
Abstract
In this study, a new imprinted electrochemical sensor for selectively detecting sunset yellow was developed based on glassy carbon electrode (GCE) modified by graphene oxide decorated with silver nanoparticles–molecular imprinted polymers (GO/AgNPs–MIPs). GO/AgNPs were firstly synthesized using self-assembly technology, and GO/AgNPs–MIPs were synthesized through surface imprinted technology by using GO/AgNPs as the substrate and sunset yellow as the template, respectively. The sensor was prepared by a drop-casting method. The synthetic materials were characterized by transmission electron microscope (TEM), Fourier transmission infrared spectra (FT-IR), and X-ray diffraction (XRD). The sensor was characterized by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The usage amount of GO/AgNPs–MIPs suspensions, solution pH, and accumulation time made an important difference in the process of detecting sunset yellow. Under optimal conditions, the peak current is linear to concentration of sunset yellow in the ranges of 0.1–0.6 and 0.6–12 μM, and the limit of detection was estimated to be 0.02 μM (S/N = 3). Finally, the proposed sensor was applied to detect sunset yellow in soft drinks with acceptable recovery, which demonstrated that the sensor could be used as a reliable and simple method for practical detection of sunset yellow.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In recent years, food safety that plays a key role in human public health has attracted considerable attention. One of the alarming problems in food field is that synthetic dyes are extensively used as food additives due to their good quality during process and storage (Ghoneim et al. 2011; Yoshioka and Ichihashi 2008). As one of the synthetic azo dyes, sunset yellow is easily found in common food products, such as carbonated drinks, fruit drinks, and candies. As a matter of fact, sunset yellow has harmful effects on human beings for its azo (N=N) functional groups and aromatic ring structure that are possible to cause allergies, diarrhea, and even kidney injury if excessively consumed (Llamas et al. 2009; Tanaka 2006). In China, the amount of sunset yellow permitted to be added in soft drinks should not be more than 0.1 g/kg (Yu et al. 2015). Considering food safety, the usage of sunset yellow in soft drinks should been strictly controlled, a sensitive and effective method for detecting sunset yellow is highly desirable.
Up to now, many methods such as high-performance liquid chromatography (Alves et al. 2008), capillary electrophoresis (Jager et al. 2005), fluorescence emission spectrophotometry (Dinç et al. 2002), mass spectrometry (Fuh and Chia 2002), and electrochemical method (Dominguez et al. 1990) have been developed for detection of sunset yellow. Among these methods, the electrochemical method has been considered to be most attractive on account of its high sensitivity, simple operation, and low cost. It is worth mentioning that the detection sensitivity and selectivity of electrochemical sensor could be enhanced by electrode modifies.
Graphene, which consists of a one-atom-thick planar sheet with sp2-bonded carbon atoms, has gained more and more applications in recent years for its excellent physicochemical properties (Stankovich et al. 2006; Geim and Novoselov 2007). Considered as a precursor of graphene, graphene oxide (GO) has received a great attention in electrochemical applications owing to its facile synthesis, high dispersibility, and coupling capability (Shen et al. 2009; Dreyer et al. 2010; Chen et al. 2012). Correspondingly, many applications have been developed based on GO, such as supercapacitor (Zhang and Zhao 2012), fuel cell (Scott 2012), and sensor (Liu et al. 2012). Additionally, GO possesses large amounts of oxygen-functional groups on both basal planes and edges, which endow GO sheets with chemical modifications to introduce special functionalities (Shen et al. 2009; Zhu et al. 2010; Liu et al. 2016a). Nevertheless, the application of GO in electrochemical sensors is confined to some extent by its poor electrical conductivity (Chen et al. 2012).
So far, metal or metal oxide nanoparticles have been extensively applied in the fabrication of sensors, such as Au (Nath and Chilkoti 2002), Pt (Rong et al. 2007), Ni (Liu et al. 2009), and Fe3O4 (Liu et al. 2008). Specifically, it is well known that silver is the best metal conductor, and Ag nanoparticles (AgNPs) have many physicochemical properties such as good biocompatibility, distinct electronic, and catalytic properties so that they are widely employed in the electrochemical sensors (Luo et al. 2006; Ren et al. 2005). Moreover, AgNPs could improve the stability of sensor and electron transfer rate of electrochemical reaction (Luo et al. 2006; Ren et al. 2005; Cui et al. 2008; Zhang et al. 2012). Accordingly, the combination of GO and AgNPs in electrochemical sensor should be worth desiring. Undoubtedly, the properties including high surface area and easy modification make GO nanosheets particularly useful as Ag nanoparticles support.
