
Abstract
A quantitative and confirmatory method has been developed for simultaneous determination of 15 aminoglycoside (AG) residues in porcine tissues (muscle, liver and kidney) by liquid chromatography tandem mass spectrometry (LC-MS/MS). The analytes were extracted from different matrices with aqueous trichloroacetic acid solution (5 %, w/v) followed by solid phase extraction (SPE) clean-up under optimised conditions. Due to the different pK a values of the compounds, two consecutive SPE steps using Oasis HLB cartridges were used to purify all 15 AGs from sample extracts, with 9 AGs quantitatively retained on Oasis HLB cartridges at pH <1 and the other 6 AGs retained at pH 8.5. The analytes were separated on a reversed-phase C18 column and eluted with water and acetonitrile containing the ion-pair reagent heptafluorobutyric acid (HFBA). The LC-MS/MS method was validated according to Decision 2002/657/EC. The optimised procedure was successfully applied to analyse 100 real porcine tissue samples (60 muscles, 20 livers and 20 kidneys) collected from local markets in southern China, demonstrating that the method is robust and useful for determination of residues of the 15 target AGs in porcine tissue samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
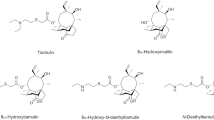
Aminoglycosides (AGs, Fig. 1) are a large class of antibiotics with chemical structures characterised by containing two or more amino sugars linked by glycosidic bonds to an aminocyclitol component. The cyclitol is 2-deoxystreptamine in most of these drugs, except for streptomycin and dihydrostreptomycin which have a streptidine instead (Oertel et al. 2004; Stead 2000). Some of AGs are naturally occurring substances isolated from various Actinobacteria (Actinomycetes), particularly members of the Streptomyces (in which case, they are named mycins) and Micromonospora (named micins) genus, while others are semi-synthetic, produced by chemical modification of the natural molecules (Stead 2000; Kaufmann et al. 2012).

Chemical structures of the aminoglycosides studied
The AGs interfere with bacterial protein synthesis by binding irreversibly to ribosomes, which causes damage to the cell membranes (McGlinchey et al. 2008). Many of these drugs are extensively used in human and veterinary medicine to treat a wide range of aerobic bacterial infections (mainly Gram-negative bacteria, but some Gram-positive organisms are susceptible) and some protozoal infections (Tawa et al. 1998; Bogialli et al. 2005). However, the application of these drugs as veterinary medicines may increase the risk of developing antimicrobial resistance among bacteria, which can consequently weaken the efficacy of these drugs as human medicines. Besides, AGs exhibit potential toxicity against the renal, vestibular, and auditory systems (Stead 2000; Hosokawa et al. 2008). To ensure more effective and controlled use of these drugs in animal husbandry, the European Union (EU) (Commission Regulation 2009), JECFA (Joint FAO/WHO 2005), USA, China, Japan (The Japan Food Chemical Research Foundation 2006) and other countries/international organisations have issued strict maximum residue levels for nine AGs in various foodstuff of animal origin. Therefore, robust, sensitive and reliable analytical methods are required to determine trace levels of AG residues in multiple animal products.
A number of analytical methods for the determination of AGs in various matrices have been reported and well documented in the literature (Stead 2000; McGlinchey et al. 2008). Chemical methods including thin-layer chromatography (TLC), capillary electrophoresis (CE), optical biosensors and resonance Rayleigh scattering (RRS), together with biological methods such as enzyme immunoassay and microbiological assay, are mainly utilised as screening methods (McGlinchey et al. 2008). Since AGs are non-volatile, and they must be derivatized to make them amenable to analysis using gas chromatography (GC) or gas chromatography-mass spectrometry (GC-MS), typically requiring lengthy derivatisation time at elevated temperatures with silylating agents (Preu et al. 1998). Due to their lack of chromophores and fluorophores, AGs are not amenable to direct ultraviolet (UV) or fluorescence detection (FLD); therefore, pre- or post-column derivatisation is often required when using liquid chromatography (LC) analysis, followed by the detection with UV or FLD (Tawa et al. 1998; Hornish and Wiest 1998; Edder et al. 1999; Posyniak et al. 2001; Viñas et al. 2007; Chen et al. 2010). Such LC-UV or LC-FLD methods generally result in poor reproducibility due to the instability and low yield of the derivatives. Because of this, liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS)-based methods are becoming the most common approach for the confirmatory determination of AG residues because of their high throughput, sensitivity, selectivity and the capability to generate sufficient structural information for analyte confirmation. However, early LC-MS/MS methods are developed to analyse only one or two different AG residues within a single experiment (Hornish and Wiest 1998; Löffler and Ternes 2003; Kaufmann et al. 2003; Gremilogianni et al. 2010; Granja et al. 2009; Heller et al. 2005; Cherlet et al. 2007). The recent introduction of better sample clean-up and improved chromatographic techniques allowed the determination of larger numbers of AG residues in a single method (Oertel et al. 2004; Kaufmann et al. 2012; Bogialli et al. 2005; Babin and Fortier 2007; Kaufmann and Maden 2005; Holthoon et al. 2009; Zhu et al. 2008; Berrada et al. 2010; Tao et al. 2012).
