodyne manual injector with 20 μL loop. The column was re-equilibrated for 15 min using initial mobile phase composition between subsequent analysis.
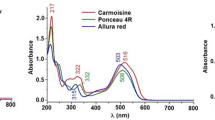
UV–VIS spectra library for dyes under investigation was created by collecting analyte spectra (200–900 nm) from the analysis of individual standards at 5 μg mL−1 concentration (Fig. 1). Four different wavelengths were selected for simultaneous detection and quantification of the analytes (420 nm for yellow dyes; 504 and 515 nm for red dyes and 630 nm for blue dyes). Identification of colourants was accomplished on the basis of their retention times and by comparison between the UV–VIS spectra of the reference compounds in the chromatograph library and the UV spectra of the detected peaks in the samples.

UV–VIS spectra (200–900 nm) of 11 synthetic food dyes collected from individual solutions (5 μg mL−1) prepared and injected in 50 mM ammonium acetate
HPLC Method Validation
The standard calibration curves of the analytes were constructed by plotting analyte concentration against peak area. Dye standards were prepared as solutions in mobile phase component A at ten concentrations in the range of 0.01–2 μg mL−1 and injected in triplicate under the same chromatographic conditions. The individual calibration concentrations were 0.01; 0.02; 0.04; 0.06; 0.08; 0.1; 0.2; 0.5; 1 and 2 μg mL−1. The calibration curves of dyes under investigation showed satisfactory linearity and correlation between concentration and peak area over the studied range with the correlation coefficients, r 2, ≥0.9998. The instrumental limits of detection (LOD) and quantification (LOQ) for all analytes were calculated using following formulas (1 and 2) (ICH guidelines 2005):
where SD is the standard deviation of y-intercept of regression lines (calculated using LINES function in MS Excel 2010), and S is the slope of the calibration plot. Retention times and full calibration data including LODs and LOQs are presented in Table 1.
The HPLC-DAD procedure was validated in terms of accuracy as well as intra- and inter-day precision. Validation study was conducted for three concentration levels of 2, 1 and 0.1 μg mL−1. During three following days, six different mixtures of dye standards for each concentration level were prepared in 50 mM CH3COONH4 and analysed per day. Average concentration measured, its standard deviation (SD) as well as accuracy (%) and precision (RSD%) were calculated for each dye standard. Inter-day accuracy (%) and precision (RSD%) was calculated for each following day (n = 6) and is given in Table 2, as well as intra-day data calculated as a mean results for all replicates (n = 18).
Validation Studies for Sample Preparation Procedures
Validation study was performed for SPE using Strata X-AW columns (Table 3) as well as for MSDP extraction for caviar/fish roe samples (Table 4). Intra-day accuracy and precision was studied by analysing samples in six replicates (n = 6). The experiments were repeated in three following days, producing results (n = 18) for inter-day accuracy and precision evaluation. Accuracy in all cases was expressed as percentage recovery of the analyte using equitation (3):
Precision was expressed as RSD% calculated as follows (4):
Intra-laboratory reproducibility was studied for both extraction procedures (SPE and MSPD). Experiments in six replicates (n = 6) were conducted by two different analysts using the same instrumentation and calibration data, but using self-prepared dyes solution for spiking procedures. In total, 30 replicates (n = 30) were analysed for each procedure (and each spiking level) and on their basis overall accuracy (Recovery %) and precision (RSD%) were determined. MSPD procedure for extraction of dyes form fish roe/caviar samples was validated at four spiking levels; at 10 μg g−1 (for the mixture of all eleven standards under investigation); and additionally at 20, 50 and 100 μg g−1 for the Brilliant Black PN and Brilliant Blue FCF, which were expected to be found in fish roe samples bought from the market to the analysis. For beverages analysis, only simple dilution was proposed as sample preparation. Therefore, validation studies for this procedure are not necessary, since HPLC-DAD method is validated (Table 2).
