
Abstract
Background
Poor sleep is prospectively linked to all-cause and cardiovascular mortality. Inflammatory processes may be an important biological mechanism linking poor sleep to cardiovascular disease. Such processes involve active participation of signaling molecules called cytokines in development of atherosclerotic plaques.
Purpose
I review evidence from experimental sleep deprivation and clinical observational studies suggesting a bidirectional relationship between sleep and inflammatory cytokines.
Results
Findings from sleep deprivation studies indicate that sleep loss is associated with increases in these cytokines. Similarly, studies in clinical populations with sleep problems, such as primary insomnia patients and those diagnosed with major depression, also show elevations in these same cytokines.
Conclusions
Bidirectional communication between the brain and the immune system is carried out through a complex network of autonomic nerves, endocrine hormones, and cytokines. Disturbed sleep appears to perturb the functioning of this network and therefore contribute to elevations in inflammatory mediators linked to cardiovascular disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
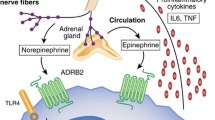
Inflammare loosely translates from Latin as “to set on fire,” which fittingly characterizes the potential of chronic inflammation to cause harm [1]. Inflammation is coordinated through the immune system, with leukocytes, cells of the immune system, relying on signaling molecules termed cytokines to communicate and coordinate inflammatory processes. In addition to the immune system, adipose and muscle tissue are also sources of inflammatory cytokines [2]. Chronic activation of inflammatory processes is thought to drive atherosclerosis and promote cardiovascular disease. Although acute inflammation can be thought of as a short-lasting, highly localized process, chronic inflammation is different, involving elevations in systemic circulating levels of inflammatory makers such as C-reactive protein (CRP) and interleukin-6 (IL-6), increased production of inflammatory cytokines by immune cells and within these immune cells, and increased gene transcription of inflammatory cytokines [1, 3, 4]. Systemic levels of inflammatory cytokines communicate distally with the brain, and the magnitude, extent, and nature of leukocyte action can be modified by the brain via autonomic nerves and neuroendocrine hormones [5–7]. It is through these pathways that poor sleep can influence inflammatory cytokines [8–10] and conversely through which the immune system, via these same cytokines, can affect sleep [11] (Fig. 1).

Sleep, inflammation, and cardiovascular disease outcomes
Characterizing Poor Sleep
Sleep processes are complicated, involving separate coordinated central and peripheral processes. Thus, clarity on the aspects of sleep and specifically poor sleep that are related to poor health is important. Poor sleep can be divided into two general categories: dysomnias and parasomnias. Dysomnias involve difficulties with sleep initiation, maintenance, and/or excessive sleepiness and include disorders such as primary insomnia, sleep apnea, periodic leg movements, and narcolepsy. Parasomnias involve abnormal behavior or physiological phenomena during sleep including sleepwalking, sleep terror, and nightmare disorders. Of all forms of poor sleep, primary insomnia is perhaps the most common sleep disorder, with a prevalence in young and middle-aged adults being approximately 6% [12] and increasing to 40% in older adults [13]. Primary insomnia is twice as likely in women as in men [12]. To be diagnosed with primary insomnia, one must report difficulty initiating or maintaining sleep, or having nonrestorative sleep for at least 1 month. Sleep difficulties must cause significant impairment or distress, and difficulties cannot be directly caused by other sleep-related disorders, medical or mental health disorders, or current medication use. These criteria do not include any requirement regarding amount of sleep; thus, an insomnia patient can have substantial amounts of sleep per night but still have insomnia symptoms if they report trouble falling asleep, maintaining sleep, or having nonrestorative sleep. Decreased or excessive amounts of sleep can be problematic. As described later, short sleep times are often related with poor health outcomes. Average sleep duration is close to 7.5 h; typically, sleep times that are considered short are less than 6 h of sleep.
In contrast to primary insomnia, “insomnia symptoms” refer not to a specific disorder but instead to the presence of difficulties initiating and maintaining sleep or nonrestorative sleep. The prevalence of insomnia symptoms is considerably higher than that of the disorder. Symptom prevalence ranges from 10–48%, depending on the criteria for chronicity and severity [12]. The prevalence of insomnia symptoms increases with age, with 50% of adults over 65 years of age reporting presence of chronic symptoms, and in terms of gender, women are approximately 50% more likely than men to report insomnia symptoms [12]. As described in the following section, studies examining poor sleep and mortality have typically examined either short sleep times or insomnia symptoms as predictors of health outcomes and have not examined whether individuals have a diagnosis of primary insomnia.
