
Abstract
In the current study, the n-hexane, chloroform, ethyl acetate, methanol, and water extracts of Phagnalon graecum Boiss. (Asteraceae), a spice plant, were investigated for their enzyme inhibitory activity against acetylcholinesterase, butyrylcholinesterase, lipoxygenase, tyrosinase, and antioxidant activities. Antioxidant activity of the extracts was determined by 2,2-diphenyl-1-picrylhydrazyl radical scavenging, ferric ion-chelation activity, and ferric-reducing antioxidant power tests. Total phenol (gallic acid equivalent) and flavonoid (quercetin equivalent) contents were calculated. Essential oil of the plant was analyzed by GC-MS, while the selected phenolic acids were analyzed by HPLC. The extracts had insignificant inhibition against the tested enzymes, whereas they displayed a remarkable antioxidant activity. Therefore, to the best of our knowledge, this study presents the first data on biological activity and phytochemical content of P. graecum and this species could be utilized as potential antioxidant applicable for food preservation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The genus Phagnalon Cass. (Asteraceae) is represented by three species in the Flora of Turkey; namely Phagnalon graecum Boiss., Phagnalon kotschyi Schultz Bip. & Boiss., and Phagnalon rupestre (L.) DC (Davis 1988). Some Phagnalon species are used as spice in Italy (Lentini and Venza 2007) or herbal medicine in some folkloric systems such as Israel (Friedman et al. 1986), Spain (Pardo De Santayana et al. 2005), Palestine (Abu-Rabia 2005), and Jordan (Hudaib et al. 2008). A limited number of phytochemical studies performed in some Phagnalon species revealed existence of quinine and some phenolic acid conjugates (Hausen and Schultz 1977; Góngora et al. 2001, 2002a, b, c, 2003, Olmos et al. 2005), some terpenes and thymol derivatives (El-Dahmy et al. 1994), and some flavonoids (Góngora et al. 2001, 2002a, b).
However, there has been no report up to date on the phytochemistry or biological activity of P. graecum. Only a recent study describing anticholinesterase and antioxidant activities of Phagnalon saxatile, a Mediterranean species growing in Italy (Conforti et al. 2010), prompted us to investigate similar enzyme inhibitory and antioxidant activities of P. graecum, which is another species of this genus growing in the Mediterranean region. Consequently, the n-hexane, chloroform, ethyl acetate, methanol, and water extracts prepared from the aerial parts of P. graecum were tested for their enzyme inhibitory activity against acetylcholinesterase (AChE), butyrylcholinesterase (BChE), lipoxygenase (LOX), and tyrosinase (TYRO) using ELISA microplate reader at 50, 100, and 200 μg/mL concentrations. Antioxidant activity was determined using three in vitro test systems; 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical-scavenging activity, ferric ion-chelating activity, and ferric-reducing antioxidant power (FRAP) at 125, 250, 500, 1,000, and 2,000 μg/mL concentrations. For phytochemical characterization of the extracts, total phenol (gallic acid equivalent) and flavonoid (quercetin equivalent) contents were established using Folin–Ciocalteau’s and aluminum chloride reagents, respectively. Additionally, high-performance liquid chromatography (HPLC) analysis for quantative determination of phenolic acids and gas chromatography–mass spectrometry (GC-MS) analysis to define essential oil composition were applied in the plant.
Materials and Methods
Plant Material
The plant sample was collected in February, 2009 in the vicinity of Belek town of Antalya province, Turkey and identified by Prof. Dr. Hayri Duman of Department of Biology, Faculty of Science and Art, Gazi University, Ankara, Turkey. The voucher specimen (GUE 2788) is preserved in the herbarium of the Department of Pharmacognosy, Faculty of Pharmacy, Gazi University, Ankara, Turkey.
