
Abstract
Food lipid major components are usually analyzed by individual methodologies using diverse extractive procedures for each class. A simple and fast extractive procedure was devised for the sequential analysis of vitamin E, cholesterol, fatty acids, and total fat estimation in seafood, reducing analyses time and organic solvent consumption. Several liquid/liquid-based extractive methodologies using chlorinated and non-chlorinated organic solvents were tested. The extract obtained is used for vitamin E quantification (normal-phase HPLC with fluorescence detection), total cholesterol (normal-phase HPLC with UV detection), fatty acid profile, and total fat estimation (GC-FID), all accomplished in <40 min. The final methodology presents an adequate linearity range and sensitivity for tocopherol and cholesterol, with intra- and inter-day precisions (RSD) from 3 to 11 % for all the components. The developed methodology was applied to diverse seafood samples with positive outcomes, making it a very attractive technique for routine analyses in standard equipped laboratories in the food quality control field.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lipids and lipophilic compounds are among the most important biomolecules in living organisms. Under this label, a diversified amount of molecules is enclosed, from fatty acids to sterols or from phospholipids to isoprenoids, all presenting important biological functions (Abumrad et al. 2012). From the nutritional point of view, three classes are of major importance: fatty acids, sterols, and lipid-soluble vitamins. The lipid-soluble vitamins are, by definition, essential in numerous metabolic paths, being only acquired through the diet and dietary supplements. Among them, vitamin E, due to its lipophilic antioxidant activity, is also important from the technological point of view as it will be a determinant of the preservation of lipophilic compound integrity within the food itself. The fatty acid composition is determinant of the equilibrium of innumerous biological functions, including immune, inflammatory, or even neurological ones. Sterol derivatives are implicated in molecular signaling, and cholesterol, in particular, if in excess, is known to contribute to the enhancement of cardiovascular diseases.
Seafood matrices were selected due to the importance of their lipids in human nutrition. Besides being regarded as the major source of long-chain omega-3 polyunsaturated fatty acids in the diet, whose benefits are increasingly highlighted (Larsen et al. 2011), some apprehension concerning its cholesterol content still coexists. Vitamin E amounts, besides contributing to the daily ingestion of this vitamin, are essential for the preservation of the remaining unsaturated lipids and can give some insight into its preservation history, particularly its oxidative status (Pazos et al. 2005). All these seafood components are usually analyzed for the purpose of nutritional evaluation (Zlatanos et al. 2006), seasonal effects (Sieiro et al. 2006), growing conditions (Miliou et al. 2006), and oxidative stability studies (Cho et al. 2001), among others.
Vitamin E is commonly extracted with non-polar organic solvents and analyzed by liquid chromatography, preferably with non-aqueous elution systems and direct fluorescence detection, requiring adequate experimental conditions due to its thermal and oxidative instability (Rupérez et al. 2001). Cholesterol, being present as free or esterified forms, requires hydrolysis for total estimation, followed by diversified chromatographic approaches for its quantification, both directly by liquid chromatography with UV or light scattering detection or after derivatization and analysis by liquid or gas chromatography (Hoving 1995).
Total fat is usually determined gravimetrically after solvent extraction, most frequently using the classical Folch or Bligh and Dyer methods (Folch et al. 1957; Bligh and Dyer 1959). Fatty acid analysis usually encompasses an initial hydrolysis of their natural ester linkages followed by derivatization to methyl esters and analysis in simple gas chromatography equipment with flame ionization detection (FID) (Casal and Oliveira 2010).
These components are usually analyzed separately, with increased organic solvent consumption and analysis time. Therefore, the aim of the present work was to provide a simple, fast, and accurate methodology able to provide the sequential analysis of all these three lipid classes in a single sample extract while making used of simple chromatographic equipment available in most analytical laboratories.
Material and Methods
Reagents
HPLC grade n-hexane was purchased from Merck (Darmstadt, Germany) and 1,4-dioxane from Sigma (Madrid, Spain). Methanol and KOH were acquired from Panreac (Spain). Boron trifluoride in methanol (14 %), butylated hydroxytoluene (BHT), and ascorbic acid were obtained from Sigma. All other chemicals were of analytical grade from diversified suppliers.
