
Abstract
The goal of this research was to evaluate the application of Quick, Easy, Cheap, Effective, Rugged, and Safe (QuEChERS) method for the determination of organochlorine, organophosphate, and carbamate pesticides in fatty animal matrices such as liver of chicken obtained from National Research Institute of Animal Production in Balice (Poland). Pesticides extraction effectiveness was evaluated at two different spiking levels (0.010 and 0.020 mg kg−1) and efficiency of the dispersive solid-phase extraction (d-SPE) clean-up step was evaluated by comparison adding different d-SPE sorbent combinations (PSA + GCB, PSA + C18, PSA + SAX, and PSA + NH2). The analysis of pesticide residues was performed by gas chromatography ion trap mass spectrometry (GC/IT-MS). The linear relation was observed from 0 to 400 ng mL−1 and the determination coefficient R 2 > 0.997 in all instances for all target analytes. Better recoveries were obtained in samples at 0.020 mg kg−1 spiking level. The recoveries were in the range 70–120 %, with relative standard deviation (RSD) values lower than 15 % at 0.020 mg kg−1 spiking level for most pesticides. Similar recovery ratios were obtained with the four different combinations of sorbents tested in the clean-up step, with better precision when the (PSA + SAX) combination was tested. Limits of detection (LODs) ranged from 0.001 to 0.005 mg kg−1 and limits of quantification (LOQs) ranged from 0.003 to 0.015 mg kg−1. The proposed method was successfully applied analyzing pesticide residues in real chicken liver samples; detectable pesticide residues were observed, but in all of the cases, the contamination level was lower than the default maximum residue levels (MRLs) set by European Union (EU), Regulation (EC) N 396/2005.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pollution by persistent chemicals is potentially harmful to the organisms at higher tropic levels in the food chain. Since diet is the main source of chronic exposure to low doses of these substances, humans are mainly exposed to these chemicals through ingestion. The chronic effects of pesticides from food intake on human health are not well defined, but there is increasing evidence of carcinogenicity and genotoxicity, as well as disruption of hormonal functions (LeDoux 2011).
Insecticides are used in henhouses to control poultry; chickens can then be accidentally exposed to these chemicals. Poultry can also be contaminated by feeding on plant materials that have been treated with pesticides during the growing and/or storage stages (LeDoux 2011). Poultry liver is considered to be one of the most important sources of mineral nutrients in humans’ diet, but due to its specific structure tends to bind chemical contaminants such as pesticides (Ghimpeteanu et al. 2012). Organochlorine pesticides (OCPs) are easy to bioaccumulate into fatty tissues as fat meat, egg yolk, or liver, due to their lipophilic nature and great stability, and because of that, they are considered as persistent organic compounds (POPs) and they can easily reach the food chain and concentrate in human and animal tissues (Cajka et al. 2012; Garrido-Frenich et al. 2007). Despite of organophosphorus pesticides (OPPs) are less persistent than OCPs, they can also reach the food chain, and liver is one of the lipophilic tissues of the animal anatomy in which pesticides can be found, especially OPPs which are metabolized in this organ (Garrido-Frenich et al. 2007).
Fifteen of the 24 chemicals targeted by the Stockholm Convention are OCPs (Stockholm 2009). Studies on the concentration of OCPs in the environmental showed that emission sources of these compounds, such as DDT, in the last 20 years have moved from industrialized countries to developing countries, due either to the late production ban in these countries or to the use in agriculture and control diseases such as malaria, typhus, and cholera (Choi et al. 2009). Today, OCPs have been banned for agricultural or domestic uses in agreement with the Stockholm Convention (Gomes-Martins et al. 2013).
Maximum residue limits (MRLs) have been set for pesticides in edible tissues (muscle, liver, and kidney) with the aim of minimizing the risk to human health associated with their consumption (Kinsella et al. 2009a). However, Regulation (EC) N 396/2005 (Regulation EC/396/2005), brought into force on 1 September 2008, defines a new fully harmonized set of rules for pesticide residues. New regulatory frameworks require sensitive and highly specific methods for the measurement of pesticide residues (Cieslik et al. 2011).