Preparation of the Extracts
The aerial parts of the plant were dried in shade at room temperature and ground in a mechanic grinder to fine powder and weighed accurately (218.54 g). The powdered plant material was firstly macerated with 500 mL of n-hexane at room temperature for 2 days and then successively with chloroform, ethyl acetate, methanol, and distilled water in the same manner. Then, the organic phases obtained in each extraction were filtered through filter paper and evaporated in reduced pressure until dryness to give the crude extracts, which were kept at +4 °C until the time bioactivity and phytochemical studies were performed. Yields (w/w) of the prepared extracts were obtained as follows; n-hexane extract: 9.38 g; chloroform extract: 13.65 g; ethyl acetate extract: 12.23 g; methanol extract: 18.30 g; water extract: 16.79 g.
Isolation of the Essential Oil
The air-dried aerial parts of the plant (38.0 g) were hydrodistilled for 3 h using a Clevenger-type apparatus to produce a trace amount of essential oil which was trapped into n-hexane.
Enzyme Inhibitory Activity Assays
AChE and BChE Inhibitory Activity
AChE and BChE inhibitory activity was measured by slightly modifying the spectrophotometric method developed by Ellman et al. (1961). Electric eel AChE (Type-VI-S, EC 3.1.1.7, Sigma) and horse serum BChE (EC 3.1.1.8, Sigma) were used, while acetylthiocholine iodide and butyrylthiocholine chloride (Sigma, St. Louis, MO, USA) were employed as substrates of the reaction. 5,5′-Dithio-bis(2-nitrobenzoic)acid (DTNB, Sigma, St. Louis, MO, USA) was used for the measurement of the cholinesterase activity. All the other reagents and conditions were the same as described in our recent publication (Senol et al. 2010). Briefly, in this method, 140 μL of sodium phosphate buffer (pH 8.0), 20 μL of DTNB, 20 μL of test solution and 20 μL of AChE/BChE solution were added by multichannel automatic pipette (Eppendorf, Germany) in a 96-well microplate and incubated for 15 min at 25°C. The reaction was then initiated with the addition of 10 μL of acetylthiocholine iodide/butyrylthiocholine chloride. The hydrolysis of acetylthiocholine iodide/butyrylthiocholine chloride was monitored by the formation of the yellow 5-thio-2-nitrobenzoate anion as a result of the reaction of DTNB with thiocholines, catalyzed by enzymes at a wavelength of 412 nm utilizing a 96-well microplate reader (VersaMax Molecular Devices, CA, USA). The measurements and calculations were evaluated by using Softmax PRO 4.3.2.LS software. Percentage of inhibition of AChE/BChE was determined by comparison of rates of reaction of samples relative to blank sample (ethanol in phosphate buffer pH = 8) using the formula \( \left( {E - S} \right)/E \times 100 \), where E is the activity of enzyme without test sample and S is the activity of enzyme with test sample. The experiments were done in triplicate. Galanthamine, the anticholinesterase alkaloid-type of drug isolated from the bulbs of snowdrop (Galanthus sp.), was purchased from Sigma (St. Louis, MO, USA) and used as reference.
LOX Inhibitory Activity
Inhibitory activity of the extracts against LOX was measured by modifying the spectrophotometric method developed by Tappel (1962). LOX (EC 1.13.11.12, type I-B), baicalein (reference drug), and linoleic acid (substrate) were purchased from Sigma (St. Louis, MO, USA). The reaction mixture of total volume of 190 μL contained sodium phosphate buffer (pH 8.0), sample and enzyme solutions, which were later incubated for 10 min at 25°C. The reaction was initiated by addition of 10 μL of linoleic acid solution. Due to the formation of (9Z, 11E)-(13S)-13-hydroperoxyoctadeca-9,11-dienoate, the change of absorbance at 234 nm was followed for 6 min. All of the reactions were performed in 96-well microplates using ELISA microplate reader (Spectramax Plus384 Molecular Devices, CA, USA). The percentage (%) inhibition was calculated as \( \left( {E - S} \right)/{ }E \times 100 \), where E is the activity of the enzyme without test compound and S is the activity of enzyme with test compound. The measurements and calculations were evaluated by using Softmax PRO 4.3.2.LS software.