There are a few studies regarding the detection of sunset yellow by modified electrodes, such as hexadecyltrimethyl ammonium bromide-functionalized graphene supported platinum nanoparticles composite modified glassy carbon electrode (Yu et al. 2015), gold nanoparticles modified carbon paste electrode (Ghoreishi et al. 2012), graphene-wrapped phosphotungstic acid hybrid modified glassy carbon electrode (Gan et al. 2012), and so on. These electrochemical modified sensors were quite sensitive and effective, but their selectivity was not enough good.
Molecular imprinting as a newly emerging recognition technology is a straightforward and versatile method to prepare molecular imprinting polymers (MIPs) that could selectively recognize template molecule (Mosbach 1994; Chen et al. 2011; Liu et al. 2016b). Although the bulk MIPs prepared by conventional method exhibit high selectivity, certain drawbacks such as low binding capacity, poor site accessibility, and slow binding kinetics are still suffered (Nicholls and Rosengren 2001; Liu et al. 2011; Liu et al. 2015b). Surface imprinting considered as a novel and feasible imprinting technology has been developed rapidly. Compared with the conventional MIPs, the surface imprinted technique carried out by fixing MIPs on a support stroma will enhance the ability of mass transfer and affinity binding of template, leading to the decrease of high diffusion barrier and binding time (Liu et al. 2015a; Kan et al. 2008). In order to improve selectivity, surface imprinting technique is beneficial to electrochemical sensor for detection of sunset yellow. The GO-supporting materials as matrix would not only maximize the availability of the nanosized surface area but also provide fast mass transport to the binding sites. Obviously, GO/AgNPs are supposed to be an excellent candidate as a supported matrix for surface imprinted materials.
Herein, a novel imprinted electrochemical sensor based on GO/AgNPs–MIPs modified glassy carbon electrode (GCE) was prepared by using sunset yellow as template molecule. The composite of GO/AgNPs–MIPs were characterized by transmission electron microscope (TEM), Fourier transmission infrared spectra (FT-IR), and X-ray powder diffraction (XRD). The prepared sensor was characterized by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). Under optimal conditions, the sensor provides a sensitive, selective, and effective electrochemical method for detecting sunset yellow in soft drinks.
Experimental
Reagents
Graphite colloidal was purchased from Shanghai Yiji Co., Ltd. Trisodium citrate dihydrate (Na3C6H5O7·2H2O), silver nitrate (AgNO3), KCl, NaH2PO4·2H2O, Na2HPO4·2H2O, acetic acid, and chitosan were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Ethylene glycol dimethacrylate (EGDMA), (3-mercaptopropyl) trimethoxysilane (MPTS), 2,2′-azobisisobutyronitrile (AIBN), methyl acrylic acid (MAA), sunset yellow, amaranth, brilliant blue G, and tratarzine were purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). Fanta drink, mirinda drink, orange juice, and mango juice were bought from a local market. All aqueous solutions were prepared by deionized water at room temperature.
Apparatus
FT-IR (4000–400 cm−1) in KBr was recorded on a NICOLET NEXUS 4700 FT-IR spectrometer (Nicolet, USA). TEM analysis was performed by using JEM-2010HR at 200 kV (JEOL, Japan). XRD measurements were carried out on a Bruker D8 diffractometer (Bruker, Germany) using an angle of 5°–80° with 7°/min. CV, linear sweep voltammetry (LSV), and EIS were tested on a CHI660C electrochemical workstation (Shanghai Chenhua Instrument Co., Ltd., China). A conventional three-electrode system was employed in the electrochemical experiments by using the bare and the modified GCEs (diameter 3 mm) as working electrodes, a platinum wire, and saturated calomel electrode (SCE) as counter electrode and reference electrode, respectively.
Synthesis of Graphene Oxide-Supported Ag Nanoparticles
Graphene oxide (GO) was prepared from graphite colloidal by a modified Hummers method (Hummers Jr. and Offeman 1958). Subsequently, GO was chemically modified using MPTS to introduce mercapto groups at the surface of GO sheets. Briefly, 50 mg of the prepared GO was added to 50 mL ethanol and then under vigorous ultrasound for 1 h. Afterward, 2 mL MPTS was added into the GO suspension. The mixture was refluxed at 60 °C for 6 h with continuous agitation; the obtained mixture was washed with ethanol several times to remove unreacted MPTS. Finally, the functionalized GO (FGO) was dried under vacuum.