For complex matrices, such as chicken liver, an extraction phase development is generally required for lipid and co-extractives removal (Garrido-Frenich et al. 2007; Lazartigues et al. 2011). Up to now, many sample preparation techniques were reported for determination of pesticide residues in foodstuffs, including liquid-liquid extraction (LLE), solid-phase extraction (SPE), accelerated solvent extraction (ASE), gel permeation chromatography (GPC), microwave-assisted extraction (MAE), matrix solid-phase dispersion (MSPD), supercritical fluid extraction (SFE), and solid-phase microextraction (SPME) (Zheng et al. 2013). However, most of the aforementioned sample preparation techniques for the determination of pesticide residues in food are rather complicated, consume a large volume of solvent, and are labor-intensive and very expensive (Park et al. 2011). Despite considerable progress in the development of methods for sample preparation and determination of pesticides has been achieved, two problems remain: the complexity and the diversity of matrices, and the low concentrations of pesticides in samples (Hou et al. 2013). In 2003, a new approach of multiresidues determination named as Quick, Easy, Cheap, Effective, Rugged, and Safe (Anastassiades et al. 2003) (QuEChERS) method has been developed based on initial extraction with acetonitrile followed by liquid-liquid partitioning step after addition of a mixture of anhydrous MgSO4 and NaCl and then cleaning up by dispersive solid-phase extraction (d-SPE) in which the extract is mixed with primary secondary amine (PSA) sorbent and anhydrous MgSO4 (Cajka et al. 2012; Hou et al. 2013). Modifications of QuEChERS method can be made by adjusting solvents, salts volumes, water content, and clean-up sorbents (Castillo et al. 2011). Several modifications of the original method are present in the literature, to adjust the method to a specific application. Recently, modified QuEChERS method has been applied on fatty complex matrices such as olives (Gilbert-Lopez et al. 2010), avocado (Rajski et al. 2013), peanut oil (Su et al. 2011), fish and fish feed (Kalachova et al. 2013; Lazartigues et al. 2011), or bovine milk (Dagnac et al. 2009); however, there is no information in the literature about determination of pesticide residues in chicken liver by QuEChERS methodology. The main advantages of the QuEChERS method are low consumption of reagents and solvents, generation of small amounts of waste, simplicity of the operations, low cost of analysis, efficient removal of matrix components, and high recoveries of the analyzed compounds.
In this work, we proposed a rapid, efficient, and reliable method based on modified QuEChERS method, evaluating the efficiency of dispersive-SPE clean-up stage by comparison different d-SPE sorbent combinations (PSA + GCB, PSA + C18, PSA + SAX, and PSA + NH2); for simultaneous gas chromatography ion trap mass spectrometry (GC/IT-MS) determination of a group of OCPs (α-HCH, hexachlorobenzene, β-HCH, lindane, δ-HCH, heptachlor, aldrin, heptachlor epoxide, γ-chlordane, α-chlordane, endosulfan, o,p′-DDE, dieldrin, endrin, 4,4′-DDD, endrin aldehyde, 4,4′-DDT, endosulfan sulfate, and methoxychlor), OPPs (diazinon, disulfoton, methyl parathion, parathion, ethion, and azinphos methyl), and carbamate pesticides (3-hydroxycarboburan, 1-naphthol, carbofuran, and carbaryl) in fatty animal matrices such as liver of chicken.
Materials and Methods
Reagents and Chemicals
Acetonitrile HPLC grade, hexane GC grade, chloroform p.a., and formic acid 98 % p.a. were purchased from Merck KGaA, Germany. Magnesium sulfate anhydrous p.a. and sodium chloride p.a. were purchased from Chempur, Poland. Trisodium citrate dihydrate p.a. and disodium hydrogencitrate sesquihydrate 99 % p.a. were obtained from Sigma-Aldrich Chemie GmbH, Germany. PSA SPE Bulk Sorbent 25 g bottle, Carbon SPE Bulk Sorbent 25 g bottle, C18 Endcapped SPE Bulk Sorbent 25 g bottle, Silica SAX SPE Bulk Sorbent 25 g bottle, and Amino NH2 Bulk Sorbent 25 g bottle were purchased from Agilent Technologies, USA. Organochlorine Pesticide Mix, Chlorobenzene Mix, 531.1 Carbamate Pesticide Mix, and Mirex internal standard (IS) were purchased from Supelco, Bellefonte, USA. Organophosphorous Pesticide Mix 1 and triphenyl phosphate (TPP) internal standard (IS) were purchased from Dr. Ehrenstorfer GmbH, Germany.