Poor Sleep, Mortality, and Cardiovascular Disease
Self-reported poor sleep is prospectively associated with all-cause mortality. Biobehavioral variables such as health status, alcohol use, smoking, and obesity are each associated with poor sleep; however, they do not fully account for the relationship. In a sample of nearly 5,000 men and women, ages 30–69, sleeping ≤6 or ≥9 h was associated with 1.7 higher relative risk of mortality over 9 years when compared to those sleeping 7 to 8 h [14]. Some of this relationship was likely due to other co-related factors; the relative risk dropped to 1.3 after controlling for age, gender, health status, smoking, inactivity, alcohol use, and social network size, yet it remained statistically significant. The Cancer Prevention Study II included nearly one million men and women, ages 30–102, and Kripke and colleagues [15] found a similar relationship in that sleeping less than 4.5 h for men and 3.5 h for women was associated with a 15% increase in mortality over 6 years, with the inclusion of 32 covariates including specific medical conditions and prescription sleeping pill use.
Objective assessment of sleep suggests that insomnia symptoms may be an even stronger predictor of mortality than sleep time. Dew and colleagues [16] followed a sample of 184 healthy older adults over 12 years and measured sleep via polysomnography instead of self-report. Insomnia symptoms, such as difficulty falling asleep and poor sleep efficiency (defined as sleep latency greater 30 min and sleep efficiency less than 80%, respectively), remained robustly associated with higher mortality after controlling for age, gender, and baseline medical status. Specifically, a sleep onset of longer than 30 min was associated with a relative risk ratio of 2.14, and a sleep efficiency of less than 80% was associated with a relative risk ratio of 1.71 for all-cause mortality. Objectively assessed sleep time had a relative hazard of 1.3, similar in magnitude to studies using self-reported sleep time. But in this study with a substantially smaller sample size than the ones mentioned in the above paragraph, the hazard ratio was not statistically significant after controlling for age, gender, and baseline medical status. Thus, self-report and polysomnographic markers of poor sleep are associated with all-cause mortality, independent of other typical risk factors, over a span of time ranging from 6 to 12 years. How long the sleep problems need to persist in order to impact mortality is unclear. Sleep duration results from the Cancer Prevention Study II were based on a single item, “on the average, how many hours do you sleep each night?” [15]. Objective sleep data reported by Dew and colleagues were based on one to two nights of sleep recordings done in a sleep laboratory [16].
Moving from all-cause mortality to cardiovascular disease-specific outcomes, chronic insomnia symptoms, assessed via 2-week sleep diaries, are associated with high blood pressure and cardiovascular disease [17], and in community samples, insomnia symptoms predict coronary artery disease (CAD) related death over 12–20 years [18, 19]. In a review by Schwartz et al. [20], the authors report six longitudinal studies indicating that insomnia complaints at baseline were associated with increased CAD-related death, even after controlling for diabetes status, hypertension, cholesterol levels, and smoking use. Insomnia symptoms at baseline are associated with higher relative risk for myocardial infarction in as little as 3 years [21] and as long as 20 years later [19]. The relative risk ratios have ranged from 1.6 to 3.9, with longer prospective windows tending to have higher risk ratios [18, 19, 21–24]. In these studies, ages at baseline have ranged from 25 to 64; it is unclear whether there is a vulnerable age range for which insomnia symptoms are particularly closely related to cardiovascular disease. Besides age, gender may also be an important moderator of sleep and cardiovascular disease. Mallon and colleagues [18] found a relationship between insomnia symptoms and cardiovascular disease-related death in men, but not women. In contrast, Eaker and colleagues [19] found a relationship between insomnia symptoms and cardiovascular mortality in a sample comprised exclusively of women. Thus, age and gender are important moderators to account for, but other biobehavioral variables are also important.
Depressive symptoms, obesity, alcohol use, and smoking are related to both poor sleep and cardiovascular disease. Although studies typically control for these variables, greater discussion of their relationships with sleep and health are in order. In depression, insomnia symptoms are highly prevalent [25], yet the association between insomnia symptoms and mortality remain after controlling for depressive symptoms [18], suggesting that poor sleep may exert its effects somewhat independently from depression.