TYRO Inhibitory Activity
Inhibition of tyrosinase (EC 1.14.1.8.1, 30 U, mushroom tyrosinase, Sigma) was determined using the modified dopachrome method with l-DOPA as substrate (Masuda et al. 2005). Assays were conducted in a 96-well microplate and an ELISA microplate reader (VersaMax Molecular Devices, CA, USA) was used to measure absorbance at 475 nm. The extracts dissolved in DMSO (50%) with 80 μL of phosphate buffer (pH 6.8), 40 μL of tyrosinase, and 40 μL of l-DOPA were put in each well. Each sample was accompanied by a blank that had all the components except for l-DOPA. Results were compared with a control consisting of 50% DMSO as well as the reference (α-kojic acid). Percentage inhibition of tyrosinase was calculated according to the formula given below:
Antioxidant Activity Assays
DPPH Free Radical-Scavenging Assay
The stable DPPH radical-scavenging activity was determined by Hatano’s method (Hatano 1995). The samples dissolved in ethanol (75%) were mixed with DPPH solution (1.5 × 10−4 M) and left for 20 min incubation in dark at room temperature. Remaining DPPH amount was measured at 520 nm using a Unico 4802 UV–visible double beam spectrophotometer (USA). Gallic acid was employed as the reference and the experiments were performed in triplicate. Inhibition of DPPH in percent (I%) was calculated as given below:\( I\% = \left[ {\left( {{A_{\text{blank}}} - {A_{\text{sample}}}} \right)/{A_{\text{blank}}}} \right] \times 100 \), where A blank is the absorbance of the control reaction (containing all reagents except the test sample), and A sample is the absorbance of the extracts/reference.
Fe+2-Ferrozine Test System for Ferric Ion-Chelating Activity
The ferric ion-chelating effect of the extracts by Fe+2-ferrozine test system was estimated by the method of Chua et al. (2008). Briefly, 740 μL of methanol and the samples were incubated with 2 mM FeCl2 solution. The reaction was initiated by the addition of 40 μL of ferrozine solution into the mixture and left standing at ambient temperature for 10 min. The absorbance of the reaction mixture was measured at 562 nm. The ratio of inhibition of ferrozine-Fe2+ complex formation was calculated as follows:
% Inhibition= [(Absorbance of control - Absorbance of test sample)/Absorbance of control] × 173 100. The control contained only FeCl2 and ferrozine. Analyses were run in triplicate and expressed as average values with S.E.M. (standard error of the mean). The reference used in this method was ethylenediamine tetraacetic acid.
Ferric-Reducing Antioxidant Power Assay (FRAP)
The ferric-reducing power was tested using the assay of Oyaizu (1986). According to the method; the samples were added to phosphate buffer (pH 6.6) and potassium ferricyanide. Later, the mixture was incubated at 50 °C for 20 min, which was followed by addition of trichloroacetic acid. After the mixture was shaken vigorously, the solution was mixed with distilled water and FeCl3 (0.1%, w/v). After 30 min incubation, absorbance was read at 700 nm using a Unico 4802 UV–visible double beam spectrophotometer (USA). Analyses were achieved in triplicate. Increasing absorbance of the reaction indicated increase in reducing power. The reference used in this method was chlorogenic acid.
Phytochemical analyses
Total Phenol and Flavonoid Contents
Phenolic content of the extracts was determined in accordance with Folin–Ciocalteau’s method (Singleton and Rossi 1965). In brief, the samples dissolved in ethanol (75%) were mixed with 750 μL of Folin–Ciocalteau’s reagent and 600 μL of sodium carbonate in test tubes. The tubes were then vortexed and incubated at 40 °C for 30 min. A number of dilutions of gallic acid were obtained to prepare a calibration curve. Afterward absorption was measured at 760 nm at a Unico 4802 UV–visible double beam spectrophotometer (USA). Total flavonoid content was calculated by aluminum chloride colorimetric method (Woisky and Salatino 1998). A number of dilutions of quercetin were obtained to prepare a calibration curve. Then, the extracts and quercetin were separately mixed with 75% ethanol, 10% aluminum chloride, 1 M sodium acetate. Following incubation for 30 min at room temperature, absorbance of the reaction mixture was measured at 415 nm with a Unico 4802 UV–visible double beam spectrophotometer (USA). The total phenolic and flavonoid contents of the extracts were expressed as gallic acid and quercetin equivalents (in milligrams per gram extract), respectively.