Samples
In the present work, we focused on the determination of pesticide residues in fatty animal matrices such as liver of chicken obtained in 2012 from National Research Institute of Animal Production in Balice (Poland). Prior to determination of pesticide residues, the determination of fat content in chicken liver samples was performed according to PN-EN ISO 734-1:2008 (PN-EN ISO 734-1:2008). Chicken liver samples resulted a 5 % fat content.
Modified QuEChERS Sample Preparation Method
A representative portion of each sample was blended, macerated, and homogenized in a mortar. The homogenized samples with no pesticides detected previously were subsequently used for recovery studies. In order to calculate the recovery, the samples were spiked with the standard solution at two fortification levels, 0.010 and 0.020 mg kg−1.
An aliquot of 2.5 g of homogenized was weighted into a 50-mL centrifuge tube. Five milliliters of water and 10 mL of acetonitrile were added to the samples and the mixture was shaken vigorously for 1 min. Spiked samples were mixed and left to stand for 15 min at room temperature prior to extraction. After that, 0.5 g Na2HCit · 1.5H2O, 1 g Na3Cit · 2H2O, 1 g NaCl, and 4 g MgSO4 were added, tube was shaken by hand immediately after salt addition, 1 mL of chloroform was added, and the shaking process was repeated for 1 min. Next, the sample was shaken vigorously for 1 min and centrifuged for 15 min at 8,700 RCF. Six milliliters of the supernatant was transferred into a 15-mL centrifuge tube containing 1 g MgSO4 and the different d-SPE sorbent combination (PSA + GCB, PSA + C18, PSA + SAX, and PSA + NH2) (150 mg PSA + 200 mg other d-SPE sorbent); two replicates were prepared for each test. The tube was shaken by hand for 1 min and centrifuged for 5 min at 5,000 RCF. Further, 4 mL of the extracts were transferred into screw cup vial and acidified with 40 μL of 5 % formic acid in acetonitrile, and finally, 100 μL of the mirex and TPP solution were added. The extracts were evaporated under the stream of N2 at temperature of 40 °C from 4 mL to dryness, then the extracts were re-dissolved in 1 mL of hexane and stored in a freezer at temperature of −26 °C for overnight; finally, the extracts were separated from the precipitates by simple decantation. The cleaned, acidified, and re-dissolved extracts were used for the multiresidue determination by gas chromatography-mass spectrometry (GC-MS); three injections were realized for each extract.
Equipment
Varian 4000 GC/MS (Varian, Inc., USA) system consisted of 3800 GC with CP-8410 autoinjector (Bruker, USA) and 4000 Ion Trap MS detector was used to perform the GC/MS analyses. CP-1177 Split/Splitless Capillary Injector was used, which the temperature was 270 °C and injection volume was 1.0 μL with the splitless time of 1.0 min for all standards and samples. Each injection was repeated three times. Chromatographic separations were done by using a Zebron Multiresidue-1 column (30 m L × 0.25 mm i.d. × 0.25 μm df; Phenomenex, Inc., USA). The GC oven was operated with the following temperature program: initial temperature 70 °C (0.30 min)–10 °C min−1–210 °C (1.0 min)–5 °C min−1–230 °C (3.0 min)–7 °C min−1–250 °C (1.70 min)–20 °C min−1–300 °C (1.0 min). The total analysis time was 30.36 min. Helium (99.999 %) (Linde Gas, Poland) was used as the GC carrier gas at a flow rate of 1.0 mL min−1. Ion trap mass spectrometer was operated in the internal ionization and in a full scan mode in the range 45–400 m/z. The trap and the transfer line temperatures were set at 180 and 220 °C, respectively. The analyses were carried out with the solvent delay of 4.50 min to prevent instrument damage. The emission current of the ionization filament was set at 15 μA. Analysis was conducted in the selected ion monitoring (SIM) mode, based on the use of one quantifier and two qualifier ions. Acquisition and processing data were performed using Varian Star Workstation software and NIST library 2.0.
MPW 350 R Centrifuge (MPW Med. Instruments, Poland) was used for samples preparation. AccuTM Thermoblock (Labnet, USA) with nitrogen (5.0) (Linde Gas, Poland) was used to evaporate the solvent and concentrate the extracts. AdventureTM Pro Balance (Ohaus, Switzerland) was used to weigh the chopped samples and solid reagents.
