
Abstract
A cloud-point extraction (CPE) method using Tergitol TMN-6 (TMN-6) non-ionic surfactant was developed for the extraction and preconcentration of propyl gallate (PG), tertiary butyl hydroquinone (TBHQ), butylated hydroxyanisole (BHA), and butylated hydroxytoluene (BHT) in edible oils. The optimum conditions of CPE were 1.5 % (v/v) Tergitol TMN-6, 1 % (w/v) NaCl, ultrasound-assist 15 min at 49 KHz, 20 min equilibrated at 45 °C, and centrifugation for 10 min at 3,000 rpm. The surfactant-rich phase was then analyzed by reversed-phase high-performance liquid chromatography (HPLC) with ultraviolet detection at 280 nm, under gradient separation, using methanol and 1.5 % (v/v) acetic acid. Under the study conditions, four synthetic phenolic antioxidants (SPAs) were successfully separated within 24 min. Limits of detection in the studied edible oils were in the range of 1.6 to 9.0 ng mL−1. The high recoveries of the spiked edible oils were obtained in the range 90–98 %. The CPE method has been shown to be a potentially useful methodology for the preconcentration of the target analytes, with a preconcentration factor of 25. This method was compared with cloud point extraction (using Triton X-114) and liquid–liquid extraction (using methanol).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Synthetic phenolic antioxidants (SPAs) play a significant role in retarding lipid oxidation reactions in food products, which are the most widely used food additives owing to their high performance, low cost, and wide availability. SPAs can interrupt the chain of free radicals involved in the autoxidation that constitutes the most common form of deterioration of fats used in the food industry. Among the SPAs, PG, TBHQ, BHA, and BHT are the most commonly used antioxidants. However, recent studies demonstrate that excessive use of PG, BHA, BHT, and TBHQ may cause a loss of sustenance and even produce toxic substances that may be harmful to people's health. The results of testing with laboratory animals suggest that BHA and BHT may be responsible for liver damage and carcinogenesis (Clayson et al. 1986; Ito et al. 1986). Because of this, in several countries and departments, permitted use levels have been established. The FDA (C.o.F.R. Food and Drug Administration 2001) and EU (Directive 1995) have established permitted food phenolic antioxidants and amounts of their allowable usage. Currently permitted for use in food SPAs are PG, TBHQ, BHA, and BHT, usually at concentrations of up to 100–200 μg g− 1 of SPAs in oils or fats, either singly or in combination. Thus, the establishment of methods for determining PG, TBHQ, BHA, and BHT levels both individually and simultaneously is necessary.
Until now, various analytical methods have been reported for the determination of synthetic phenolic antioxidants in oil and other food stuffs, including spectrophotometry (Capitan-Vallvey et al. 2004), liquid and gas chromatography (Guo et al. 2006; Saad et al. 2007; Rodil et al. 2010; Pinho et al. 2000; Perrin and Meyer 2002; Karovicova and Simko 2000; Chen et al. 2011; Bianchi et al. 1997), micellar electrokinetic chromatography (Delgado-Zamarreno et al. 2007), flow injection, and HPLC with amperometric detection (Riber et al. 2000) (Luque et al. 1999; Ruiz et al. 1999), thin-layer chromatography(Sin et al. 2006), and electroanalysis (Delgado-Zamarreno et al. 2007; Ziyatdinova et al. 2010). Among the aforementioned methods, reversed-phase high-performance liquid chromatography (HPLC) has become the most widely used for its high selectivity for the separation of synthetic antioxidants under chromatographic conditions (Ruiz et al. 1999; Aparicio et al. 2000),
Another important factor in the analysis of the SPAs is an appropriate extraction and preconcentration procedure prior to the HPLC separation of the sample. At present, preconcentration and clean-up techniques for SPAs mainly focus on the classical liquid–liquid extraction (LLE) (Saad et al. 2007), solid phase extraction (SPE), solid-phase micro-extraction (SPME) (Tombesi and Freije 2002; Yang et al. 2010), solid–liquid extraction, and solid–gas extraction. However, though these methods all have their respective advantages, the methods are time-consuming and present a threat to the environment and human health due to the use of organic solvents. Therefore, a rapid, inexpensive, efficient, and environmentally friendly method for the extraction of SPAs is in demand.