Conditions of HPLC Analysis for Phenolic Acid Determination
Chemicals used for HPLC (methanol and formic acid) were of chromatographic grade (Sigma-Aldrich, St. Louis, MO, USA). Standards of the phenolic acids used in this study; gallic acid (GA), protocatechuic acid, p-hydroxybenzoic acid, vanillic acid, caffeic acid, chlorogenic acid, syringic acid, ferulic acid, and propylparaben as internal standard (IS) were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Merck (GmbH, Darmstadt, Germany). Analysis of phenolic acids in the extracts of P. graecum was carried out with Agilent 1100 series auto sampler system from Agilent, GL Sciences Inc (Waldbronn, Germany) equipped with a system controller, DAD detector (G 1315B, 280 nm), an quaternary LC pump (G1311A). The separation was carried out with a Zorbax Eclipse XDB-C18 column (150 mm, 4.6 mm i.d. and 5 μm particle size) (Agilent, Waldbronn, Germany) with the column temperature set at 25°C. Chromatographic separation was carried out using two solvent systems: (A) methanol/water/formic acid (10:88:2,v/v/v); (B) methanol/water/formic acid (90:8:2, v/v/v), as reported elsewhere (Ozturk et al., 2007). Ultrapure water was supplied from Human UP 9000 System (18 mW) water purification system. The analyses were performed by using a linear gradient program. A gradient elution program was applied as follows: the pumping solution was 100% A between 0 and 15 min; it decreased to 85% A from 15 to 20 min, further decreasing to 50% A from 20 to 30 min, to 0% A from 30 to 35 min and at 100% A between 35–42 min. The flow-rate was adjusted as 1 mL/min and the injection volume was 10 μL. Signals were detected at 280 nm. Besides, the IS (propylparaben) technique was applied to the analysis in order to increase the repeatability. The extracts were dissolved in a mixture of methanol and water (1:1, v/v) and injected into the HPLC. Identification of the peaks was achieved as follows: All of the phenolic acid standards were dissolved individually. Propylparaben was used as an IS to increase the repeatability of the method. The integrated peak areas and their retention times were computed to get the rate of peak normalization (peak area/peak retention time) of the relevant phenolic acids, and their amounts were calculated in the related extracts via their calibration curves.
Conditions of GC-MS Analysis of the Essential Oil
The GC-MS analysis was carried out with an Agilent 5975 GC-MSD system. Innowax FSC column (60 m × 0.25 mm, 0.25 μm film thickness) was used with helium as carrier gas (0.8 mL/min). GC oven temperature was kept at 60°C for 10 min and programmed to 220°C at a rate of 4°C/min, and kept constant at 220°C for 10 min and then programmed to 240°C at a rate of 1°C/min. Split ratio was adjusted at 40:1. The injector temperature was set at 250°C. Mass spectra were recorded at 70 eV. Mass range was from m/z 35 to 450. The GC analysis was carried out using an Agilent 6890N GC system. FID detector temperature was 300°C. To obtain the same elution order with GC-MS, simultaneous auto-injection was done on a duplicate of the same column applying the same operational conditions. Relative percentage amounts of the separated compounds were calculated from the FID chromatograms.
Identification of the essential oil components was carried out by comparison of their relative retention times with those of authentic samples or by comparison of their relative retention index to a series of n-alkanes. Computer matching against commercial (Wiley GC-MS Library, Adams Library, MassFinder 3 Library; McLafferty and Stauffer 1989; Koenig et al 2004) and in-house “Başer Library of Essential Oil Constituents” built up by genuine compounds and components of known oils, as well as MS literature data (Jennings and Shibamoto 1980; Joulain and Koenig 1998; ESO 1999) was used for the identification.