Results
Optimization of the Extraction and Clean-Up Steps
Sample preparation is one of the most important steps in trace pesticide analysis, with a direct and important influence at both the quantification and detection limits achieved. The extraction efficiency strongly depends on the organic solvents used, on the nature of the sample, and on the chemical properties of the pesticide residues (Cunha and Fernandes 2011). Fatty animal matrix such as chicken liver has many matrix components that have similar properties as the pesticides of interest, thus traditional solvents extractions are not going to separate these matrix chemicals from the analytes. To minimize or eliminate co-extraction of lipids, we chose to use acetonitrile as the extraction solvent because very little fat partitions into acetonitrile (Cunha et al. 2007). The addition of chloroform was included to drive water from the acetonitrile phase and thus effectively remove both the salts and the very polar matrix components from the extract (Liu et al. 2011). The lipophilic pesticides remain or partite into the undissolved fats, which results in their lower recovery in the acetonitrile extract (Lehotay et al. 2005). Hence, at the last stage of the procedure applicability of low-temperature precipitation clean-up, freezing out, was also evaluated as a practical way to reduce the amount of co-extracts; after that, the extract was separated from the precipitates by simple decantation; the process had to be done quickly on removing the extract from the freezer to prevent the precipitated matter from re-dissolving (Walorczyk 2008).
In order to minimize matrix effects pesticides extraction effectiveness was evaluated at two different spiking levels (0.010 and 0.020 mg kg−1) and combination of different d-SPE sorbents was studied in this work to improve the clean-up procedure since the use of single sorbent could not completely remove the co-extractants and eliminate the interference peaks present in the chromatograms. PSA was chosen to combine with graphitized carbon black (GCB), C18, strong anion exchange (SAX), and NH2. In pesticide analysis, PSA is the most common sorbent used (Kinsella et al. 2009b). PSA and NH2 can act both as a polar phase and weak anion exchangers with the ability to remove fatty acids, sugars, and other matrix co-extractives. PSA removes more matrix co-extractives than NH2 per given quantity, because PSA has both a primary and secondary amine (Hercegova et al. 2007; Ru-zhen et al. 2011). GCB has been reported to be a highly effective sorbent for sample clean-up (Kinsella et al. 2009b). GCB is nonporous sorbent based on reversed phase and removes planar molecules such as natural pigments (e.g., chlorophyll, hemoglobin, and carotenoids) from sample matrices (Hercegova et al. 2007; Ru-zhen et al. 2011). C18 is the most hydrophobic sorbent based on reversed phase, because of its extreme retentive nature for non-polar compounds such as fat (Agilent 2014a; Agilent 2014b). SAX is a strong anion exchange sorbent ideally suited for the extraction of compounds such as carboxylic acids (Agilent 2014a; Agilent 2014b). NH2 is a weaker anion exchanger than sorbent such as SAX (a quaternary amine sorbent that is always charged) and is therefore a better choice for retention of very strong anions, such as sulfonic acids (Agilent 2014a; Agilent 2014b). To achieve how efficient each clean-up step removed interferences, two replicates for each test were extracted and three injections were realized by each extract for the multiresidue determination by GC-MS.
Analytical Performance
The pesticides involved in this study were identified by comparing the retention time, three ions (one quantifier and two qualifiers), and mass spectra provided by the NIST Library 2.0. Pesticide analyses and confirmation were conducted in SIM mode based on the use of one quantifier and two qualifier ions and the method was divided into as many time segments as possible to obtain the maximum signal for pesticide. Table 1 summarizes retention time and three ions (one quantifier (in bold) and two qualifiers) monitored for each analyte.
Calibration curves were constructed by plotting the ratio of the peak area, divided by the peak area of the internal standard, against the analyte concentration. Six calibration standards at concentration of between 0 and 400 ng mL−1 were prepared in hexane by adding known quantities of standard mixture solution (2 μg mL−1) and 100 μL of the internal standard (mirex and TPP). Once prepared, the standards was realized the injection of 1 μL in the gas chromatograph-mass spectrometer to obtain the calibration curve for each pesticide. Three injections were realized for each standard obtaining good parameters of reproducibility, repeatability, and linearity in the calibration curves. Table 1 shows the calibration data of the standards; as can be seen, the response of the detector was linear for each pesticide in the range tested, 0 to 400 ng mL−1, with correlation coefficients (R 2) higher than 0.997 in all instances for all target analytes. LODs were determined as 3 × s/b, where b was the slope of the regression line and s was the standard deviation of the intercept; LOQs were determined as 3 × LODs (Nardelli et al. 2010). LODs ranged from 0.001 to 0.005 mg kg−1 and LOQs ranged from 0.003 to 0.015 mg kg−1. LODs and LOQs were lower than the MRLs established by European Union (EU) (Regulation EC/396/2005).