Ag nanoparticles (AgNPs) were synthesized by a trisodium citrate reduction method according to the reference before (Kamat et al. 1998). Briefly, 0.013 g AgNO3 solid was dissolved in 150 mL deionized water. The solution was heated under continuous stirring to boiling, and then 4 mL 1 wt% trisodium citrate aqueous solution was added. When the solution turned to light yellow, the heating was stopped and stirring continued until the solution was cooled to room temperature. The finally obtained solution was yellow green and had good colloid stability, indicating the formation of AgNPs. The product was stored in a refrigerator at 4 °C.
GO/AgNPs were prepared by a simple self-assemble process, which was mainly achieved by forming covalent S–Ag bond (Yola et al. 2014) as follows: 50 mg of the prepared FGO was added to 50 mL deionized water and then under ultrasound for 1 h to obtain a uniformly dispersed solution. Afterward, 10 mL above Ag colloids were added, and the mixed solution was under ultrasound for 10 min to form a homogeneous mixture. The mixtures were aged under ambient condition for 6 h. The resulting hybrids were centrifuged and washed with deionized water for several times and dried under vacuum. Finally, GO/AgNPs were obtained.
Synthesis of GO/AgNPs–MIPs
GO/AgNPs–MIPs were prepared through a typical precipitation polymerization. In brief, 50 mg GO/AgNPs, 0.1 mmol of sunset yellow, and 0.4 mmol of MAA were added in a 100-mL three-necked flask that contained 20 mL methanol/deionized water solutions (4:1, v/v), and then under ultrasonic treatment for 10 min, it was kept stirring for 12 h in a prepolymerization process at room temperature. Next, 1 mmol EGDMA and 5 mg AIBN were added, and the mixture was purged with N2 for 15 min and sealed in nitrogen atmosphere. Then, the flask was placed in a water bath at 65 °C under stirring. In that case, the polymerization continued for 6 h. The product was washed thoroughly through Soxhlet extraction with methanol–acetic acid solution (9:1, v/v) for 96 h until no template molecule could be detected in the extraction solution. Subsequently, the polymer was washed by methanol and dried at 50 °C. The procedure for GO/AgNPs–MIPs preparation was described in Scheme 1. The non-imprinted polymers (NIPs) were synthesized in the same way except that template molecule was absent.

Illustration of the fabrication procedure for the GO/AgNPs–MIPs
Preparation of Sensors
Prior to surface modification, the bare GCE was polished with 0.3 and 0.05 μm alumina slurries and finally rinsed with deionized water, respectively. One milliliter acetic acid and 0.5 g chitosan were mixed and diluted with deionized water in a 100-mL volumetric flask. The mixture was under vigorous ultrasound until become transparent. Then, 10 mg GO/AgNPs–MIPs was added in 1 mL chitosan solution via ultrasound to form a well-distributed dispersion, and a certain amount of GO/AgNPs–MIPs dispersions was dropped onto the clean surface of GCE. Ultimately, GO/AgNPs–MIPs/GCE was obtained after naturally drying at room temperature. Other modified electrodes were prepared in a similar way.
Electrochemical Measurements
Sunset yellow was dissolved in PBS (0.1 M, pH = 5.5) solution to prepare a sunset yellow solution with desired concentration and proper volumes of the solution was transferred to a 15-mL cell. Then, the modified GCE and other two electrodes were immersed in the above solution. After accumulation for 420 s under open circuit, cyclic voltammetry (CV) or linear sweep voltammetry (LSV) was recorded in the same solution from 0.3 to 1 V at a scan rate of 50 mV s−1. Electrochemical impedance spectroscopy (EIS) was performed in a 2 mM K3Fe(CN)6 solution containing 0.1 M KCl with the frequencies ranging from 105 to 10−2 Hz at initial potential.
Results and Discussion
Characterization of Synthetic Materials
The TEM images were firstly carried out to investigate the morphology of GO, AgNPs, GO/AgNPs, and GO/AgNPs–MIPs, which are shown in Fig. 1a–d, respectively. In Fig. 1a, the TEM image of GO clearly displayed flake-like sheets with some folds that may be the key point to prevent dislocations and aggregations of GO caused by thermal fluctuations and maintain high surface area. Figure 1b shows that the prepared AgNPs were well distributed, and the mean size of AgNPs was about 10 nm. Moreover, as shown in Fig. 1c, GO had been decorated with AgNPs and exhibited irregular shape in morphology. And Fig. 1d shows that GO/AgNPs were successfully covered with the MIPs films, which indicated that the imprinted sites were uniformly generated in the GO/AgNPs hybrids.