Over the past 30 years, a procedure called cloud point extraction (CPE) has attracted a lot of attention as an alternative to traditional liquid–liquid extraction. The CPE was first introduced by Watanabe and others in 1976 (Miura et al. 1976). Cloud point of the aqueous solutions of surfactant micellar systems is a temperature at which the solution becomes turbid before separating into two phases, a surfactant-rich phase and an aqueous phase. The surfactant-rich phase is able to extract and preconcentrate analytes. The CPE methodology is based on surfactant-mediated phase separation. During the CPE procedure, targets can be successfully extracted and preconcentrated by changing the conditions affecting phase separation in a single step; the preconcentration factor can reach more than 10. Importantly, in CPE, the surfactants are less toxic, cheaper, and more environment friendly than the organic solvents.
For the aforementioned advantages, CPE has been successfully used for the preconcentration of species of widely differing characteristics as a previous step to their later determination by HPLC (Chen et al. 2011; Zhang et al. 2011a), gas chromatography (GC) (Shen and Shao 2006) (Sikalos and Paleologos 2005; Ohashi et al. 2004; Jia et al. 2008a), and atomic absorption spectrometry (Ulusoy et al. 2011; Kolachi et al. 2011; Shah et al. 2011).
Up to now, non-ionic surfactants, such as Triton X series (Mustafina et al. 2006; Ohashi et al. 2007; 2005; Purkait et al. 2005; Sun et al. 2006; Wu et al. 2007), PONPE series (Aranda et al. 2008; Luconi et al. 2000; Wuilloud et al. 2002), and Brij series (Fernandez et al. 1998; Delgado et al. 2004), are the most widely used surfactants for CPE. These surfactants have been successfully applied to extract metal ions (Moran et al. 2000; Tang et al. 2004), pesticides (Jia et al. 2008b; Zhao et al. 2011), sulphonamides (Zhang et al. 2011b), polycyclic aromatic hydrocarbons (Bai et al. 2001; Goryacheva et al. 2005; Merino et al. 2002; Sicilia et al. 1999; Sirimanne et al. 1996), dye (Liu et al. 2007; Purkait et al. 2006; Purkait et al. 2009), and so on. However, nearly all have very strong absorption in the ultraviolet region; if we do not use a UV detector, the application range of CPE will be narrow undoubtedly. In Europe, surfactants with an aromatic group are also forbidden. Therefore, using a surfactant without UV absorbance or signals is considered to be the best way to complete the CPE procedure.
Tergitol TMN-6 is polyethylene glycol trimethylnonyl ether, a non-ionic surfactant, with no aromatic group; its cloud point (CP) is about 34 °C. To our knowledge, Tergitol TMN-6 has never been applied in CPE for the determination of SPAs.
In this research, Tergitol TMN-6 was studied as the cloud point extractant in the preconcentration and treatment of the edible oils containing SPAs. Compared with Triton X-114, a much lower volume of surfactant was obtained by the CPE system with Tergitol TMN-6 under the condition of the same total surfactant concentration. Different experimental conditions were studied to determine the optimal condition for the analysis of SPAs.
Material and Methods
Reagents and Solutions
Edible Oil Samples
Edible oils were purchased from local supermarkets around Kunming city: sunflower oil, sesame, olive, coconut oil, castor oil, peanut oil, and palm kernel oil.
Chemicals and SPA Standards
Standards of PG (≥98.0 %), TBHQ (≥99.0 %), BHA (≥98.5 %), and BHT (≥99.0 %) were purchased from Sigma (St Louis, Mo, USA). The structures of the studied SPAs and their properties are shown in Table 1. Methanol (HPLC grade) was obtained from Merck (Darmstadt, Germany). Tergitol TMN-6 was purchased from Fluka (USA) and used without further purification. TX-114 was purchased from Acros (USA) and used without further purification. Acetic acid was purchased from Kedi (Tian Jin, China). Sodium chloride, sodium sulfate, and sodium carbonate were purchased from ZhiYuan (Tian Jin, China).