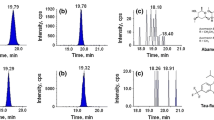
Two replicates for each test at two different spiking levels (0.010 and 0.020 mg kg−1) were extracted, and three injections were realized for each extract for the multiresidue determination by GC-MS to determine the accuracy of the method. A reagent blank (hexane) was also injected after every six sample injections to perform simple cleaning of the chromatographic system. The SIM chromatograms of the extracts demonstrated that no interference peaks were observed at the retention times of the target analytes; Fig. 1 shows selected GC-MS SIM mode chromatogram segments of spiked (0.020 mg kg−1) chicken liver samples purified by the different d-SPE sorbent combinations tested; no significant differences were seem in the chromatograms.

Selected GC-MS SIM mode chromatogram segments of spiked (0.020 mg kg−1) chicken liver samples. a (PSA + GCB) clean-up, b (PSA + C18) clean-up, c (PSA + SAX) clean-up, and d (PSA + NH2) clean-up. 1—α-HCH, 2—hexachlorobenzene, 3—carbofuran, 4—diazinon, 5—β-HCH, 6 —disulfoton, 7—lindane, 8—δ-HCH, 9—heptachlor, 10—methyl parathion, 11—carbaryl, 12—aldrin, 13—parathion, 14—heptachlor epoxide
The accuracy of the method was evaluated by the development of a recovery study by comparison adding different combinations of PSA with GCB, C18, SAX, or NH2. Comparison of the pesticide peak area to internal standard peak area ratio with the ratios obtained for calibration curve standards was performed to calculate the mean recovery values. SANCO guide establishes that a quantitative method should be demonstrated as being capable of providing mean recoveries within the range 70–120 % and relative standard deviations (RSD) lower than 20 % (Sanco, Document N. SANCO/12495/2011). Better recoveries were obtained in samples at 0.020 mg kg−1 spiking level. In all the cases, a sample free of analytes was analyzed to check the presence of these compounds; none of them gave a positive result above the LODs of the method. The recoveries were in the range 70–120 %, with relative standard deviation RSD values lower than 15 % at 0.020 mg kg−1 spiking level for most pesticides. Similar recovery ratios were obtained with the four different combinations of sorbents tested in the clean-up step. The precision of the method was evaluated in term of repeatability (intraday precision), expressed as the RSD of the spiked samples, repeatability was evaluated over two samples prepared and injected in triplicate on the same day under the same conditions; RSD was significantly better when the (PSA + SAX) combination was added during the d-SPE clean-up step, most of the studied pesticides gave RSD values lower than 15 % (Table 2).
Discussion
According to the data collected in Table 2 and looking at the chromatogram segments showed in Fig. 1, we can deduce that similar recovery ratios were obtained after addition of the four different d-SPE sorbent combinations (PSA + GCB, PSA + C18, PSA + SAX, and PSA + NH2) tested during the clean-up step in chicken liver samples, with better RSD values when the (PSA + SAX) combination was tested. Very poor (<70 %) recovery ratios were achieved at 0.010 mg kg−1 spiking level, while as it can be seen in Table 2, the second spiking level, 0.020 mg kg−1, gave more consistent results. The recoveries were in the range 70–120 %, with RSD values lower than 15 % at 0.020 mg kg−1 spiking level for most pesticides. Lindane isomers (α-HCH, β-HCH, lindane, and δ-HCH) gave slightly lower recoveries, with recovery ratios lower than 70 %, when (PSA + C18) combination was tested, it could be connected most with the use of C18 and its properties to retain some compounds, than with possible matrix effects (Lazartigues et al. 2011). For the analysis of liver, because of its high fat content, the use of C18 is reported to be beneficial (Kinsella et al. 2009a). The amount of sorbent is not a crucial factor, but too high amounts can result in cloudy extracts (Blasco et al. 2011). In d-SPE, the organic content of the extract has to be critically reviewed; when using C18. If the aqueous phase is not completely removed by phase separation and thus an excess of water is present, immediate loss of lipophilic compounds onto the sorbent material will occur (Berendsen et al. 2013). Low recoveries were achieved for hexachlorobenzene, phenomenon also observed by other authors, and justified by the use of GCB sorbent in the clean-up step, GCB removes sterols and pigments, as chlorophyll and hemoglobin, from the extracts in dispersive SPE, but it also strongly retains important pesticides with planar ring structures as hexachlorobenzene, thiabendazole, or chlorothalonil (Cunha et al. 2007; Hercegova et al. 2007; Lehotay et al. 2005).