TEM images of GO (a), AgNPs (b), GO/AgNPs (c), and GO/AgNPs–MIPs (d)
FT-IR spectrum of the GO is shown in Fig. 2a. The adsorption bands around 1725 and 1051 cm−1 were severally caused by the C=O and C–O vibrations, which were attributed to the oxygen-containing functional groups on the GO. The band at 1633 cm−1 suggested the presence of aromatic C=C groups. In addition, the bands near 1395 cm−1 were associated with O=C–O stretching vibration, and the broad absorption band caused by the O–H stretching vibration and the absorbed water molecules was observed around 3455 cm−1. Compared with GO, GO-MPTS (Fig. 2b) showed its characteristic adsorption at 2551 and 882 cm−1, which separately represented the stretching vibration of S–H and Si–O bonds. While at 1047 cm−1, the Si–O bond in MPTS was overlapped with C–O bond of GO and the absorption band was hence enhanced. The results indicated that MPTS had been successfully introduced onto GO. From Fig. 2c, the vibration of C=O and C=C became stronger after imprinting, which attributed to the polymerization of MAA and EGDMA on the surface of GO. And the intense absorption peak at 1189 cm−1 was attributed to asymmetric vibrations of ester (Esfandyari-Manesh et al. 2011). All of the above phenomena confirmed that GO/AgNPs–MIPs were successfully prepared.

FT-IR of GO (a),GO-MPTS (b), and GO/AgNPs–MIPs (c)
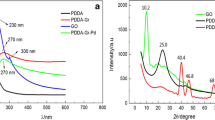
XRD patterns for GO, GO/AgNPs, and GO/AgNPs–MIPs were given in Fig. 3. Typically, GO (Fig. 3a) showed a sharp peak at 2θ = 10.76°, assigned to the (001) reflection of GO (Xu et al. 2008). Compared with the pure GO, the typical reflection of GO also could be clearly seen at 2θ = 12.43° and 10.8° in Fig. 3b, c, respectively, indicating that the ordered structure of GO is well preserved. From GO/AgNPs (Fig. 3b), the peaks at 38.18° and 44.38° were consistent with metal Ag (111) and Ag (200), suggesting that AgNPs were successfully attached on the GO surfaces. Besides these peaks, the additive peaks in GO/AgNPs–MIPs (Fig. 3c) might be corresponded to the coexistence of graphene oxide and silver in the MIPs materials.

XRD of GO (a), GO/AgNPs (b), and GO/AgNPs–MIPs (c)
Electrochemical Characteristics of Electrochemical Sensor
The electrochemical behaviors of sunset yellow on different electrodes were investigated by cyclic voltammetry in 0.1 M PBS (pH = 5.5) solution containing 50 μM sunset yellow, and cyclic voltammograms are depicted in Fig. 4. At the resulting GO/AgNPs–MIPs/GCE (Fig. 4b), comparing with GO/AgNPs/GCE (Fig. 4a), the peak current of sunset yellow decreased very fast due to the additional MIPs film hindered the electron transfer to some extent. It should be pointed out that the peak current of sunset yellow at the GO/AgNPs–MIPs/GCE was much higher than GO-MIPs/GCE (Fig. 4d), as a result of the excellent electron transfer capability of AgNPs. The result also was same with the comparison of the GO/AgNPs/GCE and GO/GCE (Fig. 4e). Compared with GO/AgNPs–NIPs/GCE (Fig. 4c), the GO/AgNPs–MIPs/GCE exhibited more sensitive response to sunset yellow, indicating that MIPs played an important role in sensing sunset yellow as well. The reason maybe that MIPs were able to provide much specific sites for sunset yellow than NIPs and thus showed excellent electrochemical response to sunset yellow. The same conclusion could be obtained by comparing GO-MIPs/GCE and GO/GCE.