The extent and nature of sleep difficulties in obese individuals likely involve some combination of sleep apnea, short sleep time, and insomnia. Sleep apnea is a disorder involving frequent interruptions in breathing during sleep, and the prevalence estimates of apnea in obese individuals have ranged between 40% and 90% [26, 27]. In addition, a number of studies have found that increased body mass index (BMI) is cross-sectionally and prospectively associated with short sleep time [28]. Insomnia is also a problem in this population, independent of apnea; polysomnographic evidence suggests that obese patients without apnea experience more nighttime wakefulness and greater daytime sleepiness than normal weight controls [29].
Alcohol use exerts complex effects on cardiovascular health and sleep. Moderate alcohol use is associated with reduced incidence of myocardial infarctions [30], but excessive intake is linked with increased all-cause and cardiovascular-related mortality [31]. Similarly, alcohol exerts mixed effects on sleep, initially hastening sleep onset, but then causing increased wakefulness later in the night [32]. Alcohol dependence is associated with poor sleep quality [33], and insomnia is associated with a 1.7 odds ratio of developing alcohol dependence over a 3- to 4-year period [34]. These relationships suggest that alcohol use may play a moderating or synergistic role in promoting cardiovascular disease in poor sleepers.
There is some evidence that smoking prevalence is nearly double in insomnia patients. As high as 40% of the insomnia patients were smokers, and of these smokers with insomnia, 45% reported smoking 5 min before bed [35]. Because of its effects on inflammation and cardiovascular disease [36], cigarette use is also likely an important health behavior relevant in poor sleepers.
The relationships between sleep and health with moderators such as age, depressive symptoms, obesity, alcohol, smoking, and gender indicate a complicated web of interrelationships. Different studies have controlled for different sets of moderators, yet overall, these moderators have not dispelled the relationship between poor sleep and mortality.
Poor Sleep and Inflammation
Exactly how poor sleep affects cardiovascular disease and mortality likely involves a complex set of direct and indirect biological and behavioral mechanisms. However, there is a convergence of evidence suggesting that sleep inflammatory processes may be an important pathway. Chronic elevations of inflammatory mediators such as IL-6, CRP, and soluble intercellular adhesion molecule (sICAM) are prospectively associated with cardiovascular disease-related outcomes [3], and these inflammatory markers are thought to play an important role in linking obesity, hypertension, and type 2 diabetes to cardiovascular disease [1]. Animal and human experimental studies examining the effects of sleep deprivation provide clear evidence that sleep loss affects inflammatory markers, and although the findings are complex, there is compelling evidence that in humans, sleep restriction is associated with increases in inflammatory markers. Furthermore, initial studies in clinical sleep-deprived populations provide corroborating evidence for this relationship.
Inflammatory Processes
Inflammation is traditionally thought of as an acute process, through which some insult, such as an infection or injury, triggers a phalanx of well-coordinated local and systemic responses. Coordination of these processes is carried out through a network of signaling molecules that include cytokines, adhesion molecules, and chemokines. Measurement of these molecules in general circulation can provide some general, systemic clues as to the state of inflammation in the body. Chronic inflammation is a prolonged state of immune activation due to factors that perpetuate immune responses to normal tissue such as invading microorganisms that are difficult to destroy or inactivate, or the prolonged presence of chemicals that repeatedly evoke immune responses.
Libby and colleagues [1, 37] have elegantly described how inflammatory processes are broadly involved in atherosclerosis, from plaque initiation and development to rupture. Atherosclerotic plaques can develop in larger arteries that are subject to great blood velocity [38]. Fatty streaks develop in the intima, the innermost layer of the endothelium, or vascular wall, due to infiltration by low-density lipoprotein (LDL) molecules. In response, endothelial cells, seeking to clear the LDL, express adhesion molecules such as vascular cellular adhesion molecule-1 and sICAM that promote tethering of leukocytes to the endothelium. Endothelial cells also produce chemokines that encourage leukocytes to migrate to the subendothelial space and to penetrate into the intima to clear the fatty deposits. At this point, these migrated leukocytes, specifically monocytes, natural killer cells, and T cells, are activated by growth factors and begin producing inflammatory cytokines such as interleukin-1 beta (IL-1b), tumor necrosis factor-alpha (TNFa) and interferon-gamma. These cytokines then stimulate leukocytes to release prodigious amounts of IL-6, which can be measured in peripheral circulation. IL-6 is secreted by immune cells, typically monocytes, as well as from adipose and muscle tissue. IL-6 is a potent stimulator of CRP. Produced in the liver, CRP turns the wheel of systemic inflammatory processes; it boosts monocyte recruitment into the intima and cellular adhesion molecule expression in the vasculature. It also promotes LDL uptake by macrophages.