Results and Discussion
The elaborated HPLC-DAD method and three different sample preparation solutions was successfully applied to the analysis of ten different beverages (including isotonic drinks, flavoured vodka, carbonated alcoholic drinks and syrups), two different hard candy products and three fish roe/caviar samples. Eight out of 11 colourants under investigation including Tartrazine (E 102), Sunset Yellow FCF (E110), Azorubin (E 122), Ponceau 4R (E 124), Allura Red AC (E 129), Patent Blue V (E 131), Brilliant Blue FCF (E 133) and Brilliant Black PN (E 151) were found in food products at concentration from 0.32 to 237.8 μg mL−1 or gram of the sample, respectively. The detailed results and most important information on method development and validation are provided in subsequent subsections devoted to HPLC-DAD method development and validation, and analysis of beverage; hard candy and fish roe/caviar samples.
HPLC-DAD Method Development and Validation
In this study, ether-linked phenyl phase (Synergi Polar-RP) was applied for chromatographic method development. Retention of dyes on this stationary phase occurs due to π−π interactions. Generally, dyes with naphthalene ring show grater retention in comparison to dyes containing benzene ring in their molecules. Addition of ammonium acetate into mobile phase caused increase in the retention of investigated food colouring agents. This was important, e.g. for Tartrazine, because in only water-organic modifier system, it elutes close to the dead volume. Another advantage of ammonium acetate additive to the mobile phase was observed in terms of peak shape and chromatographic system efficiency. Finally, separation of 11 synthetic food dyes is possible applying gradient elution mode in less than 20 min obtaining satisfactory selectivity, theoretical plate number (N/m) and peak symmetry for all of the analytes (Fig. 2). All obtained peaks were narrow with widths at their half heights from 0.075 to 0.088, which results in high efficiency of the chromatographic system with N/m from 108,800 to 1,713,800 being observed. Symmetry factors (AS) and tailing factors (tF) were from 0.799 to 0.932 and 1.050 to 1.312, respectively (Fig. 2). All these parameters proved that chromatographic system applied in this study is suitable and demonstrate satisfactory performance for analysis of synthetic dyes.

Chromatogram showing separation of 11 food dyes at concentration of 1 μg mL−1 (overlaid signals from 420, 504, 515 and 630 nm wavelengths) and table showing chromatographic parameters for individual peaks (k capacity factor, width (min) peak width at half height, A S symmetry factor, t F tailing factor, N/m theoretical plate numbers calculated by halfwidth method, α selectivity to proceeding peak, R S resolution to proceeding peak calculated by halfwidth method; Note: all chromatographic parameters were calculated automatically by ChemStation® software operating Agilent 1200 chromatograph; for more details about respective formulas used for calculation, please see the manual linked (https://www.agilent.com/cs/library/usermanuals/Public/G2070-91126_Understanding.pdf); E numbers for food dyes are explained in the text)
HPLC-DAD provides sufficient sensitivity for dyes under investigation with LODs from 0.005 to 0.013 μg mL−1 and LOQs between 0.014 and 0.038 μg mL−1 being observed (Table 1). These LOD/LOQ values are lower than those obtained by Zou et al. using HPLC-DAD and similar to those observed by the authors for HPLC-MS/MS (Zou et al. 2013). Feng et al. developed method for screening of 40 dyes in soft drinks by liquid chromatography–tandem mass spectrometry (LC-MS/MS) (Feng et al. 2011). For Sunset Yellow FCF, Allura Red AC, Azorubin, Brilliant Blue FCF and Patent Blue V, the authors obtained LOD ranging from 0.3 to 10 ng mL−1, which are similar or lower than those in our study (Feng et al. 2011). However, for Tartrazine, Amaranth and Ponceau 4R reported LODs were relatively high with values of 500, 250 and 125 ng mL−1, respectively, which means detection limits from 15.6 to 45.45-fold higher than those obtained via HPLC-DAD in our study (Feng et al. 2011).