Results and Discussion
Enzyme Inhibitory and Antioxidant Effects
The results of enzyme inhibitory activity of the extracts of P. graecum against AChE and BChE (in Alzheimer’s disease), LOX (in inflammation), and TYRO (in hyperpigmentation and melasma) are shown in Table 1. Accordingly, among the extracts, only the water extract showed a moderate inhibition against AChE having 20.89 ± 3.24 and 41.26 ± 0.07% at 100 and 200 μg/mL, respectively. However, the extracts displayed either no or very low inhibition against the tested enzymes. The extracts were tested for their antioxidant activity at 125, 250, 500, 1,000, and 2,000 μg/mL concentrations using three in vitro test systems. As shown in Table 2, the ethyl acetate, methanol, and water extracts exerted a high scavenging activity against DPPH radical. The most effective extract in this test was found to be the ethyl acetate extract (88.33 ± 0.47%) at 2,000 μg/mL. On the other hand, the water extract had the highest ferric ion-chelating capacity (75.0 ± 0.37%), followed by the n-hexane extract (66.32 ± 4.38%; Table 2). In the FRAP assay, the ethyl acetate and methanol extracts showed a remarkable activity at 2,000 μg/mL, which was similar to that of reference (chlorogenic acid) at 1,000 μg/mL (Fig. 1).

FRAP results of the P. graecum extracts and the reference (chlorogenic acid)
Total Phenol and Flavonoid Contents
Calibration equations were calculated as \( y = 2.8034x + 0.009\left( {{r^2} = 0.9796} \right){\text{and}}\;y = 2.7319x - 0.0308\left( {{r^2} = 0.9934} \right) \) for total phenol and total flavonoid contents, respectively. According to the results of total phenol and flavonoid amounts of the extracts listed in Table 2, the ethyl acetate extract possessed the highest total phenol content (219.08 ± 6.96 mg/g extract as gallic acid equivalent). On the other hand, the chloroform extract had the richest total flavonoid amount as quercetin equivalent (711.46 ± 8.71 mg/g extract), followed by the ethyl acetate extract (380.45 ± 6.14 mg/g extract).
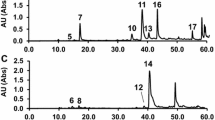
Analysis of Phenolic Acids in the Methanol and Ethyl Acetate Extracts
Determination of phenolic acids was realized by injecting the methanol and ethyl acetate extracts prepared from P. graecum into the column after addition of IS. The related chromatograms are demonstrated in Fig. 2. The amounts of the relevant phenolic acids were calculated by using their calibration equations as reported previously by one of us (Ozturk et al 2007). HPLC analysis of the extracts revealed presence of mainly two phenolic acids; ferulic and o-coumaric acids, which were only found in the ethyl acetate and methanol extracts (Table 2). According to our results, the methanol extract was rather richer in these phenolic acids then the ethyl acetate extract. Rest of the extracts did not contain any of the phenolic acids analyzed herein.

The representative chromatograms of a methanol and b ethyl acetate extracts prepared from P. graecum, and c mixture of standard phenolic acids: The phenolic acids are symbolized as 1 (gallic acid), 2 (protocatechuic acid), 3 (p-hydroxybenzoic acid), 4 (vanillic acid), 5 (caffeic acid), 6 (chlorogenic acid), 7 (syringic acid), 8 (p-coumaric acid), 9 (ferulic acid), 10 (o-coumaric acid), 11 (trans-cinnamic acid) and 12 (propylparaben as internal standard) on the chromatograms
Essential oil composition
Composition of the essential oil of P. graecum was established by GC-MS and 38 components were identified representing 86.0% of the oil. Table 3 shows the identified compounds and their percentages obtained by GC-MS as well as the retention indices listed in order of their elution from the HP Innowax capillary column. According to GC-MS data, germacrene D (21.3%) was the dominant compound in the oil, followed by hexahydrofarnesyl acetone (9.6%), β-caryophyllene (9.4%), hexadecanoic acid (6.1%), caryophyllene oxide (6.0%), and δ-cadinene (3.2%).