Cyclic voltammograms of GO/AgNPs/GCE (a), GO/AgNPs–MIPs/GCE (b), GO/AgNPs–NIPs/GCE (c), GO-MIPs/GCE (d), and GO/GCE (e) in 0.1 M PBS (pH = 5.5) containing 50 μM sunset yellow (scan rate 50 mV s−1; accumulation time 420 s)
EIS was employed to characterize the construction process of the prepared sensors in the presence of 2.0 mM K3[Fe(CN)6] solution containing 0.1 M KCl. As seen in Fig. 5, the R et of bare GCE (Fig. 5a) was much higher than GO/AgNPs (Fig. 5c), indicating that the GO/AgNPs (Fig. 5c) were excellent electric conducting material, which could form high electron conduction pathways between the electrode and the electrolyte to accelerate the electron transfer. Moreover, the R et of GO/AgNPs–MIPs (Fig. 5b) got increased significantly. It might be explained that MIPs, as an additional barrier, blocked the electron exchange between the solution and the electrode. From what has been discussed previously, MIPs were successfully composited on the surface of GO/AgNPs.

EIS recorded in 2.0 mM K3[Fe(CN)6] solution containing 0.1 M KCl: (a) bare GCE, (b) GO/AgNPs–MIPs/GCE, and (c) GO/AgNPs/GCE
Optimization Studies
Optimization of the Amount of GO/AgNPs–MIPs Suspensions
The relationship between the peak currents of 15 μM sunset yellow solution and the amount of GO/AgNPs–MIPs suspension on the GCE electrode was investigated. As shown in Fig. 6, the peak current was enhanced to maximum when the amount of GO/AgNPs–MIPs suspension increased to 4 μL, and then the peak current decreased dramatically. This might be caused by the increase of film thickness of GO/AgNPs–MIPs suspension, leading to an obvious enhancement of the interface electron transfer resistance. Therefore, 4 μL suspension of GO/AgNPs–MIPs was adopted for further study.

Effect of the amount of GO/AgNPs–MIPs suspensions on the prepared electrode response
Optimization of the pH Value
The pH value of the solution was an important factor to the current response. To achieve an optimal electrochemical signal, the solution pH was adjusted at the range of 5.0–8.5 containing 15 μM sunset yellow. As shown in Fig. 7, it was observed that the peak current got the maximum at pH 5.5. Thus, the PBS solution of pH 5.5 was selected as the supporting electrolyte in this study. Furthermore, the effect of pH value on the peak potential was examined. With increasing pH, the peak potential decreased linearly, suggesting that protonation was involved in the electrochemical reaction of sunset yellow (Zhao et al. 2014).

Effect of different pH on the prepared GO/AgNPs–MIPs/GCE (inset: the plot of peak potential versus pH)
Accumulation Time
The effect of preconcentration time on peak current of 15 μM sunset yellow solution was tested, which is presented in Fig. 8. It was clear that the peak current of sunset yellow increased gradually from 2 to 7 min, owing to the increased amount of sunset yellow on GO/AgNPs–MIPs/GCE. And the peak current reached a plateau after 7 min, meaning that 7 min was sufficient for sunset yellow to reach the saturated rebinding onto GO/AgNPs–MIPs/GCE. Consequently, 7 min was selected as the optimal accumulation time.

Effect of the incubation time on the prepared GO/AgNPs–MIPs/GCE
Calibration Curves
Under optimal conditions, the determination of sunset yellow at different concentrations was performed. Figure 9 shows that the current response was linear to the concentration of sunset yellow varying from 0.1–0.6 and 0.6–12 μM. The corresponding regression equations are I p (10−7 A) = −1.990c (μM) − 2.126 (R 2 = 0.977) and I p (10−7 A) = −0.796c (μM) − 2.633 (R 2 = 0.998), respectively. In this case, the limit of detection was estimated to be 0.02 μM (S/N = 3). It could be seen that GO/AgNPs–MIPs/GCE offered a relatively wide linear range and low detection limit, comparing with some other sensors (Ghoreishi et al. 2012; Majidi et al. 2013; Vikraman et al. 2014; Qiu et al. 2015).

The linear relationship between peak currents and concentrations of sunset yellow in the range of 0.1–16 μM
Selectivity, Reproducibility, and Stability of GO/AgNPs–MIPs/GCE
The selectivity of GO/AgNPs–MIPs/GCE was evaluated by testing the electrochemical response of 5.0 μM sunset yellow in the presence of the same amount of analogs, such as tartrazine, amaranth, brilliant blue G, and ascorbic acid. As shown in Fig. 10, the peak current of sunset yellow including brilliant blue G, ascorbic acid, and tartrazine was changed by 1.38, 6.43, and 9.03%, respectively. This indicated that GO/AgNPs–MIPs/GCE had good selectivity toward sunset yellow. However, amaranth had a serious effect on the determination of sunset yellow. In the presence of amaranth, the peak current of sunset yellow had increased by 17.8%. The phenomenon might be explained that their chemical structures were too similar to be well discriminated by MIPs. To evaluate the reproducibility, five modified GCEs were prepared by the same way and 6.0 μM sunset yellow solution was determined. As a result, the relative standard deviation (RSD) of the peak currents was 8.33% (n = 5). After stored at room temperature for a month, GO/AgNPs–MIPs/GCE still retained 85.2% of its initial current response for 6.0 μM sunset yellow. These results reflected good reproducibility and stability of the prepared sensor.