The proposed HPLC-DAD method for synthetic dyes analysis undergone validation study including inter- and intra-day accuracy (%) and precision (RSD%) measurements (Table 2). Inter-day accuracy (%) and precision (RSD%) were investigated over three following days on the basis of analysis of six different solution of dye standards (n = 6) for three concentration levels of 2, 1 and 0.1 μg mL−1. Inter-day accuracy (%) and precision (RSD%) at the highest concentration level (2 μg mL−1) were from 98.76 to 102.13% and 0.40 to 2.05% (day 1; n = 6); from 99.19 to 101.06% and 0.08 to 1.50% (day 2; n = 6); and from 99.09 to 101.72% and 0.20 to 1.86% (day 3; n = 6), respectively. At the middle concentration level (1 μg mL−1), inter-day accuracy (%) and precision (RSD%) were from 100.27 to 102.85% and 0.46 to 0.92% (day 1; n = 6); from 100.45 to 102.32% and 0.23 to 0.87% (day 2; n = 6); and from 99.78 to 102.20% and 0.54 to 1.38% (day 3; n = 6), respectively. At the low concentration level (0.1 μg mL−1), inter-day accuracy (%) and precision (RSD%) were from 101.17 to 104.78% and 1.24 to 4.13% (day 1; n = 6); from 98.30 to 102.87% and 0.59 to 3.94% (day 2; n = 6); and from 99.01 to 104.20% and 0.47 to 3.83% (day 3; n = 6), respectively. Intra-day accuracy (%) and precision (RSD%) (three following days with six replicates; n = 18) were from 99.09 to 101.72% and 0.20 to 1.86% at 2 μg mL−1, from 100.28 to 102.10% and 0.51 to 1.27% at 1 μg mL−1, and 99.38 to 103.65% and 0.96 to 3.88% at 0.1 μg mL−1, respectively. The HPLC-DAD method showed satisfactory accuracy and precision for the analysis of synthetic dyes in beverage and food samples.
Analysis of Dyes in Beverage Samples
In this study, sample preparation for beverages was practically limited to their dilution prior to chromatographic analysis. Carbonated drinks were additionally degassed in the ultrasonic bath before preparing their final solutions in 50 mM ammonium acetate. On the one hand, dilution factor (from twentyfold to hundredfold) was selected in terms of dyes concentration, and on the other hand, according to sugar content in particular beverage. In total, ten different drinks were analysed. Concertation of dyes found in the samples (recalculated to 1 mL of the beverage) were from 0.32 to 192.36 μg mL−1 of the beverage with RSD% less than 1.5% being observed for particular beverages on the basis of samples prepared in triplicates by appropriate dilution of the product. These data are shown in details in Table 5. These detected and quantified amounts of colours are in agreement with maximum level according to the Regulation (EC) No 94/36 (1994).
It should be noted that drinks containing solid components from fruits (e.g. juices) should be at least filtered prior to analysis, or, especially in case of complex matrix (e.g. milk-based drinks), solid-phase extraction clean-up ought to be introduced. SPE procedure using Strata X-AW cartridges is described further in the text.
Analysis of Dyes in Hard Candy Samples
Sample preparation for hard candies was based on SPE employing weak anion exchange retention of dyes on Strata X-AW sorbent. First, the SPE protocol was developed and evaluated for water samples spiked with known amount of standards (2 μg of each dye). Due to the presence of ionized sulfonyl groups in molecules, dyes are strongly retained in polymeric sorbent functionalized with primary-secondary amine ligands. Therefore, two possible elution approach were tested. Application for this purpose of 5% (v/v) formic acid in methanol should suppress the ionization of SO3 − groups of the dyes and cause their elution. However, this approach was insufficient and elution was carried out using 5% (v/v) aqueous ammonia solution in methanol. In this case, elution is forced by deactivating of anion exchanger ligands in SPE sorbent. Since dyes are retained strongly on Strata X-AW sorbent, there is possibility to introduce into the procedure an additional wash step using pure organic solvent (methanol). This is beneficial in terms of extraction selectivity, because majority of matrix component can be easy removed. However, in case of hard candy samples, this was not mandatory.