The obtained results were in agreement with the results achieved by Garrido-Frenich et al. (2007) performing a multiresidue analysis of pesticides in chicken liver samples by gel permeation chromatography and gas chromatography using triple quadrupole tandem mass spectrometry; satisfactory results were found in 34 pesticides, with recoveries between 70 and 115 % and RSD lower to 20 %; however, some exceptions were observed in metamidophos (65 %) and heptachlor (62 %). Recently, other authors evaluated modified QuEChERS method for the determination of pesticide residues in fatty animal matrices, and the results achieved agree closely with our results. Kalachova et al. (2013) developed and validated a new method for rapid determination of 73 target organic environmental contaminants, including 16 organochlorinated pesticides, in fish tissues and fish feed using gas chromatography coupled with triple quadrupole tandem mass spectrometry (GC-MS/MS) coupled to QuEChERS-based extraction followed by silica minicolumn clean-up; the recoveries of all target analytes in both matrices were within the acceptable range of 70–120 % and the repeatabilities of the analytical procedure were 20 % or less at all three spiking levels; the newly developed method could not be applied for several OCPs—dieldrin, endrin, endosulfan sulfate, and endosulfan (α and β isomers)—for which the silica minicolumn clean-up is not suitable owing to specific steric interactions between the sorbent and the analyte. Sapozhnikova and Lehotay (2013) developed and evaluated a multiclass multiresidue method for the analysis of 68 organic environmental contaminants, including 18 representative pesticides, congeners in catfish muscle; the method was based on a QuEChERS extraction with acetonitrile and d-SPE clean-up with zirconium-based sorbent prior to fast low-pressure gas chromatography triple quadrupole tandem mass spectrometry (LP-GC/MS-MS) analysis; the recoveries of all but one analyte were between 70 and 120 % with relative standard deviations less than 20 % (n = 5).
On the other hand, the effectiveness of extraction/partitioning conditions was systematically investigated; the addition of chloroform was introduced. During the phase separation process, the acetonitrile can be “pushed out” from water by adding very polar substances, such as salts; at the same time, the water can also be pushed out from acetonitrile by adding nonpolar ones, such as hydrophobic solvent. These two processes can be performed simultaneously to obtain mutually promoted results. The little acetonitrile left in the aqueous phase promotes the analytes to be completely extracted, while little water in the acetonitrile phase decreases the co-extraction of salts and very polar matrix components and thus the analytical selectivity is enhanced and the harmful effect to the MS instrument may be reduced (Liu et al. 2011). The effectiveness of QuEChERS extraction and dispersive-SPE clean-up tested provided colorless, clean, and low viscous final extracts, and the GC-MS analysis performed in SIM mode improved the reliability and selectivity of the chromatograms, besides enhancing the cleanliness of the chromatograms, focusing on the analytes of interest to us. In SIM mode, sensitivity is enhanced by monitoring only few selected m/z ions, thus proportionally increasing the acquisition time of the ions of interest, but spectral information has to be sacrificed (Hajslova and Cajka 2006).
Application to Real Samples
The proposed method was successfully applied for determination of pesticide residues in real chicken liver samples obtained from National Research Institute of Animal Production in Balice (Poland). Five chicken liver samples were extracted, using the d-SPE sorbent combination (PSA + SAX) at the clean-up step, and analyzed as described in the sample preparation method section; detectable pesticide residues such as lindane isomers, endosulfan isomers, methoxychlor, DDT, and its metabolities were observed, but in all of the cases the contamination level was lower than the limit of quantification and therefore lower than the default maximum residue levels (MRLs) set by European Union (EU), (Regulation (EC) N 396/2005), force since 1 September 2008. The analysis of real samples revealed the persistence, relative stability, and bioaccumulation of organochlorine pesticides, even most of them are banned for agricultural or domestic uses in agreement with the Stockholm Convention (Stockholm 2009), and the analysis were performed in animal origin samples collected from protected area.