The difficulties in multi-residue LC-MS/MS analysis of AGs in animal products are related to the extraction, clean-up, separation and detection procedures. AGs are highly polar, extremely hydrophilic compounds due to the many amino and hydroxyl groups in their chemical structures, so they are not adequately retained on conventional reversed-phase columns. As a consequence, many analytical methods use ion-pair reagents such as heptafluorobutyric acid (HFBA) (Kaufmann et al. 2012; Bogialli et al. 2005; Löffler and Ternes 2003; Kaufmann et al. 2003; Gremilogianni et al. 2010; Granja et al. 2009; Babin and Fortier 2007; Kaufmann and Maden 2005; Holthoon et al. 2009; Zhu et al. 2008; Berrada et al. 2010) or pentafluoropropionic acid (PFPA) (Cherlet et al. 2007) to improve the retention and separation by reversed-phase LC. Hydrophilic interaction chromatography (HILIC)-MS/MS has also been applied to simultaneously determine various AGs (Oertel et al. 2004; Tao et al. 2012). In general, the currently preferred detection technique for the multi-residue analysis of AGs is LC-MS/MS with electrospray ionisation in positive mode (Oertel et al. 2004; Kaufmann et al. 2012; Bogialli et al. 2005; Löffler and Ternes 2003; Kaufmann et al. 2003; Gremilogianni et al. 2010; Granja et al. 2009; Heller et al. 2005; Cherlet et al. 2007; Babin and Fortier 2007; Kaufmann and Maden 2005; Holthoon et al. 2009; Zhu et al. 2008; Berrada et al. 2010; Tao et al. 2012).
Another critical challenge with simultaneous determination of AGs at trace levels in complicated animal matrices is the extraction and clean-up procedure, which plays key roles in the overall analysis. Matrices such as the muscle, liver and kidney contain many interfering substances that need to be selectively removed. Moreover, AGs are water-soluble, highly polar and acid- and base-resistant compounds which are not extensively bound to proteins. Therefore, most extraction methods have been based on extraction into aqueous-based solvent systems including water-soluble organic solvents such as acetonitrile (Kowalski et al. 1999) or strong acids such as trichloroacetic acid (Kaufmann et al. 2012; Hornish and Wiest 1998; Chen et al. 2010; Gremilogianni et al. 2010; Heller et al. 2005; Cherlet et al. 2007; Babin and Fortier 2007; Kaufmann and Maden 2005; Holthoon et al. 2009; Zhu et al. 2008; Tao et al. 2012) and perchloric acid (Edder et al. 1999; Viñas et al. 2007) to precipitate the proteins. The extracts have been cleaned up by a variety of methods, including solid phase extraction (SPE) (Kaufmann et al. 2012; Hornish and Wiest 1998; Edder et al. 1999; Posyniak et al. 2001; Chen et al. 2010; Kaufmann et al. 2003; Gremilogianni et al. 2010; Granja et al. 2009; Cherlet et al. 2007; Kaufmann and Maden 2005; Holthoon et al. 2009; Zhu et al. 2008; Tao et al. 2012), on-line SPE (Babin and Fortier 2007), matrix solid phase dispersion extraction (Bogialli et al. 2005) and pressurised liquid extraction (Berrada et al. 2010). However, most of these purification procedures are suitable for only a few AGs. A multi-residue method for 11 AGs in the meat and liver by LC-MS/MS was reported by Kaufmann and Maden (2005). However, the clean-up procedure was quite time consuming, involving three steps: anion exchange, HLB SPE and weak cation exchange SPE. Bogialli et al. (2005) proposed matrix solid phase dispersion with heated water to extract and clean up nine AGs in bovine whole milk, which needed a special homemade extraction apparatus. Zhu et al. (2008) developed a LC-ESI-MS/MS method for the simultaneous determination of 13 AGs in the muscle, liver, kidney, honey and milk, with sample clean-up by two consecutive SPE steps using two HLB cartridges at pH <1 and pH 8.5. Kaufmann et al. (2012) carried out a single-step strong cation exchange SPE procedure to clean up 13 AGs in pork muscle, fish, veal liver and kidney. Tao et al. (2012) applied automated SPE using weak cation exchange carboxylic acid cartridges for sample clean-up and utilised a specialised column for AGs to achieve chromatographic separation for the determination of 15 AGs; however, gentamicin C2 and gentamicin C1a, two important isomers of gentamicin, were not included.