Influence of coexistent substance on the electrochemical response of GO/AgNPs–MIPs/GCE to sunset yellow. Solution composition: 5.0 μM sunset yellow + 0.1 M PBS (pH = 5.5) (a), a + 5.0 μM tartrazine (b), a + 5.0 μM amaranth (c), a + 5.0 μM brilliant blue G (d), and a + 5.0 μM ascorbic acid (e)
Analytical Application of GO/AgNPs–MIPs/GCE in Soft Drinks
Finally, in order to test the practical application feasibility, the sensor was applied to the determination of sunset yellow in several soft drinks. Prior to determination, soft drinks were under ultrasound for 10 min and then diluted from 2.0 to 12 mL with 0.1 M PBS solution (pH = 5.5). The corresponding results were presented in Table 1. As a result, the concentration of sunset yellow in 12 mL dilute fanta and mirinda drink solution was separately found to be 4.17 and 5.73 μM, which meant that the concentration of sunset yellow in 2.0 mL original fanta and mirinda drink solution was 25.02 and 34.38 μM, respectively. From this, the content of sunset yellow in fanta and mirinda drink was calculated to be about 0.011 and 0.016 g/kg, respectively, which were lower than government standard. Nevertheless, sunset yellow had not been detected in the fruit juice sample. Additionally, the standard solutions of sunset yellow were added in the sample solutions, and the recoveries were 87–111% for different samples. These suggested that the detection method had good accuracy and reliability, and the sensor showed good applicability in the detection of sunset yellow in real samples.
Conclusion
A novel GO/AgNPs composite with a MIPs outer layer for selective recognition of sunset yellow was successfully synthesized. Owing to the excellent inherent properties of GO/AgNPs, the resulting GO/AgNPs–MIPs/GCE possesses high selectivity compared with other analogs and good sensitivity toward template molecule. The prepared sensor was successfully applied to the determination of sunset yellow in soft drinks, which can be further expected to be used to fabricate various molecular imprinting-based sensors for advanced applications.
References
Alves SP, Brum DM, de Andrade ÉCB, Netto ADP (2008) Determination of synthetic dyes in selected foodstuffs by high performance liquid chromatography with UV-DAD detection. Food Chem 107:489–496
Chen D, Feng H, Li J (2012) Graphene oxide: preparation, functionalization, and electrochemical applications. Chem Rev 112:6027–6053
Chen L, Xu S, Li J (2011) Recent advances in molecular imprinting technology: current status, challenges and highlighted applications. Chem Soc Rev 40:2922–2942
Cui K, Song Y, Yao Y, Huang Z, Wang L (2008) A novel hydrogen peroxide sensor based on Ag nanoparticles electrodeposited on DNA-networks modified glassy carbon electrode. Electrochem Commun 10:663–667
Dinç E, Baydan E, Kanbur M, Onur F (2002) Spectrophotometric multicomponent determination of sunset yellow, tartrazine and allura red in soft drink powder by double divisor-ratio spectra derivative, inverse least-squares and principal component regression methods. Talanta 58:579–594
Dominguez FB, Diego FG, Mendez JH (1990) Determination of sunset yellow and tartrazine by differential pulse polarography. Talanta 37:655–658
Dreyer DR, Park S, Bielawski CW, Ruoff RS (2010) The chemistry of graphene oxide. Chem Soc Rev 39:228–240
Esfandyari-Manesh M, Javanbakht M, Atyabi F, Mohammadi A, Mohammadi S, Akbari-Adergani B, Dinarvand R (2011) Dipyridamole recognition and controlled release by uniformly sized molecularly imprinted nanospheres. Mater Sci Eng C 31:1692–1699
Fuh M-R, Chia K-J (2002) Determination of sulphonated azo dyes in food by ion-pair liquid chromatography with photodiode array and electrospray mass spectrometry detection. Talanta 56:663–671