Only some species of the genus Phagnalon have been investigated so far in terms of phytochemistry and biological activity. Nevertheless, our literature survey pointed out to absence of phytochemical and bioactivity data on P. graecum, which encouraged us to perform the current study. Up to date, there has been only one report on AChE and BChE inhibitory activity of a single Phagnalon species; namely P. saxatile (Conforti et al. 2010), which also encouraged us to work on P. graecum in a similar manner. The methanol extract of P. saxatile showed no inhibition against AChE, while it displayed 100% inhibition on BChE at 1,000 μg/mL, which was tested using the same method used in our study. Although the extract per se did not show anti-AChE activity, its main constituents including caffeic acid, apigenin, apigenin-7-O-β-glucopyranoside, 3,5-dicaffeoylquinic acid, and luteolin-4'-O-glucopyranoside had 100% inhibitory activity against AChE at 100 μg/mL, except for chlorogenic and methylchlorogenic acids. All of the active compounds against AChE were found to have 100% inhibition at 100 μg/mL towards BChE, whereas only luteolin-4'-O-glucopyranoside displayed lower inhibition (50.3%) at the same concentration. It should be noted that cholinesterase inhibitory data of P. saxatile seems to differ with our anti-cholinesterase results of P. graecum. However, this dissimilarity might be apparently resulting from their different phytochemical contents. Considering the same report on P. saxatile, its methanol extract exerted a very high DPPH radical-scavenging activity (over 90% at 100, 250, 500, and 1,000 μg/mL), which is compliant with our results on the methanol extract of P. graecum. The extract and its phenolic components also showed remarkable antioxidant activity in β-carotene bleaching assay and inhibition of nitric oxide production in that study (Conforti et al. 2010). Since the phenolic compounds had the high antioxidant activity in P. saxatile, the phenolic acids in the methanol extract of P. graecum identified in our analysis could be assumed to be responsible for the marked antioxidant activity of this extract.
There has been only one other study on antioxidant activity related to other Phagnalon species. In that study, Góngora et al. (2003) described xanthine oxidase inhibitory activity of the caffeoyl conjugates of prenylhydroquinone glucoside and quinic acid isolated from P. rupestre. In the conclusion of this study, the authors stated that the caffeoyl moiety is vital for both the antiperoxidative and radical-scavenging activities, and the methylation of the quinic carboxyl group enhanced the effectiveness on xanthine oxidase inhibitory activity.
Although various derivatives of phenolic compounds were identified in different Phagnalon species (Hausen and Schultz 1977; Góngora et al. 2001, 2002a, b, 2003; Olmos et al. 2005), none of the phenolic acids (ferulic and o-coumaric acids), that we identified in P. graecum, has been reported to exist in other Phagnalon species up to date.
Among all species of Phagnalon, only the essential oil composition of Phagnalon sordidum was elucidated (Salgues 1953), which was dominated by α-pinene and azulogenene, a sesquiterpene derivative commonly found in roman chamomile. Conversely, the essential oil of P. graecum examined herein did not contain azulogenene and a quite low amount of α-pinene (0.2%) (Table 3).
Conclusion
Our results on enzyme inhibitory and antioxidant activities of P. graecum indicated that the plant has a strong antioxidant activity and quite prosperous in total phenol and flavonoid contents, which can be considered to be a rich source of potential antioxidants. The water extract having moderate AChE inhibitory activity may be considered to be tested for further phytochemical analysis in order to elucidate its possible active constituents. To the best of our knowledge, we herein report the first study describing AChE, BChE, LOX, and TYRO enzyme inhibitory and antioxidant activities of P. graecum as well as its phenolic content and essential oil composition.