Over time, encouraged by IL-6, CRP, and other inflammatory mediators, macrophages ingest copious amounts of LDL and convert to foam cells, which become lodged in the plaque. Simultaneously, T cells promote smooth muscle cells and endothelial cells to replicate. This causes considerable congestion in the plaque, which starts to bulge. Repeated stimulation of inflammatory processes drive forward the growth of the atherosclerotic lesion and also promote the eventual thinning of a fibrous cap that, when sufficiently degraded, leads to rupture of the plaque into circulation. Libby et al. [1] suggest that it is inflammatory processes that link hypertension, diabetes, and obesity to atherosclerosis.
The extent of ongoing inflammatory processes can be measured by monitoring the levels of markers such as IL-6, CRP, and sICAM; each is prospectively associated with cardiovascular disease. CRP is an independent risk predictor of cardiovascular disease events such as myocardial infarction, stroke, coronary arterial disease, and peripheral arterial disease after controlling for traditional risk factors such as total cholesterol, smoking, BMI, diabetes status, and hypertensive status [3, 39]
A number of biobehavioral factors related to poor sleep are associated with inflammatory processes as well. O’Connor and colleagues [40] have reviewed a number of these variables and their association with circulating cytokines. For example, obesity, indexed via BMI, positively correlates with CRP and IL-6 levels, and adipose tissue itself is an important source of IL-6 [2, 41]. As mentioned earlier, obesity is also associated with poor sleep; little is known about the interaction of poor sleep and obesity on inflammatory processes. The direction of effects on inflammation can vary as well. Mild to moderate alcohol use is associated with reduced atherosclerosis and CRP levels [31]. However, these benefits are reversed with chronic high levels of alcohol intake; risk of all-cause mortality and cardiovascular disease-specific outcomes increases and CRP levels rise as well [42]. Identifying important covariates is important to determine the nature and extent of the effects of poor sleep. In the next section, I discuss findings from experimental protocols, demonstrating that sleep loss affects inflammatory processes.
Poor Sleep and Inflammation: Experimental Paradigms
Sleep deprivation studies indicate that short-term sleep loss affects inflammatory markers, with the nature and magnitude of the effects depending on the extent of the deprivation. Deprivation studies can involve total sleep deprivation, in which participants are awake throughout the nighttime, or partial sleep deprivation, also called sleep restriction. In such protocols, participants might be awake for 4 h and then sleep for 4 h during the nighttime period.
Total deprivation studies lasting longer than one night show a pattern of increased levels of inflammatory markers such as CRP [8]. Similarly, multiple nights of partial sleep deprivation each night, ranging from five to 12 nights, also elicit increased expression of IL-6 messenger RNA (mRNA) [43], IL-6 levels [44, 45], and CRP levels [8, 43, 44].
For example, Meier-Ewert and colleagues [8] compared ten consecutive nights of 8.2 versus 4.2 h of scheduled sleep per night in a small sample (n = 10) of young and healthy men and women. Those who were sleep-deprived showed a 5-fold increase in CRP, increasing from 0.05 mg/dl on day 1 to 0.27 mg/dl on day 10, whereas the non-sleep-deprived group showed no change in CRP levels. IL-6 is the major stimulator of CRP production in the liver. Vgontzas and colleagues [45] found that seven nights of 6 versus 8 h of sleep led to increased levels of IL-6 in men and women in the partial deprivation group. Thus, extended periods of brief deprivation lead to increases in IL-6 levels, which likely then promote increases in CRP.
When the deprivation lasts only one night or just part of one night, the direction of inflammatory measures is mixed, and the duration of effects appears shorter-lived. IL-6 and CRP levels have either not changed [46, 47] or decreased [48]. When deprivation is so brief, the effects of sleep on inflammation may be highly dynamic and brief. Irwin and colleagues have found that in the morning following partial sleep deprivation, monocyte expression and mRNA levels of IL-6 and TNFa are briefly increased at 8 am, but not later in the day [10]. There may be a differential effect by gender as well, with women having extended elevations in monocyte expression of IL-6 and TNFa into the evening [49]. Thus, one night of deprivation can lead to rapidly changing effects on inflammatory processes, and more extended sleep deprivation protocols, involving either partial or total deprivation, suggest a progressive increase in inflammatory markers.