The existing literature provides little information about content of pesticide residues observed by other authors in animal liver samples. Garrido-Frenich et al. 2007 developed a multiresidue analysis of pesticides in animal liver by gel permeation chromatography and gas chromatography using triple quadrupole tandem mass spectrometry; α-endosulfan and endosulfan sulfate were detected in three different lamb samples, while endosulfan sulfate was detected only in one pork sample; no pesticides were detected in chicken liver samples; however, these positive samples did not exceed the default MRL value established by the EU. Garcia de Llasera and Reyes-Reyes (2009) validated a MSPD method for the extraction of organophosphorus pesticides from bovine samples, the proposed method was applied to the analysis of 20 real liver samples: 10 samples of healthy appearance and 10 samples of unhealthy appearance; the results showed that only two pesticides, chlorfenvinphos and chlorpyrifos, were detected in two different unhealthy samples, these positive samples did not exceed the LOD concentrations of these pesticides using the developed method.
Conclusions
An easy and quick method was developed to determine residues of 29 organochlorine, organophosphate, and carbamate pesticides in chicken liver. The method involves QuEChERS method extraction coupled with d-SPE clean-up and freezing out steps. The efficient clean-up procedure proposed allows the normal course of routine analysis because it significantly reduces the analysis time and the cost of the GC-MS instrument maintenance. d-SPE sorbent combination (PSA + SAX) was chosen as the most appropriate for the pesticide residues determination. Acceptable linearity, precision, and recoveries were obtained. The method was applied for analysis of chicken liver samples, detectable pesticide residues such as lindane isomers, endosulfan isomers, methoxychlor, DDT, and its metabolities were observed, but in all of the cases the contamination level was lower than the default maximum residue levels (MRLs) set by European Union (EU), Regulation (EC) N 396/2005. Results showed that modified QuEChERS method developed herein is simple, efficient, and reliable and exhibited acceptable levels of sensitivity and accuracy to fulfill the requirements of multiple pesticide residue analysis applied to chicken liver samples.
References
Agilent (2014a) Agilent sampliQ products for sample preparation. Agilent Technologies. http://www.chem.agilent.com/Library/brochures/5989-9334EN.pdf. Accessed Jun 2014
Agilent (2014b) Agilent bond elut silica-based SPE selection guide. Agilent Technologies. https://www.chromspec.com/pdf/e/ag78.pdf. Accessed Jun 2014
Anastassiades M, Lehotay S, Stajnbaher D, Schenck FJ (2003) JAOAC Int 86(22):412–431
Berendsen BJA, Stolker LAM, Nielen MWF (2013) Trends Anal Chem 43:229–239
Blasco C, Masia A, Gabriel-Morillas F, Pico Y (2011) JAOAC Int 94(3):991
Cajka T, Sandy C, Bachanova V, Drabova L, Kalachova K, Pulkrabova J, Hajslova J (2012) Anal Chim Act 743:51–60
Castillo M, Gonzalez C, Miralles A (2011) Anal Bioanal Chem 400:1315–1328
Choi MPK, Kang YH, Peng XL, Ng KW, Wong MH (2009) Chemosphere 77:714–719
Cieslik E, Sadowska-Rociek A, Molina-Ruiz JM, Surma-Zadora M (2011) Food Chem 125:773–778
Cunha SC, Fernandes JO (2011) J Chromatogr A 1218:7748–7757
Cunha SC, Lehotay S, Mastovska K, Fernandes JO, Oliveira MBPP (2007) J Sep Sci 30:620–632