Therefore, the main objective of this study was to develop a multi-residue LC-ESI-MS/MS method for the simultaneous determination and quantification of 15 AGs including apramycin, streptomycin, dihydrostreptomycin, amikacin, spectinomycin, kanamycin A, neomycin, paromomycin, tobramycin, gentamicin (consisting of C1, C1a and isomers C2/2a/2b), hygromycin B, netilmicin and sisomicin in porcine tissues (muscle, liver and kidney). Samples were extracted by homogenisation with 5 % trichloroacetic acid followed by SPE clean-up before analysis. Experimental conditions for sample preparation and instrumental analysis were optimised to achieve best performance. The optimised method was then applied to determine AGs in porcine tissue samples collected from local markets in southern China.
Experiment
Chemicals and Reagents
Unless otherwise specified, all reagents were of analytical reagent quality or better. Apramycin (APRA), amikacin (AMIK), spectinomycin (SPEC), kanamycin A (KANA), neomycin (NEO), paromomycin (PARO), streptomycin (STREP), dihydrostreptomycin (DISTREP), tobramycin (TOBRA) and gentamicin (consisting of GENT C1, GENT C1a and GENT C2/2a/2b) were obtained from Dr. Ehrenstorfer GmbH (Augsburg, Germany), while hygromycin B (HYGRO) aqueous solution (54 mg mL−1), netilmicin (NETIL) and sisomicin (SISO) were purchased from Sigma-Aldrich (St. Louis, MO, USA), European Pharmacopoeia (Strasbourg, France) and Toronto Research Chemicals Inc. (Toronto, Canada), respectively. HFBA (>99.5 %) was supplied by Sigma-Aldrich (USA). HPLC grade acetonitrile, methanol, glacial acetic acid and n-hexane were obtained from Merck (Germany). Analytical grade trichloroacetic acid (TCA), sodium hydroxide (NaOH) and hydrochloric acid (HCl) were supplied by Guangzhou Chemical Company (Guangzhou, China). Ultrapure water (18.2 MΩ cm−1) was generated by a Milli-Q® Advantage A10 water purification system (Millipore, France). The cartridges used for solid phase extraction were Oasis HLB cartridges (3 mL/60 mg) from Waters (Milford, MA, USA). Filter membranes (0.45 μm) provided by Jinteng Laboratory Facilities Co. Ltd. (Tianjin, China) were used to filter the sample solutions before injection onto the LC-MS/MS system.
Standard Solutions
The analytical standard solutions were prepared in a dilution solution comprising acetonitrile/water/acetic acid (20:78:2, v/v/v). Individual stock standard solutions of all 15 AGs (1000 μg mL−1) were prepared by accurately weighing 50 mg of reference substance (calculated as dry free base, except HYGRO, for which 926 μL of the aqueous standard solution was transferred into a 50-mL volumetric flask), dissolved with the dilution solution and made up to volume with the solution. These stock standard solutions were stable for 1 year when stored in plastic tubes at 2–4 °C. Since the solid standard of gentamicin consists of GENT C1, C1a, C2, C2a and C2b, with proportion 29.1 % (C1), 21.3 % (C1a) and 49.6 % (sums of C2, C2a and C2b), so there are three stock standard solutions of gentamicin, corresponding to 1000 μg mL−1 for GENT C1, C1a and C2 (sums of C2, C2a and C2b), respectively. Tuning solutions of each analyte (10 μg mL−1) were prepared by dilution of the stock standard solutions. Working mixed standard solutions for fortification/validation experiments were prepared by diluting individual stock standard solutions to the appropriate concentrations. The working mixed standard solutions were stored in plastic tubes at 2–4 °C and remained stable for 1 month.
Sample Collection and Preservation
Porcine tissue (muscle, liver and kidney) samples were collected from local markets in Shenzhen (Guangdong, China). For each sample, around 500 g tissue was first minced using a Foss Tecator 2094 homogeniser (Höganäs, Sweden) and then packaged into a plastic container, capped and stored at −20 °C before analysis. Samples were returned to cold storage immediately after sub-sampling. Special care was taken while sample handling to prevent accidental contamination or loss of target analytes.
Sample Preparation
Extraction
Five-gramme aliquots of the homogenised tissue samples were weighed into 50-mL polypropylene centrifuge tubes. For validation purposes, samples were spiked with the working mixed standard solutions at appropriate concentrations and let stand for 1 h. A volume of 10 mL of 5 % TCA (w/v) was added to each centrifuge tube. The mixture was homogenised thoroughly at 10,000 rpm for 1 min using a high-speed blender (IKA® T25 digital Ultra-Turrax® with IKA® works S25N-25F dispersing, IKA, Germany) and then centrifuged at 5 °C, 8000 rpm (6953×g) for 5 min (Beckman Coulter Allegra™ X-22R, USA). The extraction procedure was repeated with 10 mL of 5 % TCA, and the TCA supernatants were combined into another centrifuge tube. A 5 mL volume of 0.2 mol L−1 HFBA and 5 mL n-hexane were added to the extracts. After vibration mixing using a platform shaker (IKA KS 260 control, IKA, Germany) at 360 rpm for 30 min and additional centrifuging at 5 °C, 6953×g for 5 min, the upper n-hexane phase was removed and then the residual aqueous extracts were cleaned up as described below.