All reagents were of analytical reagent grade.
Preparation of SPA Standards and Tergitol TMN-6
A stock standard solution containing 1 mg mL-1 of each SPA (PG, TBHQ, BHA, and BHT) was prepared in methanol. The flask was shaken until a homogenous and clear solution was formed. The stock solution was covered with aluminum foil and stored in a refrigerator (4 °C) and away from light for a maximum of 1 month. Before use, standard working solutions were prepared by diluting appropriate amounts of the stock solution in MeOH. Tergitol TMN-6 was used without further purification, to prepare 10 % (v/v) aqueous solutions.
Instruments
Chromatographic separation and evaluation were performed with an HPLC system (consisting of a vacuum degasser, an autosampler, a quatpump, and a diode-array detector; Agilent 1200 Series, Agilent Technologies, USA) equipped with a reversed-phase C18 analytical column of 150 mm × 4.6 mm (Agilent TC-C18). Empower software was used for spectra recording of the studied SPAs and used for spectra confirmations of peaks in the studied samples. An ultrasonic cleaner (Shanghai, China) was used to assist the CPE procedure. A water-bathing constant temperature vibrator (Jintan crystal glass experimental instrument, SHY-2A, Jin Tan, China) was used to implement CPE. A centrifuge (Shanghai surgical instrument factory, 80-2, Shanghai, China) was used for complete phase separation.
Extraction from Aqueous Samples
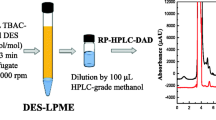
Aliquots of the solution (2.0 mL) containing different concentrations of SPAs (PG, TBHQ, BHA, and BHT) were placed in centrifugal vials. TMN-6 stock solution (1.5 mL) and 0.1 g of NaCl were consequently added, and the mixtures were diluted with doubly distilled water to 10 mL. The centrifugal vials were left in an ultrasonic cleaner ultrasound-assist 15 min at 49 KHz and thermostatically controlled bath at 45 °C for 20 min; centrifugation for 10 min at 3,000 rpm was adopted for complete separation of the surfactant-rich phase. The water phase was then carefully removed, using a syringe with a long needle, achieving a final volume of 0.4 mL by adding MeOH. Therefore, a preconcentration of about 25 times was finally obtained. A 10-μL aliquot of the surfactant-rich phase solution was injected into the HPLC system.
Extraction from Edible Oils
An edible oil sample spiked with 50 mg/kg of each SPA was used to test the applicability of CPE method to edible oil samples. The time of interaction between SPAs and the matrix of edible sample before analysis was at least 12 h. A mixture of water, MeOH, and TMN-6 (157:40:3, V/V/V) was used for extraction of SPAs from edible oil. A 2.0 ± 0.05-g sample was extracted in the presence of 1 mL of MeOH at 25 °C for 10 min. A portion of the upper liquid phase was transferred into a centrifugal vial through a paper filter. The residue was reextracted with another 1 mL of MeOH, and the supernatant was isolated and collected with the first extraction fraction. The surfactant-rich phase was separated as described in “Extraction from aqueous samples” by adding 0.1 g of NaCl.
HPLC Conditions
The separation and determination of SPAs (PG, TBHQ, BHA, and BHT) were carried out by injecting the extracts into the HPLC–UV (DAD) after filtering. The HPLC separation was performed on a reversed-phase system with the gradient elution using methanol and water with 1.5 % acetic acid. The gradient elution was performed as follows: 40 % methanol (0–5 min), ramped to 70 % methanol (5–10 min), and then ramped to 90 % methanol (10–25 min). Next, the system was allowed to stabilize for 1–2 min under the initial conditions. The prepared mobile phase was filtered and degassed using ultrasonic agitation. The flow rate was set at 1.0 mL min−1. The column temperature was maintained at 40 °C, and the injection volume was set to 10 μL. SPAs were recorded at the wavelength of 280 nm.
Analytical Performance of the Method
In order to determine the recovery, repeatability, and reproducibility of this method, SPAs were spiked into blank edible oil at three different concentrations (25, 50, and 75 mg kg-1), and five replicates were analyzed per concentration level in two independent analytical runs under the established chromatographic conditions.