Standards
Tocopherol (α-) was purchased from Sigma. A 5-mg/mL standard solution was prepared in ethanol and kept at−20 °C. Its accurate concentration was evaluated by UV spectrophotometry according to its molar absorptivity in ethanol (3,265; Podda et al. 1996). Dilutions in n-hexane were performed as required for calibration or other purposes. The remaining vitamin E tocopherols (-β, -γ, -δ) and tocotrienols (α-, -β, -γ, -δ) were from Sigma or Matreya (PA, USA), being used only for identification purposes as their presence was not confirmed in the samples analyzed. The internal standard for vitamin E quantification was tocol (2-methyl-2-(4,8,12-trimethyltridecyl) chroman-6-ol), obtained from Matreya Inc.. A 100-μg/mL solution was prepared in n-hexane and kept at−20 °C.
Cholesterol and cholesteryl palmitate were obtained from Sigma-Aldrich (Spain). Accurate stock solutions of cholesterol (1.5 mg/mL) and its ester (2.4 mg/mL) were prepared in n-hexane and kept at−20 °C. Working solutions were prepared for calibration purposes by dilution in n-hexane.
Triundecanoin was used as the internal standard for fat estimation, based on the total fatty acid amounts, and was purchased from Sigma. A 10-mg/mL solution was prepared in n-hexane. A commercial standard solution with 37 fatty acid methyl esters (FAME) was used for the calibration of the FID signals (Supelco 37 FAME mix, USA).
Samples
For the present work, common octopus (Octopus vulgaris), european squid (Loligo vulgaris), atlantic chub mackerel (Scomber scombrus), and sardine (Sardine pilchardus) were chosen due to their elevated representativeness among seafood in Mediterranean countries. All samples were bought in daily markets, transported to the lab on ice, and washed with demineralized water. The edible parts (arms of octopus, mantle in squid, and muscle in mackerel and sardine samples) were separated, minced, and frozen at−20 °C. Sample portions were lyophilized (Telstar Cryodos-80, Terrassa, Barcelona), preserved in the dark at 4 °C, and finely ground before analysis.
Extractive Methodology
In Situ and Modified Folch Method Comparison
Preliminary assays for the development of the extractive method were performed using octopus samples. Both defrosted (1.0 g) and lyophilized (0.2 g) samples were tested, with the addition of the internal standards tocol (20 μL) and triundecanoin (20 μL) solutions, BHT (50 μL, 10 mg/mL in methanol), ascorbic acid (50 mg), and two or three glass pearls. The in situ method was performed under alkaline conditions with KOH 2 M in methanol due to their milder heating condition required (80 °C, 10 min., under N2). After extraction with n-hexane (2 × 3 mL), the extract was used for all the chromatographic analysis (vitamin E and cholesterol simultaneously by HPLC/diode array detector (DAD)/fluorescence detector (FD) and fatty acids by GC/FID). Folch extraction was performed as the control (Folch et al. 1957), with few modifications. Briefly, 3 mL of methanol and 6 mL of dichloromethane were used and the samples kept overnight under refrigeration after a brief agitation under vortexing. The non-lipid material was removed by washing with aqueous 0.9 % KCl once using one fifth of the collected volume. Hydrolysis/methylation was executed on the extracted lipids as described above followed by all the chromatographic analysis. Vitamin E was also analyzed before the hydrolysis/methylation step (taken to dryness and reconstituted in n-hexane) for comparison. A second derivatization method, using boron trifuoride (BF3, 14 % in methanol) after the initial KOH hydrolysis, was also tested for the fatty acid analysis.
Modified Folch and Smedes Method Comparison
An accurate lyophilized sample amount was weighted in a glass tube (between 200 and 300 mg) followed by the addition of the internal standards tocol (20 μL) and triundecanoin (20 μL) solutions, BHT (50 μL, 10 mg/mL in methanol), ascorbic acid (50 mg), and two or three glass pearls. Lipids were extracted by a modified Folch procedure (Folch et al. 1957), as described above, and by the non-chlorinated Smedes method (Smedes 1999). In the latter, 3.2 mL of propan-2-ol and 4 mL of ciclohexane were added, briefly agitated by vortexing, and kept overnight under refrigeration. The non-lipid material was removed by washing with 5.5 mL of aqueous 0.9 % KCl once, taking into account the inexistence of water in the lyophilized samples. These volumes assure that the solvent ratios recommended by the authors are preserved and can be adjusted when using fresh samples. The organic phases were dehumidified with anhydrous sodium sulfate and divided into two portions as described below.