SPE Clean-up and Concentration
A HLB cartridge was pre-conditioned with 3 mL methanol, 3 mL water and 3 mL of 0.2 mol L−1 HFBA by gravity. A 5 mL volume of the extract was transferred onto the cartridge at a flow rate of 1 mL min−1. The total effluent was collected into another tube and adjusted to pH 8.5 ± 0.2 with 100 g L−1 NaOH (about nine drops) and 0.2 mol L−1 HCl. Afterwards, the cartridge was dried by a vacuum pump (Visiprep™ SPE vacuum manifold DL, 24-port model, Supelco, USA) for 5 min. Another HLB cartridge was pre-conditioned with 3 mL methanol, 3 mL water, 3 mL of 0.2 mol L−1 HFBA and 3 mL of pH 8.5 aqueous NaOH by gravity. Then, the pH 8.5 ± 0.2 effluent was loaded onto the column at 1 mL min−1. After the sample had been passed through, the two HLB cartridges were joined with vacuum joints. The two tandem cartridges were rinsed with 5 mL water and then dried at less than 15 mmHg for 10 min. AG residues were finally eluted with 6 mL acetonitrile/0.15 mol L−1 HFBA (4:1, v/v), and the eluate was evaporated to 0.3 mL under a gentle steam of nitrogen at 40 °C. Finally, the residue was reconstituted to 1 mL with 20 mmol L−1 HFBA. The resulting solution (filtered through a 0.45-μm membrane first if turbid) was transferred into an LC autosampler vial for LC-ESI-MS/MS analysis.
LC-ESI-MS/MS Analysis
LC Condition
Liquid chromatography was performed using an Agilent 1100 Series HPLC system (Agilent Technologies, USA) equipped with an automatic degasser, a quaternary pump and an autosampler. Chromatographic separation was carried out using an Atlantis® dC18 column (150 mm × 2.1 mm, particle size 5 μm) at 30 °C. The flow rate of mobile phase was maintained at 0.4 mL min−1, and the injection volume was 30 μL. Mobile phase A was acetonitrile containing 20 mmol L−1 HFBA, mobile phase C was acetonitrile/water (5:95, v/v) containing 20 mmol L−1 HFBA and mobile phase D was acetonitrile/water (50:50, v/v) containing 20 mmol L−1 HFBA. The gradient elution program is summarised in Table 1.
MS/MS Parameters
The HPLC system was connected to an API 3000 triple quadrupole tandem mass spectrometer (Applied Biosystems/MDS Sciex, Toronto, Canada), equipped with a turbo ion spray source and a syringe pump. The electrospray ionisation mode (positive) was adopted. Optimisation of the ionisation parameters for each analyte was achieved by infusing each compound separately at a flow rate of 10 μL min−1. For sample analysis, the instrument was operated in multiple reaction monitoring (MRM) mode and two transitions were monitored for each compound. The higher intensity transition was selected for quantitation, and the resolution was set at 0.7 U. The analyst 1.4.1 software was used for instrument control and data acquisition.
Mass parameters for each analyte including precursor ion (Q1), product ion (Q3), declustering potential (DP), entrance potential (EP), cell exit potential (CXP) and collision energy (CE) are summarised in Table 2. The focusing potential (FP) and dwell time (DT) for all analytes were 350 V and 40 ms, respectively. The MS ion source parameters including nebuliser gas (NEB), curtain gas (CUR), collision gas (collision-activated dissociation (CAD)), ion spray voltage (IS) and ion source temperature (TEM) were 12 psi, 8 psi, 6 L min−1, 3500 V and 500 °C, respectively.
Method Validation
The LC-MS/MS method was validated in compliance with European Commission Decision 2002/657/EC. According to the criteria, the performance characteristics including recovery, repeatability, decision limit (CCα), detection capability (CCβ), calibration curves, stability and specificity, together with limit of detection (LOD) and limit of quantification (LOQ) in porcine tissue (muscle, liver and kidney) matrices, were evaluated. The sums of gentamicin C2, C2a and C2b were determined as GENT C2 due to the same molecule and mass fragment of them. The robustness of the method was demonstrated by continuous verification of the validation parameters during actual application of the method for survey samples.
Calibration Curves
Calibration was performed using matrix-matched standards prepared by adding the appropriate amounts of the aminoglycoside-mixed standard solutions to each blank matrix at six concentration levels, corresponding to 0, 0.25, 0.5, 1.0, 2.5 and 5.0 times MRL or suggested level (for compounds HYGRO, AMIK, TOBRA, NETIL and SISO, of which there was no MRL in porcine tissue matrices) for muscle; 0, 0.2, 0.4, 0.8, 2.0 and 4.0 times MRL or suggested level for liver; and 0, 0.2, 0.5, 1.0, 2.5 and 5.0 times MRL or suggested level for kidney. Those samples fortified with AGs were operated with entire extraction and purification procedure and finally injected to the LC-ESI-MS/MS system. The calibration curves were constructed using linear regression of the peak areas from the six concentration levels versus the concentration of analytes. Thus, there were always 15 different matrix-matched calibration curves for each test sample (one calibration curve per analyte).