Results and Discussion
HPLC Chromatograms of the Extracted SPAs in Edible Oil Sample
Figure 1 shows typical HPLC chromatograms of extracted and preconcentrated SPAs. Figure 1a is the chromatogram of blank edible oil. Figure 1b is the chromatogram of standard SPAs without CPE, and Fig. 1c is the chromatogram of SPAs after its CPE from edible oil spiked with it. The preconcentration effect of CPE is clearly demonstrated in Fig. 1b, c. TMN-6 does not interfere with the determination of SPAs.

HPLC–UV chromatograms: a blank edible oil, b standard (500 μg mL−1), and c edible oil spiked with SPAs (20 μg mL−1). Peak no.:(1) PG, (2) TBHQ, (3) BHA, (4) BHT. HPLC conditions—gradient separation using methanol and 1.5 % (v/v) acetic acid; injection volume 10 μl; flow rate 1 mL min−1; wavelength 280 nm
Optimization of the CPE Conditions
Effect of TMN-6 Concentration
As shown in Fig. 2, the cloud point of TMN-6 aqueous varies with its differing concentrations. When its concentration is higher than 1 % (v/v), its CP has a slow decrease as the concentration increases; when its concentration is higher than 3 % (v/v), the influence of its concentration to CP can be ignored, and CP is stable at about 35 °C. When its concentration is lower than 1 % (v/v), its CP dropped to 34 °C (0.25 %, v/v), and as its concentration decreases beyond 0.5 %, its CP increases rapidly to 80 °C at 0.08 % (v/v) concentration, which may be caused by the saltation of micelle status.

Cloud point of TMN-6 solution as a function of surfactant concentration
The variation of extraction efficiency with TMN-6 concentration was examined within the range of 1–10 % (v/v). As shown in Fig. 3, at a lower concentration of surfactant, the extraction efficiency of the four SPAs was low. Content quantitative extraction was observed for a TMN-6 concentration of 1.5 % (v/v), and the recovery remained constant above that. However, the volume of the surfactant-rich phase increases from approximately 0.2 to 0.7 mL with increasing concentration of TMN-6 from 1.5 % to 10 % (v/v). To avoid the decrease of the concentration factor, a TMN-6 concentration of 1.5 % (v/v) was used in the following experiments.

Effect of TMN-6 concentration on the recovery. Other extraction conditions—equilibrium temperature 45 °C, equilibrium time 20 min, concentration of sodium chloride solution of 5 % (w/v), ultrasound-assist 15 min at 49 KHz, centrifugation for 10 min at 3,000 rpm
Effect of Salt Type and Concentration
The CPE behavior was also determined as functions of salt types and concentrations. Addition of salt can accelerate phase separation and lower the cloud point of the surfactant solution. Also, the fractional coacervate phase volume decreases with salt concentration. This is explained by the fact that more water goes into the dilute phase due to a salting-out effect. The relevant electrolytes are usually in high concentrations (Carabias-Martinez et al. 2000; Purkait et al. 2004). Different salts (Na2SO4, NaCl, and Na2CO3) were tested. The CPE systems had different behaviors with salt type. When NaCl was added to the system, the highest recovery efficiency was obtained as shown in Fig. 4.

Effect of salt type on the recovery: 5 % (w/v) of NaCl, Na2SO4, and Na2CO3, respectively. Other extraction conditions—TMN-6 concentration 1.5 % (v/v), equilibrium temperature 45 °C, equilibrium time 20 min, ultrasound-assist 15 min at 49 KHz, centrifugation for 10 min at 3,000 rpm
The extraction efficiencies of four SPAs increased as the concentration of NaCl increased from 0.2 % to 1 % (w/v). Further increase in concentration of NaCl had no significant effect on the extraction efficiency.
Thus, a NaCl concentration of 1 % (w/v) was employed for further experiments.
Effect of Equilibration Temperature and Equilibration Time
When the CPE procedure was processed at the equilibration temperature of the surfactant, the best extraction efficiency was achieved. A temperature range of 35–60 °C was studied. Maximum extraction efficiency was observed in the range of 40–50 °C; beyond 50 °C, a little decrease of the efficiency was obtained. Accordingly, 45 °C was used in the CPE procedure.