Satisfactory recovery, intra- and inter-day repeatability and intra-laboratory reproducibility were obtained for spiked water samples (2 μg of each dye standard) (Table 3). Intra-day accuracy (Recovery %) and precision (RSD%) were from 98.5 to 104.3% and 2.3 to 7.1% (day 1; n = 6), from 95.4 to 102.4% and 3.8 to 8.1% (day 2; n = 6) and from 96.6 to 103.4% and 2.3 to 6.7%, respectively. On the basis of all 18 replicates analysed in 3 days (n = 18), inter-day repeatability was calculated obtaining recovery in the range of 96.7–103.3% with RSD% values ranging from 2.8 to 6.9%. As for intra-laboratory reproducibility (experiments conducted by two different analysts, n = 12 in total), recoveries were in the range of 95.5–101.9% with RSD% values ranging from 3.0 to 6.5%. In total, 30 different samples were analysed with overall accuracy and precision ranging from 96.3 to 102.8% and 2.9 to 6.7%, respectively. Detailed validation data of SPE procedure are provided in Table 3. Since sample preparation for hard candies involves dissolving 100 mg of the grinded down sweet in deionized water and then performing SPE, there was no need for additional recovery evaluation for this particular protocol. Accordingly, recoveries summarized in Table 3 are also applicable to the hard candy samples. Two different hard candy products of various manufacturers were analysed. Two dyes (E131 and E133) were detected at concentrations ranging from 15.7 to 29.6 μg g−1 of the product. One of the hard candy products contained both dyes (E131 and E133) in spite of the fact that only one colourant (E131) was declared on the label. Nevertheless, detected amounts of dyes in hard candies are lower than maximum level (300 μg g−1) established and approved for confectionery by EU Regulation (EC) No 94/36 (1994). Detailed data on dyes analysis in hard candy samples are provided in Table 6.
Analysis of Dyes in Fish Roe/Caviar Samples
Sample preparation method development for caviar/fish roe food products is more challenging. Due to the complex matrix of fish roe, extraction of dyes using selective solid adsorbents or ion exchangers prior to HPLC analysis is necessary. Ultrasound-assisted extraction (UAE) applying different solvents, such as water, methanol, acetonitrile, water-methanol, water-acetonitrile, acidified water-acetonitrile or alkalized water-acetonitrile, was tested for initial dyes extraction before SPE on Strata X-AW. Nevertheless, all of preliminarily tested condition (including also different variants in terms of extraction duration and temperature) failed to extract colourants with high recovery rates from caviar/fish roe samples. This problem could be explained by the fact that the dyes bind strongly to the food matrix (e.g. to proteins). To overcome these limitations, MSPD with Strata X-AW sorbent is proposed.