Recovery and Precision
Porcine muscle, liver and kidney known to be compliant served as blank matrices. Recoveries and precision (intra-day, inter-day) were calculated from the determination of seven aliquots of each sample fortified at three levels (0.5, 1.0 and 1.5 times MRL or 0.5, 1.0 and 1.5 times suggested level for no MRL substances). Seven blank samples for each matrix were included in each series, taken through the entire extraction and purification procedure. After being processed, five of these blank samples were used to prepare matrix-matched recovery standards for recovery calculation. Aminoglycoside-mixed standard solutions were added to the dried extracts to obtain concentrations corresponding to 0.25, 0.5, 1.0, 1.5 and 2.0 times MRL/suggested level for muscle, liver and kidney. The analyses were performed by the same operator in triplicate within a 1-month period. The recoveries were calculated by the measured content/the fortified level × 100, and the precision was expressed as the relative standard deviation (RSD).
Decision Limit (CCα) and Detection Capability (CCβ)
The decision limit (CCα) is the lowest concentration at which a method can discriminate with a statistical certainty of 1 − α that the analyte is present. For all 15 AGs, CCα was established by the following: 21 blank matrix samples of porcine muscle, liver and kidney spiked at the MRL/suggested level were analysed. The average measured concentration at the MRL/suggested level plus 1.64 times the corresponding standard derivation (SD) is defined as CCα (α = 5 %).
The detection capability (CCβ) is the concentration at which the method is able to detect truly contaminated samples with a statistical certainty of 1 − β. For all 15 AGs, CCβ was calculated by analysing 21 porcine muscle, liver and kidney spiked with the analytes at CCα and then the CCα value plus 1.64 times the corresponding SD is equal to CCβ (β = 5 %).
Limit of Detection and Limit of Quantification
The limit of detection (LOD) and limit of quantification (LOQ), defined as three and ten times the S/N ratio for each compound, respectively, were calculated using the results from at least six aliquots of spiked negative samples (porcine muscle, liver and kidney). Afterwards, negative samples (porcine muscle, liver and kidney) were fortified with the concentration of calculated LOD and LOQ (six replicates) at each level to confirm.
Specificity
The specificity of the method was demonstrated by testing all matrices available in our research. Briefly, 20 muscle, 20 kidney and 20 liver (n = 60) samples were studied, which were fortified with identical concentration (40–500 μg kg−1) for 15 AGs. The results were evaluated by the presence/absence of interfering substances around the AGs’ retention time.
Stability
It is well known that insufficient stability of the analytes during storage or analysis may give rise to significant deviations of the result of analysis (EC Decision 2002). Consequently, stability must be taken into account during the validation of residue analysis. In order to simplify the operation, some important factors were investigated: (1) stability in different solvents, where all AGs (1000 μg mL−1) were dissolved in acetonitrile, acetonitrile/water (50:50, v/v) or acetonitrile/water/acetic acid (20:78:2, v/v/v); (2) stability under different storage conditions, where the stock solutions (1000 μg mL−1) and lower concentration solutions (e.g. 100 ng mL−1) were stored at room temperature (about 25 °C), 4 °C or −20 °C; and (3) stability in different containers, where the stock solutions (1000 μg mL−1) and lower concentration solutions (100 ng mL−1) were placed in glass or plastic tubes. The measured values were compared to those of freshly prepared standard solutions.
Results and Discussion
Optimisation of Sample Preparation Procedure
The sample pre-treatment procedures were studied step by step based on porcine muscle matrix, including extraction solvent, centrifuge conditions and solid phase extraction conditions. An IKA® T25 digital Ultra-Turrax® high-speed blender with IKA® works S25N-25F dispersing was used for sample homogenisation.
For centrifugation at 6953×g, the temperature of 5, 10 and 20 °C was studied. The centrifuge time required producing clear supernatants prolonged with temperature, and therefore, 5 °C was chosen. Considering pork muscle containing a lot of fat, a de-fatting step was introduced to remove the remaining fat in the HFBA-acidified TCA extracts. A 5 mL volume of n-hexane was added to extract the fat from the HFBA-acidified TCA extracts before loading onto the HLB cartridge and compared to no n-hexane addition series. The result showed that the recoveries of all 15 AGs improved when 5 mL of n-hexane was added for de-fatting.