An accurate volume, corresponding to approximately two thirds of the total extract volume, was transferred to a Pyrex tube and the solvent was evaporated under a nitrogen stream (60 °C). Hydrolysis was performed with 1.5 mL of KOH (0.5 M in methanol) at 100 °C (10 min) in a dry heating block (Stuart SBH200D/3). After attaining room temperature, methylation was completed by the addition of 1.5 mL of BF3 and heating for further 30 min at 100 °C. Once cooled, NaCl (1 %, 2.5 mL) and n-hexane (2 × 2 mL) were added and the mixture was mixed by vortexing, followed by centrifugation (5 min, 4,000×g; Heraeus Sepatech, Germany). The n-hexane layers were transferred to a centrifuge tube and water remains were adsorbed in anhydrous sodium sulfate followed by centrifugation for 5 min at 13,000×g (Heraeus Sepatech Biofuge Pico, Germany). A supernatant portion was directly transferred to a clear glass vial (12 × 32 mm, sealed with red PTFE-faced silicone septa and screw caps with hole; Supelco, USA). This vial was positioned in the HPLC automatic injector for the quantification of cholesterol and thereafter transferred to the gas chromatograph autosampler for fatty acid analysis.
The remaining lipid extract (1/3) was transferred to an amber glass vial, equivalent to the one described above, taken to dryness under a gentle nitrogen stream (40 °C), resuspended in about 600 μL of HPLC grade n-hexane, and placed in the same HPLC autosampler for vitamin E quantification. This final volume can be readjusted on a direct dependency of the expected concentration range.
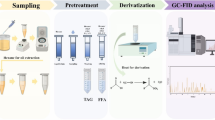
The final overall extraction methodology is represented in Fig. 1.

Overall procedure used for the simultaneous quantification of α-tocopherols (a), cholesterol (b), and fatty acids (c). Chromatograms obtained with an octopus sample
HPLC Chromatographic Conditions for Cholesterol and Vitamin E
The liquid chromatograph consisted of a Jasco integrated system (Japan) equipped with an LC-NetII/ADC data unit, a refrigerated autosampler (AS-2057 Plus), a PU-980 Intelligent Pump, and a multiwavelength DAD (MD-910, recorded at 210 nm), connected in series to a FD (FP-2020 Plus; λ exc = 290 nm and λ em = 330 nm; gain, 10). The chromatographic separation was achieved on a Supelcosil™ LC-SI column (75 × 3.0 mm, 3 μm; Supelco, Bellefonte, PA) operating at constant room temperature (23 °C). A mixture of n-hexane and 1,4-dioxane (97.5:2.5, v/v) was used as eluent at a flow rate of 0.8 mL/min. Data were analyzed with the ChromNAV Control Center–JASCO Chromatography Data Station (Japan). The compounds were identified by chromatographic comparisons with authentic standards, by co-elution, and by their UV spectra. Cholesterol was quantified at 210 nm, with calibration based on external standard solutions. Tocopherol was evaluated by the internal standard method based on the fluorescence data.
Gas Chromatographic Conditions for Fatty Acid Analysis
Gas chromatography was performed on a Chrompack CP 9001 chromatograph (Chrompack, Middelburg, the Netherlands) equipped with a split–splitless injector, a FID, and a Chrompack CP-9050 autosampler. The temperatures of the injector and detector were 250 and 270 °C, respectively. Separation was achieved on a 50-m × 0.25-mm i.d. CP-Sil 88 column (0.19-μm film; Chrompack-Varian). Helium was used as the carrier gas at an internal pressure of 120 kPa. The column temperature was 140 °C, for a 5 min hold, and then programmed to increase to 220 °C at a rate of 5 °C/min and then held for 15 min. The total analysis time was 35 min, but a further 5-min elution is advised for complete elution of possible interference compounds. The split ratio was 1:50 and the injected volume was 1.2 μL.
The results are expressed in relative percentage of each fatty acid, calculated by internal normalization of the chromatographic peak area. Fatty acid identification (from C11:0 to C22:6 n-6) was accomplished by comparing the relative retention times of FAME peaks with standards from diversified suppliers and from literature data. Peaks were corrected using empirical response factors obtained by the standard FAME solution. The response factors for FAME not present in the mixture were estimated according to the identity and retention time relative to similar FAME. Total fat was estimated on the basis of the total fatty acid methyl ester area counts in comparison with the undecanoic methyl ester.