The equilibration time was also investigated in the rage of 5–60 min. It was found that the maximum extraction efficiency was presented between 15 and 20 min. Therefore, 15 min was chosen as the optimum equilibration time.
Effect of Ultrasound-Assist CPE
Ultrasound-assist plays an important role in phase separation and recovery. The ultrasound-assist CPE time was defined as the time before heating, and it was varied within the range of 0–60 min. The extraction efficiency increased with an increase of the extraction time in the range of 0–15 min, and it remained almost constant after 15 min. Therefore, 15 min was chosen as the most appropriate ultrasound-assist CPE time. Comparing ultrasound-assist CPE with no-assist, recovery of analytes with ultrasound-assist is higher.
Figure 5 describes the phase separation condition of ultrasound-assist CPE and no-assist CPE at the 10-min equilibration time; it indicates that the phase separation time was reduced when using the ultrasound-assisted approach. Therefore, ultrasound-assist CPE was used for all further experiments

Comparison of ultrasound-assist CPE with no-assist CPE in terms of phase separation condition. Other extraction conditions—TMN-6 concentration 1.5 % (v/v), concentration of sodium chloride solution of 5 % (w/v), equilibration temperature 45 °C, equilibrium time 20 min, centrifugation for 10 min at 3,000 rpm
Effect of MeOH on Efficiency for Extracting SPAs
MeOH is the most frequently used organic solvent to extract SPAs from foodstuffs (Saad et al. 2007). As the ability of an aqueous solution of non-ionic surfactant to extract SPAs from edible oils was not adequate, MeOH was added in order to raise the extraction efficiency of the cloud point procedure. Recovery percentages ranging from 93 % to 97 % were obtained using 20 % (V/V) MeOH.
Comparison with Triton X-114 CPE and LLE (Using MeOH)
Different surfactants and extracting agents demonstrate different efficiencies of extraction for certain targets based on various factors. Thus, the most commonly used surfactant, Triton X-114(Chen et al. 2011), in CPE and extracting agent, MeOH, were selected to compare with TMN-6 CPE method performance. The remaining results are shown in Fig. 6. TMN-6 had an advantage over Triton X-114-CPE and MeOH-LLE in terms of recovery.

Comparison of TMN-6 CPE with TX-114CPE and LLE-MeOH in terms of recovery. Other extraction conditions—TMN-6 concentration 1.5 % (v/v); concentration of sodium chloride solution of 5 % (w/v); equilibration temperature 45 °C; equilibrium time 20 min; centrifugation for 10 min at 3,000 rpm; 2.5 % (v/v) TX-114, 0.5 % (w/v) NaCl, and 40-min equilibration time at 50 °C; 9-mL MeOH for LLE
Analytical Characteristic
The analytical characteristic data of the present system for the four SPAs are summarized in Table 2. A linear calibration graph for the peak areas of SPAs versus concentrations of SPAs was obtained for concentrations in the range from 1.0 to 500.0 μg mL−1. The correlation coefficients for SPAs were all more than 0.99. The limit of detection calculated from a signal/noise ratio (s/n = 3) was 1.6 ng mL−1 for PG, 9.0 ng mL−1 for TBHQ, 2.0 ng mL−1 for BHA, and 5.5 ng mL−1 for BHT. The results in the current method were lower than or similar to those values obtained by other methods reported (Saad et al. 2007; Chen et al. 2011; Sin et al. 2006). The recovery, repeatability, and reproducibility varied from 90 % to 98 %, from 1.1 % to 2.1 %, and from 1.5 % to 2.1 % (CVR), respectively. Validation of precision also has been done considering intra-day and inter-day variation. Data are listed in Table 3.
Application of This Method in Commercial Matrices of Foods
To evaluate the effectiveness of the established method in larger range of edible oils, it was applied to the analysis of a total of 14 samples of edible oils (Table 4). In 13 samples, SPAs were found at concentration levels near the limit of detection. Furthermore, the total content of SPAs in all of the samples was below the critical value defined in Chinese National Standard for all of the SPA-positive samples.