The influence of HFBA concentration used for pre-conditioning of HLB cartridge was investigated by testing HFBA at 0.02, 0.05, 0.10, 0.15, 0.20, 0.25, 0.30 and 0.40 mol L−1. It was observed that the recoveries of most AGs increased with HFBA concentration and achieved a maximum value at 0.20 mol L−1. Since there was no further significant improvement of recovery at higher concentrations, the 0.20 mol L−1 HFBA was adopted.
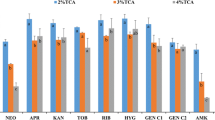
Since the 15 AGs have different pK a values, the pH of the solution loaded onto the columns plays a key role in method performance. Thus, HFBA-acidified TCA extracts were adjusted to pH 0.5, 1.0, 1.5, 2.0, 7.0, 7.5, 8.0, 8.5, 9.0, 9.5, 10.0, 10.5 and 11.0 in duplicate and then loaded onto conditioned HLB columns to assess the performance under different pH conditions. Only nine AGs had optimum extraction yields at pH 1.0, while high recoveries for the other six AGs (SPEC, HYGRO, STREP, DISTREP, AMIK and KANA) were found at pH 8.5 (the results are shown in Fig. 2). After many trials, it was impossible to quantitatively retain and elute all 15 AGs on only one HLB column. The optimised overall performance could be obtained with two coupled HLB columns, each under different pH conditions. Additionally, the HFBA-acidified TCA extracts without pH adjustment showed similar recovery behaviour to those adjusted to pH 1.0. Therefore, the n-hexane de-fatted and HFBA-acidified TCA extracts were directly loaded onto the first HLB cartridge, from which the effluent was collected and adjusted to pH 8.5 for clean-up on the second HLB cartridge.

Recoveries of target drugs upon SPE clean-up in porcine muscle extracts with different pH conditions
Considering that the pH value of effluent collected from the first column was adjusted to 8.5 before applying to the second column, the pre-conditioning of the second HLB cartridge was investigated under different pH conditions. The second cartridge was pre-conditioned sequentially with 3 mL methanol, 3 mL water and 3 mL aqueous NaOH with different pH values. The optimum conditions were found to be 3 mL methanol, 3 mL water, 3 mL of 0.2 mol L−1 HFBA and 3 mL of pH 8.5 aqueous NaOH.
The elution procedure of the two HLB cartridges was also investigated by comparing the elution efficiency of different solvents (at different volumes), 6 mL acetonitrile, 6 mL methanol/0.2 mol L−1 HFBA (4:1, v/v), 3 mL methanol plus 3 mL methanol/ammonium hydroxide (9:1, v/v) and 6 mL acetonitrile/0.2 mol L−1 HFBA (4:1, v/v). From the results obtained, 6 mL of acetonitrile/0.2 mol L−1 HFBA (4:1, v/v) showed the best elution efficiency (Fig. 3). Finally, different compositions of the acetonitrile/HFBA mixture were investigated, of which elution efficiencies were compared. The best elution efficiency was found using 6 mL of acetonitrile/0.15 mol L−1 HFBA (4:1, v/v), and so, this was applied for elution of the 15 AGs from the two tandem HLB cartridges.

The elution efficiency of different solvents in the SPE clean-up procedure
Several other preparation methods for AGs in foods of animal origin have been published, using different commercially available SPE cartridges (Varian AccuBond ODS-C18, Waters Oasis MCX, Waters Oasis WCX, Supelclean™ LC-WCX). These alternatives were also evaluated, but it was observed that not all of the target AGs in our study were quantitatively retained or eluted on the columns.
Mass Spectrometry and HPLC Separation
Each aminoglycoside tuning solution was directly injected into the electro ion spray source by a syringe pump. Full scan and CAD tests were operated to set up an appropriate MRM method. Protonated molecular ions [M+H]+ were the dominant ions produced for most of the AGs in positive ESI mode and were selected as precursor ions. Exceptions were SPEC and STREP, which produced intensive water adducts [M+H2O+H]+.
According to EU criteria, a minimum of three identification points (IPs) is required for the confirmation of group B substances in foodstuffs (EC Decision 2002). Our method fulfilled this requirement with the use of two MRM transitions (one precursor and two product ions) for each compound, which count for four IPs.
The LC gradient program had a run time of 18 min. Good peak shapes and separation were achieved, and all 15 AGs had sufficient data points in both mixed standard solutions and fortified samples. MRM chromatograms are shown in Fig. 4.

MRM chromatograms of a spiked pork sample with 1.0 times MRL/suggested level for 15 aminoglycosides at 50–1000 μg kg−1 obtained by LC-ESI-MS/MS
Method Performance and Validation
Table 3 summarises all performance characteristics of this procedure. The peak areas and concentrations were fitted to a linear equation in the range of zero to five times MRL or suggested level. For each compound, three calibration curves were obtained. There were no significant differences between the curves. The correlation coefficients (r) for the calibration curves were all greater than 0.99. As can be observed in Table 3, the recoveries ranged from 47 to 93 % with overall precisions of 2.9–15.4 %. The CCα and CCβ values were in the range of 52–20,545 and 54–21,298 μg kg−1 for all analytes, respectively. The LOD and LOQ for all AGs ranged from 0.9 to 9.0 μg kg−1 and from 3.0 to 30 μg kg−1, respectively. Furthermore, the calculated LOQ levels were confirmed by spiking negative matrices. The results are shown in Fig. 5.