Intra- and inter-day accuracy and precision as well as intra-laboratory reproducibility was evaluated for the proposed MSPD procedure (Table 4). Samples were spiked with mixture of 11 dyes at 10 μg g−1 level and left at room temperature for 30 min in order to allow dyes to bind to the matrix and to evaporate the solvent (methanol). Chromatogram obtained after MSPD extraction of spiked fish roe sample is presented (Fig. 3). Intra-day accuracy (Recovery %) and precision (RSD%) were from 75.6 to 99.7% and 2.8 to 13.7% (day 1; n = 6), from 76.4 to 99.1% and 1.7 to 16.7% (day 2; n = 6) and from 76.8 to 100.4% and 4.9 to 18.7%, respectively. On the basis of all 18 replicates analysed in 3 days (n = 18), inter-day repeatability was calculated obtaining recovery in the range of 76.3–99.7% with RSD% values ranging from 3.6 to 15.3%. As for intra-laboratory reproducibility (experiments conducted by two different analysts, n = 12 in total), recoveries were in the range of 83.9–102.8% with RSD% values ranging from 3.9 to 17.2%. In total, 30 different samples were analysed with overall accuracy and precision ranging from 79.3 to 101.0% and 3.7 to 16.6%, respectively (Table 4(a)). The validation study was extended for Brilliant Black PN (E151) and Brilliant Blue FCF (E133) (Table 4(b)), which were expected to be found in fish roe samples bought from the market. Overall recovery (n = 30) of Brilliant Black PN from samples spiked at 20, 50 and 100 μg g−1 level were 85.7% with RSD% 7.4%; 85.8% with RSD% 7.1; and 90.6% with RSD% 5.6%, respectively. Similarly, overall recovery (n = 30) of Brilliant Blue FCF from samples spiked at 20, 50 and 100 μg g−1 level were 91.1% with RSD% 9.4%; 85.1% with RSD% 4.2; and 84.6% with RSD% 3.9%, respectively (Table 4(b)). Finally, the procedure was employed for analysis of coloured fish roe products available to consumers. Chromatograms and correlations between spectra of library standards and detected peaks is shown (Fig. 4). Each product samples were extracted and analysed via HPLC-DAD in triplicates and on this basis concentration of E133 and E151 dyes per 1 g of foodstuff were calculated and are presented in Table 6. Detected amounts of dyes in fish roe samples (from 67.3 to 237.8 μg g−1) are lower than maximum level (300 μg g−1) established and approved by EU Regulation (EC) No 94/36 (1994) for this kind of food product.

Chromatogram of fish roe sample spiked at 10 μg g−1 level (overlaid signals from 420, 504, 515 and 630 nm wavelengths) after proposed MSPD extraction procedure

MSPD-HPLC-DAD chromatogram regions (630 nm) of selected from the market fish roe samples with identified peaks of Brilliant Blue FCF (E 133) and Brilliant Black PN (E 151). Identification of dyes was accomplished on the basis of retention times and spectra correlation between library standards and spectra within detected peaks (shown on figures). Caviar sample #1—caviar from lumpfish roe (Sweden); Caviar sample #2—caviar from capelin roe and herring roe (Sweden); and Caviar sample #3—caviar from lumpfish roe (Germany). Average concentration (μg g−1) and RSD% on the basis of triplicates are given in Table 6
To the authors best knowledge, this is the second analytical procedure devoted to artificial colourants analysis in caviar/fish roe samples. According to Web of Science search (queries in topic area: ‘caviar and dye’ or ‘fish roe and dye’ or ‘caviar and colourants’), only one article by Kirschbaum et al. is found (Kirschbaum et al. 2006). The method elaborated by the authors relays on ultrasound-assisted extractions with 1 M aqueous ammonia followed by extraction of dyes using polyamide A sorbent. The extraction steps were repeated until the resulting aqueous layer (1st step) or polyamide (2nd step) were colourless (for more details, please see referenced article) (Kirschbaum et al. 2006). This all makes the procedure reported by Kirschbaum et al. more labour-intense than the one developed in this study’s MSPD approach.
Conclusions
In this study, we propose cost-effective and reliable analytical method for food dyes analysis. Ether-linked phenyl stationary phase provide satisfactory selectivity and chromatographic performance for 11 dyes separation using mobile phase of simple composition. Considering low values of LODs and LOQs obtained via HPLC-DAD, this relatively inexpensive technique may be useful and alternative platform to LC-MS/MS. Weak anion exchange SPE polymeric sorbent (STRATA X-AW) can be easily and successfully utilized in MSPD procedure for extraction of colouring factors in fish roe samples. Developed procedures were successfully applied for analysis of beverages, hard candies and fish roe samples available to consumers. In all cases detected, the amounts of dyes were in agreement with the maximum levels established by the European Union.
It is also worth to highlight that the scope of analytical procedures developed in this study may be easily expanded. SPE or MSPD extraction/clean-up procedures can be applied in order to cover broader spectrum of food types. In some cases, only initial extraction of dyes should be taken into optimization.