Conclusion
A simultaneous cloud point method has been developed for determination of four SPAs in edible oils. It was demonstrated that the proposed procedure is comparable to, or even better than, most works that are available for monitoring of PG, TBHQ, BHA, and BHT in mixtures or alone. In addition, the ultrasound-assist CPE based on TMN-6/NaCl system is an environment- friendly, one-stop extraction, and simple preconcentration way for the determination of trace amount of SPAs prior to HPLC detection. This method gives reasonably low detection limit (ng mL−1 levels), higher recoveries, and also good standard deviations. Moreover, the method is simple, has high sensitivity, consumes much less solvent than traditional methods, and is environment friendly.
References
Aparicio A, San Andres MP, Vera S (2000) Hrc-J High Resolut Chromatogr 23:324–328
Aranda PR, Gil RA, Moyano S, De Vito IE, Martinez LD (2008) Talanta 75:307–311
Bai DS, Li JL, Chen SB, Chen BH (2001) Environ Sci Technol 35:3936–3940
Bianchi L, Colivicchi MA, Della Corte L, Valoti M, Sgaragli GP, Bechi P (1997) J Chromatogr B: Biomed Sci Appl 694:359–365
C.o.F.R. Food and Drug Administration (2001) In: C.o.F.R. Food and Drug Administration (ed) Washington, DC
Capitan-Vallvey LF, Valencia MC, Nicolas EA (2004) Anal Chim Acta 503:179–186
Carabias-Martinez R, Rodriguez-Gonzalo E, Moreno-Cordero B, Perez-Pavon JL, Garcia-Pinto C, Laespada EF (2000) J Chromatogr A 902:251–265
Chen M, Xia Q, Liu M, Yang Y (2011) J Food Sci 76:C98–C103
Clayson DB, Iverson F, Nera E, Lok E, Rogers C, Rodrigues C, Page D, Karpinski K (1986) Food Chem Toxicol: An Int J Publ British Ind Biol Res Assoc 24:1171–1182
Delgado B, Pino V, Ayala JH, Gonzalez V, Afonso AM (2004) Anal Chim Acta 518:165–172
Delgado-Zamarreno MM, Gonzalez-Maza I, Sanchez-Perez A, Carabias Martinez R (2007) Food Chem 100:1722–1727
C. Directive (1995) In: C. Directive (ed) Brussels, pp 1
Fernandez AE, Ferrera ZS, Rodriguez JJS (1998) Anal Chim Acta 358:145–155
Goryacheva IY, Shtykov SN, Loginov AS, Panteleeva IV (2005) Anal Bioanal Chem 382:1413–1418
Guo L, Xie M-Y, Yan A-P, Wan Y-Q, Wu Y-M (2006) Anal Bioanal Chem 386:1881–1887
Ito N, Fukushima S, Tamano S, Hirose M, Hagiwara A (1986) J Nat Cancer Inst 77:1261–1265
Jia GF, Lv CG, Zhu WT, Qiu J, Wang XQ, Zhou ZQ (2008a) J Hazard Mater 159:300–305
Jia G, Lv C, Zhu W, Qiu J, Wang X, Zhou Z (2008b) J Hazard Mater 159:300–305
Karovicova J, Simko P (2000) J Chromatogr A 882:271–281
Kolachi NF, Kazi TG, Khan S, Wadhwa SK, Baig JA, Afridi HI, Shah AQ, Shah F (2011) Food ChemToxicol 49:2548–2556
Liu W, Zhao W-j, Chen J-b, Yang M-m (2007) Anal Chim Acta 605:41–45
Luconi MO, Silva MF, Olsina RA, Fernandez LP (2000) Talanta 51:123–129
Luque M, Rios A, Valcarcel M (1999) Anal Chim Acta 395:217–223
Merino F, Rubio S, Perez-Bendito D (2002) J Chromatogr A 962:1–8
Miura J, Ishii H, Watanabe H (1976) Bunseki Kagaku 25:808–809
Moran G, Harimanow L, Finlayson R, Hook J (2000) Abstr Pap Am Chem Soc 219:U88–U88