Assessment of other proinflammatory cytokines, such as TNFa or IL-1b, has been hampered by measurement difficulties. Both TNFa and IL-1b are difficult to measure in plasma, due to low baseline circulating levels. Deprivation-induced changes in TNFa have been equivocal, with results showing an increase in circulating levels of TNFa in men but not women [45]. Levels of soluble TNFa receptors in circulation have been used as a proxy for estimating levels of TNFa, and here too, results have been mixed, with sleep deprivation inducing either an increase [50] or no change in receptor levels [44].
Unlike measurement of circulating levels of cytokines, stimulation assays test the capacity of immune cells to respond in the face of a threat. In these assays, whole blood or peripheral blood mononuclear cells (PBMC; refers to lymphocytes and monocytes) are stimulated in vitro with a substance known to provoke an inflammatory response. There can be a considerable output of TNFa, for example, in response to lipopolysaccharide (LPS), a component of gram-negative bacteria. Born and colleagues [47] found that stimulated levels of TNFa and IL-1b were enhanced during sleep deprivation as compared to sleeping. Monocytes are the primary immune cell producers of IL-1b and TNFa, and Born and colleagues found that increased stimulated levels of TNFa and IL-1b were due to increased numbers of monocytes in circulation. In other words, when blood was drawn and stimulated with antigen, there were substantially more monocytes in those samples drawn during sleep deprivation as opposed to normal sleep, and it was the increased numbers of monocytes in the sample that accounted for the increased stimulated production of IL-1b and TNFa. Other approaches have worked around this by examining cytokine production on a per cell basis, examining whether individual cells produce more inflammatory cytokine following sleep deprivation. Irwin and colleagues [10] found that a single night of partial sleep deprivation led to increases in the capacity of stimulated monocytes to produce TNFa and IL-6 on a per cell basis. Thus, extended periods of total deprivation, or brief, or partial sleep deprivation can produce increases in circulating levels of IL-6 and CRP, but the effects on other inflammatory cytokines such as TNFa are less clear. Lastly, there is some initial evidence that a single night of partial sleep deprivation briefly potentiates the functional capacity of immune cells to produce IL-6 and TNFa.
What inferences can we make about chronic sleep loss from these experimental studies on acute sleep deprivation studies? Acute deprivation studies provide evidence for a mechanistic relationship between sleep disruption and cytokines. Because these studies are typically done with young, healthy sleeping participants, caution must be taken in generalizing the findings to clinical populations. Acute studies do provide hypotheses for how sleep and cytokine regulation might be disrupted in clinical samples. Ideally, deprivation studies in clinical samples could characterize the mechanistic effects of sleep loss in sleep dysregulated populations, but such studies have rarely been done.
In studies reviewed that have examined sleep deprivation and cytokines [8, 10, 43–45, 47–50], nearly all of these studies are with participants under 40 years of age, with mean ages typically in the mid-20s. Each of these studies has taken care to ensure that participants do not have current sleep problems, but reporting of other important moderators has been less consistent. For example, of the nine studies discussed, five excluded individuals with depression [10, 44, 48–50], five excluded individuals with alcohol problems [8, 10, 44, 49, 50], five excluded smokers [10, 44, 47–49], and five reported BMI status [8, 10, 45, 48, 49]. Although researchers report thoroughly assessing sleep disorders, illicit substance use, and medication use in deprivation studies, broader characterization of study samples on these other behavioral variables is important for future studies.