References
Abu-Rabia A (2005) Alt Compl Ther 11:295
Chua MT, Tung YT, Chang ST (2008) Bioresour Technol 99:1918
Conforti F, Rigano D, Formisano C, Bruno M, Loizzo MR, Menichini F, Senatore F (2010) J Enzym Inh Med Chem 25:97
Davis PH (1988) Phagnalon Cass. In: Davis PH (ed) Flora of Turkey and the East Aegean Islands, vol 5. Edinburgh University Press, Edinburgh, pp 78–79
El-Dahmy SI, Abdel Aal M, Abd El-Fatah H, Fid F (1994) Acta Pharm Hung 64:115
Ellman GL, Courtney KD, Andres V, Featherstone RM (1961) Biochem Pharmacol 7:88
ESO 2000 (1999) The complete database of essential oils. Boelens aroma chemical information service, The Netherlands
Friedman J, Yaniv Z, Dafni A, Palewitch D (1986) J Ethnopharmacol 16:275
Góngora L, Giner RM, Máñez S, Del Carmen-Recio M, Ríos JL (2001) J Nat Prod 64:1111
Góngora L, Giner RM, Máñez S, Del Carmen-Recio M, Ríos JL (2002a) Planta Med 68:561
Góngora L, Mánez S, Giner RM, Del Carmen-Recio M, Gray AI, Ríos JL (2002b) Phytochemistry 59:857
Góngora L, Giner RM, Máñez S, Del Carmen-Recio M, Schinella G, Ríos JL (2002c) Life Sci 71:2995
Góngora L, Máñez S, Giner RM, Del Carmen-Recio M, Schinella G, Ríos JL (2003) Planta Med 69:396
Hatano T (1995) Natural Med 49:357–363
Hausen BM, Schultz KH (1977) Planta Med 32:287
Hudaib M, Mohammad M, Bustanji Y, Tayyem R, Yousef M, Abuirjeie M, Aburjai T (2008) J Ethnopharmacol 120:63
Jennings WG, Shibamoto T (1980) Quantitative analysis of flavor and fragrance volatiles by glass capillary GC. Academic, New York
Joulain D, Koenig WA (1998) The atlas of spectra data of sesquiterpene hydrocarbons. EB, Hamburg
Koenig D, Joulain D, Hochmuth DH (2004) Terpenoids and related constituents of essential oils. MassFinder 3. Hochmuth DH (Ed). Convenient and Rapid Analysis of GC-MS, incorporating W.A. Hamburg, Germany.
Lentini F, Venza F (2007) J Ethnobiol Ethnomed 3:15
Masuda T, Yamashita D, Takeda Y, Yonemori S (2005) Biosci Biotechnol Biochem 69:197
McLafferty FW, Stauffer DB (1989) The Wiley/NBS registry of mass spectral data. Wiley, New York
Olmos A, Máñez S, Giner RM, Del Carmen-Recio M, Ríos JL (2005) Nitric oxide. Biol Chem 12:54
Ozturk N, Tuncel M, Tuncel NB (2007) J Liq Chrom Rel Technol 30:587
Oyaizu M (1986) Jap J Nutr 44:307
Pardo De Santayana M, Blanco E, Morales R (2005) J Ethnopharmacol 98:1
Salgues R (1953) Materia Veg 1:139
Senol FS, Orhan I, Yilmaz G, Cicek M, Sener B (2010) Food Chem Toxicol 48:781
Singleton VL, Rossi JA Jr (1965) Am J Enol Viticult 16:144
Tappel AL (1962) Methods in enzymology. Academic, New York, p 539
Woisky R, Salatino A (1998) J Apicol Res 37:99
Acknowledgment
F.S. Senol expresses her genuine gratitude to the Scientific and Technological Research Council of Turkey (TUBITAK) for the scholarship provided during her Ph.D. program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Erdogan Orhan, I., Senol, F.S., Demirci, B. et al. Phytochemical Characterization of Phagnalon graecum Boiss. by HPLC and GC-MS with its Enzyme Inhibitory and Antioxidant Activity Profiling by Spectrophotometric Methods. Food Anal. Methods 6, 1–9 (2013). https://doi.org/10.1007/s12161-012-9386-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-012-9386-6