MRM chromatograms (quantitative ion transition shown) of apramycin in three porcine tissue matrices fortified at corresponding LOQ levels
Scrutiny of the background noise in all three matrices demonstrated that there were no interfering peaks that could be detected at the expected retention time window (±2.5 %) for the target analytes.
The optimum conditions for storage of all AGs were as follows: the compounds (1000 μg mL−1) should be dissolved with acetonitrile/water/acetic acid (20:78:2, v/v/v) and stored in plastic tubes at 4 °C. The stock solutions and lower concentration solutions (100 ng mL−1) are stable in plastic tubes at 4 °C for at least 1 year and for 1 week, respectively.
Method Application
About 100 porcine tissue samples (60 muscles, 20 livers and 20 kidneys) were collected from local supermarkets in different cities of Guangdong Province (southern China). All the samples were transferred to our laboratory and analysed in triplicate according to the optimised procedure. The confirmatory and quantitative results of the real samples are summarised in Table 4, including the ion ratio of two transitions of each analyte in incurred samples and the matrix-matched standard solutions. The results of relative ion abundance for detected AGs in incurred samples and matrix-matched standard solutions indicate that the deviations of the ion ratio meet the relevant requirements of EU (EC Decision 2002). Only KANA and STREP were simultaneously found in two porcine kidneys at a concentration of 991 ± 67 μg kg−1 (KANA) and 346 ± 30 μg kg−1 (STREP), and 29,114 ± 2475 μg kg−1 (KANA) and 14,600 ± 1343 μg kg−1 (STREP), respectively.
Conclusions
A LC-MS/MS method was developed and validated for the quantitative determination of 15 AGs in porcine muscle, liver and kidney. Baseline separation of all 15 AGs was achieved within an 18-min gradient elution program. The method showed good sensitivity, and the performance characteristics of the method comply with EU recommendations. Sufficient identification points were generated for the confirmation of the identity of all 15 target AGs according to EU criteria. The method was successfully applied to determine AGs in porcine tissue samples from local markets in southern China. In terms of overall data and consideration, this method should be an efficient approach for multi-residue analysis of AGs in animal tissues.
References
Babin Y, Fortier S (2007) A high-throughput analytical method for determination of aminoglycosides in veal tissues by liquid chromatography/tandem mass spectrometry with automated cleanup. J AOAC Int 90:1418–1426
Berrada H, Moltó JC, Mañes J, Font G (2010) Determination of aminoglycoside and macrolide antibiotics in meat by pressurized liquid extraction and LC-ESI-MS. J Sep Sci 33:522–529
Bogialli S, Curini R, Corcia AD, Laganà A, Mele M, Nazzari M (2005) Simple confirmatory assay for analyzing residues of aminoglycoside antibiotics in bovine milk: hot water extraction followed by liquid chromatography–tandem mass spectrometry. J Chromatogr A 1067:93–100
Chen YQ, Hu XZ, Xiao XL (2010) Sample preparation for determination of neomycin in swine tissues by liquid chromatography-fluorescence detection. Anal Lett 43:2496–2504
Cherlet M, Baere SD, Backer PD (2007) Quantitative determination of dihydrostreptomycin in bovine tissues and milk by liquid chromatography-electrospray ionization-tandem mass spectrometry. J Mass Spectrom 42:647–656
Commission Regulation (EU) No 37/2010 of 22nd December, 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Official Journal of the European Union.
EC Decision 2002/657 (2002) Official Journal of the European Communities, L 221: 8–36
Edder P, Cominoli A, Corvi C (1999) Determination of streptomycin residues in food by solid-phase extraction and liquid chromatography with post-column derivatization and fluorometric detection. J Chromatogr A 830:345–351
Granja RHMM, Niño AMM, Zucchetti RAM, Niño REM, Patel R, Salerno AG (2009) Determination of streptomycin residues in honey by liquid chromatography–tandem mass spectrometry. Anal Chim Acta 637:64–67
Gremilogianni AM, Megoulas NC, Koupparis MA (2010) Hydrophilic interaction vs ion pair liquid chromatography for the determination of streptomycin and dihydrostreptomycin residues in milk based on mass spectrometric detection. J Chromatogr A 1217:6646–6651
Heller DN, Peggins JO, Nochetto CB, Smith ML, Chiesa OA, Moulton K (2005) LC/MS/MS measurement of gentamicin in bovine plasma, urine, milk, and biopsy samples taken from kidneys of standing animals. J Chromatogr B 821:22–30
Holthoon FLV, Essers ML, Mulder PJ, Stead SL, Caldow M, Ashwin HM, Sharman M (2009) A generic method for the quantitative analysis of aminoglycosides (and spectinomycin) in animal tissue using methylated internal standards and liquid chromatography tandem mass spectrometry. Anal Chim Acta 637:135–143
Hornish RE, Wiest JR (1998) Quantitation of spectinomycin residues in bovine tissues by ion-exchange high-performance liquid chromatography with post-column derivatization and confirmation by reversed-phase high-performance liquid chromatography–atmospheric pressure chemical ionization tandem mass spectrometry. J Chromatogr A 812:123–133
Hosokawa S, Nakamura K, Fujita Y, Horiuchi R, Yamamoto K (2008) Determination of isepamicin in human plasma by HPLC with fluorescence detection after derivatization using 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate. Biol Pharm Bull 31:1866–1869
Joint FAO/WHO food standards programmed codex committee on residues of veterinary drugs in foods. Sixteenth session. CX/RVDF 06/16/13 (Part 2) October 2005.