Method Validation
Validation of the analytical method was based on the following parameters: linearity, limits of detection and quantification, precision (within-day and between-day variability), recovery, and stability.
Results and Discussion
Method Development and Validation
In Situ Versus Modified Folch Extraction and Fresh Versus Lyophilized Samples
Direct hydrolysis/derivatization procedures, commonly designated as one-step or in situ methodologies, have been described for fatty acid analysis in several food matrices without the need of previous lipid extraction. These methods are simple and fast to perform, achieving frequently higher recovery rates than the classical extractive procedures when efficient matrix disruption is assured (Carrapiso and García 2000). In order to verify the feasibility of this approach, preliminary assays were conducted with direct alkaline hydrolysis/derivatization, as described in “In situ and modified Folch method comparison,” aiming to simultaneously hydrolyze the fatty acids and cholesterol esters while converting the fatty acids to their methyl esters and preserving vitamin E. A resume of the results achieved during these first stages of method development is presented in Table 1 on a comparative basis.
When the direct (in situ) approach was used, lower amounts of cholesterol, vitamin E, and total fat were detected when compared directly with those obtained with previous Folch extraction despite the similarity of the fatty acid profiles. An identical observation was already reported for meat (Juárez et al. 2008), with smaller fat extraction efficiency and lower recovery values for in situ methods. Also, the only vitamin E compound detected, α-tocopherol, was present in small amounts on both procedures, and the internal standard tocol was also partially lost when compared to direct HPLC injections. This loss was probably a consequence of the aggressive conditions used for hydrolysis despite being performed in the presence of antioxidants (BHT and ascorbic acid) and under N2. Finally, the fatty acid derivatization efficiency on fresh samples was also reduced in the direct approach. The presence of water in the reaction media can decrease the efficiency of fatty acid methylation under alkaline conditions.
Except for vitamin E, all these observations in the direct approach could be a straight consequence of incomplete hydrolysis and/or methylation under alkaline conditions. The presence of water, to be avoided under these conditions, could be the main justification in the defrosted samples, but not in the lyophilized ones (Table 1). Several authors use an organic solvent in the reaction media under the explanation that it improves lipid solubilization. Still, on the basis of the detailed experiments undertaken by Araújo et al. (2008), the organic solvent should only be added after the reaction, as in our experiment.
The alternative use of acidic conditions was considered as it provides higher efficiency in the methylation of fatty acids, even in the presence of water. However, acidic conditions require higher temperature/times for completeness, incompatible with vitamin E determination. This acidic approach was already tested by Meier et al. (2006), describing a one-step methodology for the determination of fatty acids and cholesterol in marine tissues. Despite being adequate for the fatty acids, the decomposition peaks of cholesterol formed under acidic conditions are responsible for carryover peaks in gas chromatography. Therefore, the alternative use of acidic methylation was not considered as an option in this work.
Based on the preliminary results achieved, direct hydrolysis was discarded as it requires further optimization in a straight dependency of the matrix characteristics. Despite being possible to attain ideal condition for these samples in particular, it would require further tests for different matrices. As our objective was to provide a methodology as universal as possible, previous lipid extraction is therefore more advisable.
The classical Folch method, among others, frequently with small adaptations, is a methodology with proven results over decades in a huge amount of diversified matrices. The recognized Bligh and Dyer method, derived from the former and frequently used for marine tissues, is known to produce lower estimates of lipid content for high lipid matrices (Iverson et al. 2001). Again, as we were aiming to develop a method that could be applied both to lean and fatty tissues, this method was not tested under this work. Although involving some analytical labor and solvent consumption, the same lipid extract obtained after Folch extraction can be used for all the determinations, reducing their global impact in the overall determinations. With regard to vitamin E, it should be determined before the hydrolysis/methylation or, instead, in a separate lipid extract fraction taken aside. This was devised as the best alternative, and the presence of the internal standard (tocol) avoided the need of accuracy in this volume measurement. As vitamin E analysis under normal-phase HPLC requires solvent switch to hexane, this was achieved by solvent evaporation under nitrogen stream, easily performed for an elevated number of samples on adequate evaporative devices.