References
Ahlström L-H, Eskilsson CS, Björklund E (2005) Determination of banned azo dyes in consumer goods. TrAC-Trend Anal Chem 24(1):49–56. doi:10.1016/j.trac.2004.09.004
Baggiani C, Anfossi L, Baravalle P, Giovannoli C, Giraudi G, Barolo C, Viscardi G (2009) Determination of banned Sudan dyes in food samples by molecularly imprinted solid phase extraction-high performance liquid chromatography. J Sep Sci 32:3292–3300. doi:10.1002/jssc.200900126
Bonan S, Fedrizzi G, Menotta S, Elisabetta C (2013) Simultaneous determination of synthetic dyes in foodstuffs and beverages by high-performance liquid chromatography coupled with diode-array detector. Dyes Pigments 99(1):36–40. doi:10.1016/j.dyepig.2013.03.029
Burrows AJD (2009) Palette of our palates: a brief history of food coloring and its regulation. Compr Rev Food Sci F 8:394–408. doi:10.1111/j.1541-4337.2009.00089.x
Chen D, Li X, Tao Y, Pan Y, Wu Q, Liu Z, Peng D, Wang X, Huang L, Wang Y, Yuan Z (2013) Development of a liquid chromatography–tandem mass spectrometry with ultrasound-assisted extraction method for the simultaneous determination of sudan dyes and their metabolites in the edible tissues and eggs of food-producing animals. J Chromatogr B 939:45–50. doi:10.1016/j.jchromb.2013.08.028
Culzoni MJ, Schenone AV, Llamas NE, Garrido M, Di Nezio MS, Fernández Band BS, Goicoechea HC (2009) Fast chromatographic method for the determination of dyes in beverages by using high performance liquid chromatography—diode array detection data and second order algorithms. J Chromatogr A 1216:7063–7070. doi:10.1016/j.chroma.2009.08.077
De Andrade FI, Guedes MIF, Vieira ÍGP, Mendes FNP, Rodrigues PAS, Maia CSC, Ávila MMM, de Matos RL (2014) Determination of synthetic food dyes in commercial soft drinks by TLC and ion-pair HPLC. Food Chem 157:193–198. doi:10.1016/j.foodchem.2014.01.100
Del Giovine L, Piccioli Bocca A (2003) Determination of synthetic dyes in ice-cream by capillary electrophoresis. Food Control 14:131–135. doi:10.1016/S0956-7135(02)00055-5
Feng F, Zhao Y, Yong W, Sun L, Jiang G, Chu X (2011) Highly sensitive and accurate screening of 40 dyes in soft drinks by liquid chromatography–electrospray tandem mass spectrometry. J Chromatogr B 879:1813–1818. doi:10.1016/j.jchromb.2011.04.014
Fuh M-R, Chia K-J (2002) Determination of sulphonated azo dyes in food by ion-pair liquid chromatography with photodiode array and electrospray mass spectrometry detection. Talanta 56:663–671. doi:10.1016/S0039-9140(01)00625-7
Huang H-Y, Chuang C-L, Chiu C-W, Chung M-C (2005) Determination of food colorants by microemulsion electrokinetic chromatography. Electrophoresis 26:867–877. doi:10.1002/elps.200410279
International Conference On Harmonisation Of Technical Requirements For Registration Of Pharmaceuticals For Human Use (2005) Validation Of Analytical Procedures: Text And Methodology Q2(R1);
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed 23.10.2016)
Jia W, Chu X, Ling Y, Huang J, Lin Y, Chang J (2014) Simultaneous determination of dyes in wines by HPLC coupled to quadrupole orbitrap mass spectrometry. J Sep Sci 37:782–791. doi:10.1002/jssc.201301374
Kaur A, Gupta U (2012) The review on spectrophotometric determination of synthetic food dyes and lakes. GU J Sci 25(3):579–588 http://gujs.gazi.edu.tr/article/view/1060000729