Gan T, Sun J, Cao S, Gao F, Zhang Y, Yang Y (2012) One-step electrochemical approach for the preparation of graphene wrapped-phosphotungstic acid hybrid and its application for simultaneous determination of sunset yellow and tartrazine. Electrochim Acta 74:151–157
Geim AK, Novoselov KS (2007) The rise of graphene. Nature Mater 6:183–191
Ghoneim MM, El-Desoky HS, Zidan NM (2011) Electro-Fenton oxidation of sunset yellow FCF azo-dye in aqueous solutions. Desalination 274:22–30
Ghoreishi SM, Behpour M, Golestaneh M (2012) Simultaneous determination of sunset yellow and tartrazine in soft drinks using gold nanoparticles carbon paste electrode. Food Chem 132:637–641
Hummers WS Jr, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80:1339–1339
Jager AV, Tonin FG, Tavares MFM (2005) Optimizing the separation of food dyes by capillary electrophoresis. J Sep Sci 28:957–965
Kamat PV, Flumiani M, Hartland GV (1998) Picosecond dynamics of silver nanoclusters. Photoejection of electrons and fragmentation. J Phys Chem B 102:3123–3128
Kan X, Zhao Y, Geng Z, Wang Z, Zhu J-J (2008) Composites of multiwalled carbon nanotubes and molecularly imprinted polymers for dopamine recognition. J Phys Chem C 112:4849–4854
Liu X, Aizen R, Freeman R, Yehezkeli O, Willner I (2012) Multiplexed aptasensors and amplified DNA sensors using functionalized graphene oxide: application for logic gate operations. ACS Nano 6:3553–3563
Liu Y, Teng H, Hou H, You T (2009) Nonenzymatic glucose sensor based on renewable electrospun Ni nanoparticle-loaded carbon nanofiber paste electrode. Biosens Bioelectron 24:3329–3334
Liu Z, Wang J, Xie D, Chen G (2008) Polyaniline-coated Fe3O4 nanoparticle–carbon-nanotube composite and its application in electrochemical biosensing. Small 4:462–466
Liu Y et al (2015a) Synthesis of hydrophilic surface ion-imprinted polymer based on graphene oxide for removal of strontium from aqueous solution. J Mater Chem A 3:1287–1297
Liu Z et al (2015b) Monodisperse magnetic ion imprinted polymeric microparticles prepared by RAFT polymerization based on γ-Fe2O3@meso-SiO2 nanospheres for selective solid-phase extraction of Cu(II) in water samples. Rsc Adv 5:52369-52381
Liu Y, Hu X, Meng M, Liu Z, Ni L, Meng X, Qiu J (2016a) RAFT-mediated microemulsion polymerization to synthesize a novel high-performance graphene oxide-based cadmium imprinted polymer. Chem Eng J 302:609-618
Liu Y, Zhong G, Liu Z, Meng M, Liu F, Ni L (2016b) Facile synthesis of novel photoresponsive mesoporous molecularly imprinted polymers for photo-regulated selective separation of bisphenol. A Chem Eng J 296:437-446
Liu Y et al (2011) Selective adsorption behavior of Pb(II) by mesoporous silica SBA-15-supported Pb(II)-imprinted polymer based on surface molecularly imprinting technique J Hazard Mater 186:197-205
Llamas NE, Garrido M, Di Nezio MS, Band BSF (2009) Second order advantage in the determination of amaranth, sunset yellow FCF and tartrazine by UV–vis and multivariate curve resolution-alternating least squares. Anal Chim Acta 655:38–42
Luo X, Morrin A, Killard AJ, Smyth MR (2006) Application of nanoparticles in electrochemical sensors and biosensors. Electroanal 18:319–326
Majidi MR, Baj RFB, Naseri A (2013) Carbon nanotube–ionic liquid (CNT–IL) nanocomposite modified sol-gel derived carbon-ceramic electrode for simultaneous determination of sunset yellow and tartrazine in food samples. Food Anal Method 6:1388–1397
Mosbach K (1994) Molecular imprinting. Trends Biochem Sci 19:9–14
Nath N, Chilkoti A (2002) A colorimetric gold nanoparticle sensor to interrogate biomolecular interactions in real time on a surface. Anal Chem 74:504–509
Nicholls IA, Rosengren JP (2001) Molecular imprinting of surfaces. Bioseparation 10:301–305
Qiu X, Lu L, Jing L, Yu Y, Wang W, Min J, Ling B (2015) An enhanced electrochemical platform based on graphene oxide and multi-walled carbon nanotubes nanocomposite for sensitive determination of sunset yellow and tartrazine. Food Chem 190:889–895