Kaufmann A, Butcher P, Kölbener P (2003) Trace level quantification of streptomycin in honey with liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Sp 17:2575–2577
Kaufmann A, Maden K (2005) Determination of 11 aminoglycosides in meat and liver by liquid chromatography with tandem mass spectrometry. J AOAC Int 88:1118–1125
Kaufmann A, Butcher P, Maden K (2012) Determination of aminoglycoside residues by liquid chromatography and tandem mass spectrometry in a variety of matrices. Anal Chim Acta 711:46–53
Kowalski P, Oledzka I, Okoniewski P, Switala M, Lamparczyk H (1999) Determination of streptomycin in eggs yolk by capillary electrophoresis. Chromatographia 50:101–104
Löffler D, Ternes TA (2003) Analytical method for the determination of the aminoglycoside gentamicin in hospital wastewater via liquid chromatography–electrospray-tandem mass spectrometry. J Chromatogr A 1000:583–588
McGlinchey TA, Rafter PA, Regan F, McMahon GP (2008) A review of analytical methods for the determination of aminoglycoside and macrolide residues in food matrices. Anal Chim Acta 624:1–15
Oertel R, Neumeister V, Kirch W (2004) Hydrophilic interaction chromatography combined with tandem-mass spectrometry to determine six aminoglycosides in serum. J Chromatogr A 1058:197–201
Posyniak A, Zmudzki J, Niedzielska J (2001) Sample preparation for residue determination of gentamicin and neomycin by liquid chromatography. J Chromatogr A 914:59–66
Preu M, Guyot D, Petz M (1998) Development of a gas chromatography–mass spectrometry method for the analysis of aminoglycoside antibiotics using experimental design for the optimisation of the derivatisation reactions. J Chromatogr A 818:95–108
Stead DA (2000) Current methodologies for the analysis of aminoglycosides. J Chromatogr B 747:69–93
Tao YF, Chen DM, Yu H, Huang LL, Liu ZY, Cao XQ, Yan CX, Pan YH, Liu ZL, Yuan ZH (2012) Simultaneous determination of 15 aminoglycoside(s) residues in animal derived foods by automated solid-phase extraction and liquid chromatography–tandem mass spectrometry. Food Chem 135:676–683
Tawa R, Matsunaga H, Fujimoto T (1998) High-performance liquid chromatographic analysis of aminoglycoside antibiotics. J Chromatogr A 812:141–150
The Japan Food Chemical Research Foundation (2006) The Japanese positive list system for agricultural chemical residues in foods (enforcement on May 29)
Viñas P, Balsalobre N, Hernández-Córdoba M (2007) Liquid chromatography on an amide stationary phase with post-column derivatization and fluorimetric detection for the determination of streptomycin and dihydrostreptomycin in foods. Talanta 72:808–812
Zhu WX, Yang JZ, Wei W, Liu YF, Zhang SS (2008) Simultaneous determination of 13 aminoglycoside residues in foods of animal origin by liquid chromatography–electrospray ionization tandem mass spectrometry with two consecutive solid-phase extraction steps. J Chromatogr A 1207:29–37
Acknowledgments
This work has been financially supported by the International Atomic Energy Agency (Research Contract No. 15598).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants performed by any of the authors.
Conflict of Interest
Zhou Zhu declares that he has no conflict of interest. Guihua Liu declares that she has no conflict of interest. Fu Wang declares that he has no conflict of interest. James Jacob Sasanya declares that he has no conflict of interest. Andrew Cannavan declares that he has no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhu, Z., Liu, G., Wang, F. et al. Development of a Liquid Chromatography Tandem Mass Spectrometric Method for Simultaneous Determination of 15 Aminoglycoside Residues in Porcine Tissues. Food Anal. Methods 9, 2587–2599 (2016). https://doi.org/10.1007/s12161-016-0446-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0446-1