Khalikova MA, Šatínský D, Solich P, Nováková L (2015) Development and validation of ultra-high performance supercritical fluid chromatography method for determination of illegal dyes and comparison to ultra-high performance liquid chromatography method. Anal Chim Acta 874:84–96. doi:10.1016/j.aca.2015.03.003
Kirschbaum J, Krause C, Brückner H (2006) Liquid chromatographic quantification of synthetic colorants in fish roe and caviar. Eur Food Res Technol. doi:10.1007/s00217-005-0157-0
Kiseleva MG, Pimenova VV, Eller KI (2003) Optimization of conditions for the HPLC determination of synthetic dyes in food. J Anal Chem 58(7):685–690. doi:10.1023/A:1024716322120
Kucharska M, Grabka J (2010) A review of chromatographic methods for determination of synthetic food dyes. Talanta 80:1045–1051. doi:10.1016/j.talanta.2009.09.032
Li J, Ding X, Zheng J, Liu D, Guo F, Liu H, Zhang Y (2014) Determination of synthetic dyes in bean and meat products by liquid chromatography with tandem mass spectrometry. J Sep Sci 37:2439–2445. doi:10.1002/jssc.201400349
Li J, Ding X-M, Liu D-D, Guo F, Chen Y, Zhang Y-B, Liu H-M (2013) Simultaneous determination of eight illegal dyes in chili products by liquid chromatography–tandem mass spectrometry. J Chromatogr B 942–943:46–52. doi:10.1016/j.jchromb.2013.10.010
Li XQ, Zhang QH, Ma K, Li HM, Guo Z (2015) Identification and determination of 34 water-soluble synthetic dyes in foodstuff by high performance liquid chromatography–diode array detection–ion trap time-of-flight tandem mass spectrometry. Food Chem 182:316–326. doi:10.1016/j.foodchem.2015.03.019
Liu R, Hei W, He P, Li Z (2011) Simultaneous determination of fifteen illegal dyes in animal feeds and poultry products by ultra-high performance liquid chromatography tandem mass spectrometry. J Chromatogr B 879:2416–2422. doi: 10.1016/j.jchromb.2011.06.037
Otero P, Saha SK, Hussein A, Barron J, Murray P (2016) Simultaneous determination of 23 azo dyes in paprika by gas chromatography-mass spectrometry. Food Anal Methods. doi:10.1007/s12161-016-0648-6
Pardo O, Yusà V, León N, Pastor A (2009) Development of a method for the analysis of seven banned azo-dyes in chilli and hot chilli food samples by pressurised liquid extraction and liquid chromatography with electrospray ionization-tandem mass spectrometry. Talanta 78:178–186. doi:10.1016/j.talanta.2008.10.052
Petigara Harp B, Miranda-Bermudez E, Barrows JN (2013) Determination of seven certified color additives in food products using liquid chromatography. J Agric Food Chem 61:3726–3736. doi:10.1021/jf400029y
Prado MA, Vilas Boas LF, Bronze MR, Godoy HT (2006) Validation of methodology for simultaneous determination of synthetic dyes in alcoholic beverages by capillary electrophoresis. J Chromatogr A 1136:231–236. doi:10.1016/j.chroma.2006.09.071
Qi P, Lin Z-h, G-y C, Xiao J, Liang Z-a, Luo L-n, Zhou J, X-w Z (2015) Fast and simultaneous determination of eleven synthetic color additives in flour and meat products by liquid chromatography coupled with diode-array detector and tandem mass spectrometry. Food Chem 181:101–110. doi:10.1016/j.foodchem.2015.02.075
Rebane R, Leito I, Yurchenko S, Herodes K (2010) A review of analytical techniques for determination of Sudan I–IV dyes in food matrixes. J Chromatogr A 1217:2747–2757. doi:10.1016/j.chroma.2010.02.038
Regulation (EC) No 1333/2008 Of The European Parliament And Of The Council of 16 December 2008 on food additives. http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32008R1333&from=EN (accessed 23.10.2016a)