Cholesterol hydrolysis efficiency was verified using cholesteryl palmitate against free sterol, both subjected to the complete methodology. The alkaline conditions applied were adequate, enabling 98 % recovery. Concerning fatty acid hydrolysis and methylation, efficiency should be increased as higher recoveries were achieved with direct nonadecanoic methyl ester injection in comparison with the undecanoic methyl ester formed after triundecanoin hydrolysis/methylation under the assayed conditions. This was achieved by a further classic methylation with a weak Lewis acid (BF3), followed by hexane extraction (data not shown). This final extract was initially used for cholesterol quantification, followed by gas chromatography, without the need to exchange vials or septa, as a slight concentration in the latter will not interfere with the fatty acid quantification due to the presence of an internal standard.
With regard to the sample pretreatment, invariably higher amounts of cholesterol and vitamin E were detected with lyophilized subsamples when compared with fresh ones due to the accomplishment of a higher sample dispersion.
Modified Folch Extraction Versus Smedes Extraction
Concerning the Folch method itself and those derived from it, the common use of chloroform is a negative parameter, with several alternatives tested over the years for its replacement. Dichloromethane, a less hazardous solvent with fewer restrictions and drawbacks, was used under the present work due to the proven inexistence of differences in the lipid extraction efficiency (Cequier-Sánchez et al. 2008). Still, the use of non-chlorinated solvents is a greener approach, and the method described by Smedes (1999), already used under several research areas, was devised as a good alternative. Besides the environmental advantage, this extractive method is faster to perform since lipids are extracted into the upper phase, easier to remove, simplifying and reducing analytical labor. Therefore, in parallel with Folch extraction, Smedes method was also tested and validated.
Regarding cholesterol validation features (Table 2), linearity range was tested within the expected levels for the samples under evaluation. The high correlation coefficients achieved (r 2 > 0.99), even when standards were extracted, demonstrate the accuracy of the methodology. Concerning vitamin E, validation was accomplished only for α-tocopherol, once it was the only detected tocochromanol in the samples analyzed. As for cholesterol, a good linearity was observed (r 2 > 0.99).
For sensitivity evaluation, the limits of detection (LOD) and quantitation (LOQ) were calculated. The LOD was defined as the lowest concentration of each compound that gave an average signal-to-noise ratio >3 over five replicate injections (Table 2). The LOQ was defined as the lowest concentration of each compound that gave peak area counts or peak-to-IS area ratios with a relative standard deviation (RSD) <2.5 % over five replicate injections. The concentration was experimentally confirmed in real samples. Cholesterol quantitation limit (Table 2) achieved under UV detection, despite being in the milligrams per 100 g range, is adequate for application in most animal source foods.
Regarding total fat estimation, it was achieved by the total sum of fatty acids methyl esters between lauric (C12:0) and docosahexaenoic (C22:6n3) acids, including unknown ones (<5 %), in comparison with the undecanoic methyl ester formed from triundecanoin internal standard, after correction for the FID responses. Still, this estimation can only be used on a comparative level as it will be systematically low when compared to those obtained by gravimetric methodologies, as these include also non-fatty acid lipid components. This approach can provide important information on a comparative basis, being therefore included in the methodology, but for accurate total lipid amount quantification, other alternatives should be devised.
In the absence of an adequate reference material for all these parameters, accuracy was evaluated by recovery assays, enabling to infer on possible matrix effects. For the purpose, known amounts of both cholesterol and α-tocopherol were simultaneously spiked into aliquots of a previously characterized octopus sample, ranging from 20 to 60 % of their initial content. The spiked samples were prepared in triplicate, at two spiked levels, embracing the range expected in similar samples. Excellent recoveries were achieved for both compounds, as depicted in Table 2.
The method precision was evaluated by analyzing six subsamples on the same day (intra-day) and comparing with the values achieved on a different day (inter-day: n = 12). Again, the low relative variation achieved for cholesterol and vitamin E (Table 2) confirms the adequacy of the proposed method, but it is recommended to assure total absence of water in the final extracts in order to achieve retention time consistency. The apparent higher precision achieved with the Smedes extractive method can be a consequence of its easier laboratorial performance, with easier collection of the organic phase. The intra-day RSD achieved for the fatty acid relative percentage varied between 4.3 % for palmitic acid and 8.9 % for linoleic acid. The inter-day RSD was similar, ranging between 4.1 and 11.3 % for the same methyl esters. The variability for the main omega-3 fatty acids EPA (eicosapentanoic acid, C20:5n-3) and DHA (docosahexaenoic acid, C22:6n-3) was always below 6 %, as was the global fat estimation based on total fatty acid area counts.