Dagnac T, Garcia-Chao M, Pulleiro P, Garcia-Jares C, Llompart M (2009) J Chromatogr A 1216:3702–3709
Garcia de Llasera MP, Reyes-Reyes ML (2009) Food Chem 114:1510–1516
Garrido-Frenich A, Plaza-Bolaños P, Martinez-Vidal JL (2007) J Chromatogr A 1153:194–202
Ghimpeteanu OM, Toca C, Rosu GB, Mitranescu E, Barbuceanu F, Militaru M (2012) Scientific Works. C Series. Veterinary Medicine, Vol. LVIII Issue 4
Gilbert-Lopez B, Garcia-Reyes JF, Lozano A, Fernandez-Alba AR, Molina-Diaz A (2010) J Chromatogr A 1217:6022–6035
Gomes-Martins J, Amaya-Chavez A, Waliszewski SM, Colin-Cruz A (2013) Chemosphere 92:233–246
Hajslova J, Cajka T (2006) Gas chromatography-mass spectrometry (GC-MS). In: Pico Y (ed) Food toxicants analysis. Elsevier, Oxford, pp 419–473
Hercegova A, Domotorova M, Matisova E (2007) J Chromatogr A 1153:54–73
Hou X, Han M, Dai XH, Yang XF, Yi S (2013) Food Chem 138:1198–1205
Kalachova K, Pulkrabova J, Cajka T, Drabova L, Stupak M, Hajslova J (2013) Anal Bioanal Chem 405:7803–7815
Kinsella B, Lehotay SJ, Mastovska K, Lightfield AR, Furey A, Danaher M (2009a) Anal Chim Act 637:196–207
Kinsella B, O’Mahony J, Malone E, Moloney M, Cantwell H, Furey A, Danaher M (2009b) J Chromatogr A 1216:7977–8015
Lazartigues A, Wiest L, Baudot R, Thomas M, Feidt C, Cren-Olive C (2011) Anal Bioanal Chem 400:2185–2193
LeDoux M (2011) J Chromatogr A 1218:1021–1036
Lehotay S, Mastovska K, Yun SJ (2005) JAOAC Int 88(2):630–638
Liu G, Rong L, Guo B, Zhang M, Li S, Wu Q, Chen J, Chen B, Yao S (2011) J Chromatogr A 1218:1429–1436
Nardelli V, dell’Oro D, Palermo C, Centonze D (2010) J Chromatogr A 1217:4996–5003
Park JY, Choi JH, Abd El-Aty AM, Kim BM, Oh JH, Do JA, Kwon KS, Shim KH, Choi OJ, Shin SC, Shim JH (2011) Food Chem 128:241–253
PN-EN ISO 734-1:2008, Oilseed crush — determination of oil content — Part 1: method of extraction with hexane
Rajski L, Lozano A, Ucles A, Ferrer C, Fernandez-Alba AR (2013) J Chromatogr A 1304:109–120
Regulation EC/396/2005, Official Journal of the European Union, Regulation EC/396/2005
Ru-zhen Y, Jin-hua W, Ming-lin W, Rong Z, Xiao-yu L, Wie-hua L (2011) J Chromatogr Sci Vol 49:702–708
Sanco, Document N. SANCO/12495/2011. Method validation and quality control procedures for pesticide residues analysis in food and feed
Sapozhnikova Y, Lehotay SJ (2013) Anal Chim Act 758:80–92
Stockholm Convention (2009) http://chm.pops.int/Convention/ThePOPs/ListingofPOPs/tabid/2509/Default.aspx Accessed Jun 2014
Su R, Xu X, Wang X, Li D, Li X, Zhang H, Yu A (2011) J Chromatogr A 879:3423–3428
Walorczyk S (2008) J Chromatogr A 1208:202–214
Zheng HB, Zhao Q, Mo JZ, Huang YQ, Luo YB, Yu QW, Feng YQ (2013) J Chromatogr A 1300:127–133
Acknowledgments
The authors would like to thank TALENTIA Fellowship Program by the Ministry of Economy, Innovation, Science and Employment in Andalusia, Spain, for their financial support.
Conflict of interest
Juan Manuel Molina-Ruiz declares that he has no conflict of interest. Ewa Cieslik declares that she has no conflict of interest. Izabela Walkowska declares that she has no conflict of interest. This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Molina-Ruiz, J.M., Cieslik, E. & Walkowska, I. Optimization of the QuEChERS Method for Determination of Pesticide Residues in Chicken Liver Samples by Gas Chromatography-Mass Spectrometry. Food Anal. Methods 8, 898–906 (2015). https://doi.org/10.1007/s12161-014-9966-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-014-9966-8