Mustafina A, Elistratova J, Burilov A, Knyazeva I, Zairov R, Amirov R, Solovieva S, Konovalov A (2006) Talanta 68:863–868
Ohashi A, Ogiwara M, Ikeda R, Okada H, Ohashi K (2004) Anal Sci 20:1353–1357
Ohashi A, Ito H, Kanai C, Imura H, Ohashi K (2005) Talanta 65:525–530
Ohashi A, Hashimoto T, Imura H, Ohashi K (2007) Talanta 73:893–898
Perrin C, Meyer L (2002) Food Chem 77:93–100
Pinho O, Ferreira I, Oliveira M, Ferreira MA (2000) Food Chem 68:353–357
Purkait MK, Vijay SS, DasGupta S, De S (2004) Dyes Pigments 63:151–159
Purkait MK, Banerjee S, Mewara S, DasGupta S, De S (2005) Water Res 39:3885–3890
Purkait MK, DasGupta S, De S (2006) J Hazard Mater 137:827–835
Purkait MK, DasGupta S, De S (2009) Desalination 244:130–138
Riber J, de la Fuente C, Vazquez MD, Tascon ML, Batanero PS (2000) Talanta 52:241–252
Rodil R, Benito Quintana J, Basaglia G, Pietrogrande MC, Cela R (2010) J Chromatogr A 1217:6428–6435
Ruiz MA, Garcia-Moreno E, Barbas C, Pingarron JM (1999) Electroanalysis 11:470–474
Saad B, Sing YY, Nawi MA, Hashim N, Ali ASM, Saleh MI, Sulaiman SF, Talib KM, Ahmad K (2007) Food Chem 105:389–394
Shah F, Kazi TG, Afridi HI, Naeemullah, Arain MB, Baig JA (2011) J Hazard Mater 192:1132–1139
Shen JC, Shao XG (2006) Anal Chim Acta 561:83–87
Sicilia D, Rubio S, Perez-Bendito D, Maniasso N, Zagatto EAG (1999) Anal Chim Acta 392:29–38
Sikalos TI, Paleologos EK (2005) Anal Chem 77:2544–2549
Sin DWM, Wong YC, Mak CY, Sze ST, Yao WY (2006) J Food Compos Anal 19:784–791
Sirimanne SR, Barr JR, Patterson DG Jr, Ma L (1996) Anal Chem 68:1556–1560
Sun ZM, Liang P, Ding Q, Cao J (2006) Anal Sci 22:911–913
Tang AN, Jiang DQ, Yan XP (2004) Anal Chim Acta 507:199–204
Tombesi NB, Freije H (2002) J Chromatogr A 963:179–183
Ulusoy HI, Akcay M, Ulusoy S, Gurkan R (2011) Anal Chim Acta 703:137–144
Wu D, Wu Q, Bian R, Wang Y (2007) Sep Sci Technol 42:2693–2704
Wuilloud GM, de Wuilloud JCA, Wuilloud RG, Silva MF, Olsina RA, Martinez LD (2002) Talanta 58:619–627
Yang TJ, Tsai FJ, Chen CY, Yang TCC, Lee MR (2010) Anal Chim Acta 668:188–194
Zhang W, Duan C, Wang M (2011a) Food Chem 126:779–785
Zhang WJ, Duan CM, Wang ML (2011b) Food Chem 126:779–785
Zhao W-j, Sun X-k, Deng X-n, Huang L, Yang M-m, Zhou Z-m (2011) Food Chem 127:683–688
Ziyatdinova GK, Gainetdinova AA, Budnikov GK (2010) J Anal Chem 65:929–934
Acknowledgement
This work was supported by the Analysis Test Research Center of Kunming University of Science and Technology, Yunnan Province, China. Innovation Fund for Small and Medium Technology Based Firms (No. 10C26215305131). Thank you to Daniel Paddon for the assistance with editing this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, M., Hu, X., Tai, Z. et al. Determination of Four Synthetic Phenolic Antioxidants in Edible Oils by High-Performance Liquid Chromatography with Cloud Point Extraction Using Tergitol TMN-6. Food Anal. Methods 6, 28–35 (2013). https://doi.org/10.1007/s12161-012-9413-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-012-9413-7