The performances achieved by the present methodology are similar or higher than those described in the literature. Cholesterol and tocopherol analysis, for instance, were already validated simultaneously by Cayuela et al. (2003) in fresh meat samples, but lower recovery and repeatability were achieved, particularly for tocopherol (68 ± 15 %). The absence of adequate internal standard and the quantification of α-tocopherol after hydrolysis are probably the basis of these achievements. When the direct analysis of vitamin E is performed, higher recoveries are attained (Özogul et al. 2011), simultaneously with higher repeatability, when using adequate internal standards (Cunha et al. 2006; Semeraro et al. 2009).
Chromatograms obtained with the FD and DAD (210 nm) for the same standard solution containing all vitamin E compounds and internal standard plus cholesterol are represented in Fig. 2. Adequate baseline separation was obtained, with no interferences between cholesterol and tocol, the internal standard used for vitamin E quantification. On the subject of choosing the ideal detector for each compound, fluorescence detection is highly selective, significantly improving the sensitivity for vitamin E determination (Fig. 2a). In contrast, the direct UV detection of cholesterol is based on the isolated double bond in the structure, which needs high excitation energy and short wavelengths. It is therefore a relatively nonspecific detection and easily interfered by a coexisting unsaturated double bond in the matrix or solvent system. Its alternative analyses by gas chromatography, however, require derivatization and the use of diverse chromatographic conditions from those used in the fatty acid quantification.

Chromatograms (HPLC) from a standard mixture of vitamin E and cholesterol obtained with the fluorescence detector (a) and DAD (b): α-tocopherols (1); α-tocotrienol (2); β-tocopherol (3); γ-tocopherol (4); β-tocotrienol (5); γ-tocotrienol (6); δ-tocopherol (7); tocol (IS); δ-tocotrienol (8). Chol cholesterol
All the chromatographic results in the present methodology can be obtained in 40 min, imposed by the fatty acid analysis by gas chromatography, as both vitamin E and cholesterol take only 8 min each and are injected under the same chromatographic conditions.
Applicability of the Method
As previously indicated, this method was developed for the analysis of seafood, in particular those with an elevated representativeness in the Mediterranean area. As example, the results achieved for some octopus, squid, Atlantic chub mackerel, and sardine samples are represented in Table 3. The chromatograms on Fig. 1 correspond to an octopus sample.
The results obtained are all in accordance with literature data despite the known differences achieved between seasons, sex, and species. Fatty acid and total lipid data are quite abundant in the literature, namely, for octopus (Zlatanos et al. 2006; Sieiro et al. 2006; Miliou et al. 2006; Ozogul et al. 2008), squid (Zlatanos et al. 2006; Ozogul et al. 2008), Atlantic chub mackerel (Özogul et al. 2007; Passi et al. 2002), or sardine (Bandarra et al. 1997; Zlatanos 2007; Leonardis and Macciola 2004). Their richness in polyunstaurated fatty acids (PUFAs) is clear in the present study, although of smaller significance in mackerel. The total fat estimation gave reduced fat amounts for the cephalopods, while both sardine and mackerel are regarded as fatty fishes. Literature data on cholesterol is not so abundant, particularly regarding the edible portions, but a recent review was published for seafood (Ochlenschläger et al. 2006), highlighting the differences between species, as obtained in the samples analyzed in the present study, particularly for squid (255 mg/100 g FW). With regard to α-tocopherol, the main lipid-soluble vitamin, data are scarcer. Indeed, only a few examples are found, and the amounts are highly influenced by oxidation (Cho et al. 2001; Özogul et al. 2011; Passi et al. 2002), being reduced after cooking (Gotoh et al. 2011). Under the present study, sardine presented clearly higher tocopherol amounts than the remaining samples. Therefore, besides the nutritional importance, their amounts can be regarded as important quality parameters associated with oxidation, particularly important on highly polyunsaturated foods, as seafood.
Conclusions
The methodology validated enables the sequential quantification of the most important food lipids using a single sample extract and easily available laboratorial equipment. Both Folch and Smedes extraction methods grant accurate results, but the latter presents analytical and environmental advantages. Despite being presented here for seafood, the methodology can be easily applied to different food matrices, providing that the extractive methods are applied correctly and the internal standards and hexane dilutions are used in correct proportion. The proposed method has some practical advantages: consistent extraction procedure, reduced sample amounts, reduced consumption of organic solvents, combined with a low overall cost and standard chromatographic equipment. Moreover, the total analysis time is substantially reduced when compared with the evaluation of these parameters separately, with increased productivity. It can be an attractive technique for routine analyses in standard equipped laboratories.