Regulation (EC) No 94/36/1994 Of The European Parliament And Of The Council of 30 June 1994 on colours for use in foodstuff. http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:31994L0036&from=EN (accessed 23.11.2016b)
Rejczak T, Tuzimski T (2015) A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem 13:980–1010. doi:10.1515/chem-2015-0109
Rovina K, Prabakaran PP, Siddiquee S, Shaarani SM (2016) Methods for the analysis of Sunset YellowFCF (E110) in food and beverage products- a review. TrAC-Trend Anal Chem 85:47–56. doi:10.1016/j.trac.2016.05.009
Šuleková M, Hudák A, Smrčová M (2016) The determination of food dyes in vitamins by RP-HPLC. Molecules 21:1368. doi:10.3390/molecules21101368
Sun H, Sun N, Li H, Zhang J, Yang Y (2013) Development of multiresidue analysis for 21 synthetic colorants in meat by microwave-assisted extraction–solid-phase extraction–reversed-phase ultrahigh performance liquid chromatography. Food Anal Methods 6:1291–1299. doi:10.1007/s12161-012-9542-z
Sun H-w, Wang F-c, Ai L-f (2007) Determination of banned 10 azo-dyes in hot chili products by gel permeation chromatography–liquid chromatography–electrospray ionization-tandem mass spectrometry. J Chromatogr A 1164:120–128. doi:10.1016/j.chroma.2007.06.075
Tsai C-F, Kuo C-H, Shih DY-C (2015) Determination of 20 synthetic dyes in chili powders and syrup-preserved fruits by liquid chromatography/tandem mass spectrometry. J Food Drug Anal 23:453–462. doi:10.1016/j.jfda.2014.09.003
Tuzimski T (2011) Determination of sulfonated water-soluble azo dyes in foods by SPE coupled with HPTLC-DAD. J Planar Chromatogr – Modern TLC 24(4):281–289. doi:10.1556/JPC.24.2011.4.2
Tuzimski T, Woźniak A (2008) Application of solid-phase extraction and planar chromatography with diode-array detection to the qualitative and quantitative analysis of dyes in beverages. J Planar Chromatogr – Modern TLC 21(2):89–96. doi:10.1556/JPC.21.2008.2.3
Yamjala K, Nainar MS, Ramisetti NR (2016) Methods for the analysis of azo dyes employed in food industry—a review. Food Chem 192:813–824. doi:10.1016/j.foodchem.2015.07.085
Yan H, Qiao J, Pei Y, Long T, Ding W, Xie K (2012) Molecularly imprinted solid-phase extraction coupled to liquid chromatography for determination of Sudan dyes in preserved beancurds. Food Chem 132:649–654. doi:10.1016/j.foodchem.2011.10.105
Zhu C, Wei J, Dong X, Guo Z, Liu M, Liang X (2014) Fast analysis of malachite green, leucomalachite green, crystal violet and leucocrystal violet in fish tissue based on a modified QuEChERS procedure. Se Pu 32(4):419–425. doi:10.3724/SP.J.1123.2014.01016
Zou T, He P, Yasen A, Li Z (2013) Determination of seven synthetic dyes in animal feeds and meat by high performance liquid chromatography with diode array and tandem mass detectors. Food Chem 138:1742–1748. doi:10.1016/j.foodchem.2012.11.084
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Tomasz Tuzimski declares that he has no conflict of interest. Tomasz Rejczak declares that he has no conflict of interest
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rejczak, T., Tuzimski, T. Application of High-Performance Liquid Chromatography with Diode Array Detector for Simultaneous Determination of 11 Synthetic Dyes in Selected Beverages and Foodstuffs. Food Anal. Methods 10, 3572–3588 (2017). https://doi.org/10.1007/s12161-017-0905-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-017-0905-3