Ren X, Meng X, Chen D, Tang F, Jiao J (2005) Using silver nanoparticle to enhance current response of biosensor. Biosens Bioelectron 21:433–437
Rong L-Q, Yang C, Qian Q-Y, Xia X-H (2007) Study of the nonenzymatic glucose sensor based on highly dispersed Pt nanoparticles supported on carbon nanotubes. Talanta 72:819–824
Scott K (2012) Freestanding sulfonated graphene oxide paper: a new polymer electrolyte for polymer electrolyte fuel cells. Chem Commun 48:5584–5586
Shen J, Hu Y, Shi M, Lu X, Qin C, Li C, Ye M (2009) Fast and facile preparation of graphene oxide and reduced graphene oxide nanoplatelets. Chem Mater 21:3514–3520
Stankovich S et al (2006) Graphene-based composite materials. Nature 442:282–286
Tanaka T (2006) Reproductive and neurobehavioural toxicity study of tartrazine administered to mice in the diet. Food Chem Toxicol 44:179–187
Vikraman AE, Thomas D, Cyriac ST, Kumar KG (2014) Kinetic and thermodynamic approach in the development of a voltammetric sensor for sunset yellow. J Electrochem Soc 161:B305–B311
Xu C, Wang X, Zhu J, Yang X, Lu L (2008) Deposition of Co3O4 nanoparticles onto exfoliated graphite oxide sheets. J Mater Chem 18:5625–5629
Yola ML, Gupta VK, Eren T, Şen AE, Atar N (2014) A novel electro analytical nanosensor based on graphene oxide/silver nanoparticles for simultaneous determination of quercetin and morin. Electrochim Acta 120:204–211
Yoshioka N, Ichihashi K (2008) Determination of 40 synthetic food colors in drinks and candies by high-performance liquid chromatography using a short column with photodiode array detection. Talanta 74:1408–1413
Yu L, Shi M, Yue X, Qu L (2015) A novel and sensitive hexadecyltrimethyl ammonium bromide functionalized graphene supported platinum nanoparticles composite modified glassy carbon electrode for determination of sunset yellow in soft drinks. Sensors Actuators B Chem 209:1–8
Zhang J, Zhao X (2012) Conducting polymers directly coated on reduced graphene oxide sheets as high-performance supercapacitor electrodes. J Phys Chem C 116:5420–5426
Zhang Y et al (2012) One-pot green synthesis of Ag nanoparticles-graphene nanocomposites and their applications in SERS, H2O2, and glucose sensing. RSC Adv 2:538–545
Zhao L, Zeng B, Zhao F (2014) Electrochemical determination of tartrazine using a molecularly imprinted polymer—multiwalled carbon nanotubes—ionic liquid supported Pt nanoparticles composite film coated electrode. Electrochim Acta 146:611–617
Zhu Y, Murali S, Cai W, Li X, Suk JW, Potts JR, Ruoff RS (2010) Graphene and graphene oxide: synthesis, properties, and applications. Adv Mater 22:3906–3924
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Changchun Qin declares that he has no conflict of interest. Wenlu Guo declares that he has no conflict of interest. Yan Liu declares that she has no conflict of interest. Zhanchao Liu declares that he has no conflict of interest. Jian Qiu declares that he has no conflict of interest. Jianbo Peng declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 21207051), Ph.D. Programs Foundation of Ministry of Education of China (No. 20123227120015), Natural Science Foundation of Jiangsu Province (BK20150483), special financial grant from the China Postdoctoral Science Foundation (2014T70488), Natural Science Fund for Colleges and Universities in Jiangsu Province (Nos. 16KJB530002, 15KJB550003), Society Development Fund of Zhenjiang (No. SH2013110), and Programs of Senior Talent Foundation of Jiangsu University (No. 15JDG024).
Rights and permissions
About this article

Cite this article
Qin, C., Guo, W., Liu, Y. et al. A Novel Electrochemical Sensor Based on Graphene Oxide Decorated with Silver Nanoparticles–Molecular Imprinted Polymers for Determination of Sunset Yellow in Soft Drinks. Food Anal. Methods 10, 2293–2301 (2017). https://doi.org/10.1007/s12161-016-0753-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0753-6