References
Abumrad NA, Piomelli D, Yurko-Mauro K, Merrill A, Clandinin MT, Serhan CN (2012) Adv Nutr 3:60
Araújo P, Nguyen T-T, Frøyland L, Wang J, Kang JX (2008) J Chrom A 1212:106
Bandarra NM, Batista I, Nunes ML, Empis JM, Christie WW (1997) J Food Sci 62:40
Bligh EG, Dyer WJ (1959) Can J Biochem Phys 37:911
Carrapiso AI, García C (2000) Lipids 35:1167
Casal S, Oliveira M (2010) Fatty acids analysis by gas chromatography (GC). In: Cazes J (ed) Encyclopedia of chromatography. 3rd edn
Cayuela JM, Garrido MD, Banón SJ, Ros JM (2003) J Agric Food Chem 51:1120
Cequier-Sánchez E, Rodriguez C, Ravelo A, Zarate R (2008) J Agric Food Chem 56:4297
Cho SY, Joo DS, Choi HG, Nara E, Miyashita K (2001) Fish Sci (Tokyo, Jpn) 67:738
Cunha SC, Amaral JS, Fernandes JO, Oliveira MBPP (2006) J Agric Food Chem 54:3351
Folch J, Less M, Sloane GH (1957) J Biol Chem 226:497
Taylor & Francis, London Podda M, Weber C, Traber MG, Packer L (1996) J Lipid Res 37:893
Gotoh N, Mashimo D, Oka T, Sekiguchi K, Tange M, Watanabe H, Noguchi N, Wada S (2011) Food Chem 129:279
Hoving EB (1995) J Chrom B 671:341
Iverson SJ, Lang SLC, Cooper MH (2001) Lipids 36:1283
Juárez M, Polvillo O, Contò M, Ficco A, Ballico S, Failla S (2008) J Chrom A 1190:327
Larsen R, Eilertsen K-E, Elvevoll EO (2011) Biotechnol Adv 29:508
Leonardis A, Macciola V (2004) Mol Nutr Food Res 48:209
Meier S, Mjøs SA, Joensen H, Grahl-Nielsen O (2006) J Chrom A 1104:291
Miliou H, Fintikaki M, Tzitzinakis M, Kountouris T, Verriopoulos G (2006) Aquaculture 256:311
J Ochlenschläger (2006) In: Luten JB, Jacobsen C, Bekaret K, Sæbø A, Oehlenschläger J (eds) Seafood research from fish to dish quality, safety and processing of wild and farmed fish. Wageningen Academic Press, Wageningen, pp 41–57
Özogul Y, Özogul F, Alagoz S (2007) Food Chem 103:217
Özogul Y, Duysak O, Özogul F, Özkütük AS, Türeli C (2008) Food Chem 108:847
Özogul F, Özogul Y, Kuley E (2011) Food Sci Technol Res 17:595
Passi S, Cataudella S, Di Marco P, De Simone F, Rastrelli L (2002) J Agric Food Chem 50:7314
Pazos M, Sánchez L, Medina I (2005) J Agric Food Chem 53:4000
Rupérez FJ, Martin D, Herrera E, Barbas C (2001) J Chrom A 935:45
Semeraro A, Altieri I, Patriarca M, Menditto A (2009) J Chrom B 877:1209
Sieiro MP, Aubourg SP, Rocha F (2006) Eur J Lipid Sci Technol 108:479
Smedes F (1999) Analyst 124:1711
Zlatanos S, Laskaridis K (2007) Food Chem 103:9725
Zlatanos S, Laskaridis K, Feist C, Sagredos A (2006) Mol Nutr Food Res 50:967
Acknowledgments
This work has been supported by FCT (Fundação para a Ciência e a Tecnologia) and FEDER through the COMPETE program under the projects FCT PTDC/AGR-AAM/102316/2008 and PEst-C/EQB/LA0006/2011.
Disclamer
No disclamers.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cruz, R., Casal, S., Mendes, E. et al. Validation of a Single-Extraction Procedure for Sequential Analysis of Vitamin E, Cholesterol, Fatty Acids, and Total Fat in Seafood. Food Anal. Methods 6, 1196–1204 (2013). https://doi.org/10.1007/s12161-012-9526-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-012-9526-z