Primary Insomnia, Insomnia Symptoms, and Inflammation
Sleep deprivation studies provide evidence that experimentally induced short sleep and/or insomnia symptoms can lead to brief elevations in inflammatory markers. Observational studies with sleep-impaired populations suggest that chronic sleep difficulties are also associated with elevations in inflammatory markers. Two studies with primary insomnia patients offer additional evidence that insomnia is associated with inflammatory markers. In a small study with 11 primary insomnia patients (eight of whom were women), Burgos and colleagues [51] found that IL-6 circulating levels were higher than in 11 gender-matched healthy sleepers at 3:00, 5:00, and 7:00 am. Area under the curve (AUC) analyses indicated that insomnia patients had greater values across the day as well. Unfortunately, it is unclear if adiposity was tightly controlled in these analyses as the insomnia group had a mean BMI = 24 ± 4 whereas the control group had a BMI of 21 ± 2 (adipose tissue is a potent source of inflammatory cytokines). The authors did note that no participants were obese (BMI > 30). Smoking and alcohol use across the insomnia and healthy sleep groups were not described. In another study with 11 primary insomnia patients (five women) and 11 control participants, Vgontzas and colleagues found that circulating IL-6 levels were higher between 2:00 to 11:00 pm in insomnia patients versus controls [52]. Because they matched the groups on age and BMI, these findings suggest that differences in body fat cannot fully account for group differences in inflammatory markers. Furthermore, current major depression, current smoking, and alcohol dependence were exclusion criteria, suggesting that these variables did not account for group differences.
In this same study, healthy sleepers had peak circulating levels of IL-6 at 2:00 am, but in insomnia patients, peak IL-6 levels were reached considerably earlier at 7:00 pm, suggesting a circadian shift in IL-6 levels. Circulating levels of TNFa were not significantly different between the groups. Rather than conceptualizing IL-6 as a sleep disruptor however, Vgontzas and colleagues suggest that increased daytime levels of IL-6 promote fatigue and sleepiness during the day, and as such, higher levels from 2–11 pm promotes daytime fatigue during a time of day in which people are typically awake. However, the increased day and evening levels of IL-6 were not associated with improved sleep at night in insomnia patients, suggesting a clear sleep behavior dysregulation. Other studies examining sleep and cytokines in depressed patients suggest that IL-6 is clearly associated with disrupted sleep [53, 54]. Taken together, these preliminary studies suggest that a diagnosis of primary insomnia is associated with increased levels of inflammatory markers. Although the sample sizes were small and although attempts were made to minimize confounds, it is possible that other factors beside sleep in these samples may have been accounting for the relationship. More studies are needed to better understand the effects of circadian variation of markers such as IL-6 on daytime functioning and nighttime sleep.
Larger studies examining insomnia symptoms and inflammatory measures are clearly needed; one recent community-based study [55] provides some corroboration that sleep and inflammation are correlated but that gender affects the relationship. In a study of 210 healthy young men and women, Suarez found that difficulty falling asleep was related to higher morning levels of CRP and IL-6, but only in women. BMI status was similar in both men and women (~25 kg/m2). However, they did find that poorer sleep quality scores were correlated with increasing BMI scores in women. The findings from the study described by Suarez are consistent with experimental sleep deprivation studies that show that women have enhanced inflammatory processes, specifically NFkB signaling [10] as well as more prolonged increased monocyte expression of IL-6 and TNFa [49] following one night of partial sleep deprivation.
Unfortunately, observational studies do not address whether insomnia symptoms specifically cause increases in inflammatory processes in insomnia patients; other sleep-related difficulties, such as sleep apnea, could play an important role. Apnea itself is associated with cardiovascular disease and increased inflammatory markers [56]. Sleep apnea is characterized by frequent episodes in which breathing ceases for greater than 10 s. These patients also report nonrestorative sleep, snoring, waking, and gasping for air [57]. Insomnia symptoms are not required for the diagnosis, but there is some controversy about the comorbidity between insomnia and apnea. Some studies indicate that there is no relationship between apnea and sleep–wake disturbance (insomnia symptoms) [58], whereas others indicate that apnea patients spend more time in bed awake, although their overall sleep time is not reduced [59]. In fact, a number of studies indicate that insomnia symptoms (trouble falling asleep, staying asleep, or waking too early) are highly prevalent in sleep apnea patients, with rates as high as 50% [60–62], yet the reverse does not appear true. Results from a national cooperative study with approximately 5,000 patients from 11 sleep disorder centers found that only 6% of those with insomnia had sleep-disordered breathing [63]. Thus, for insomnia patients, sleep apnea may not play a major role in accounting for the relationship between poor sleep and poor health outcomes. However, by the same token, the relationship between sleep apnea and poor health outcomes may be at least partially due to insomnia, given its high prevalence in apnea patients.
Inflammation, Sickness Behaviors, and Insomnia
Inflammatory cytokines induce a pattern of behavioral and affective changes similar to depressive symptoms. This pattern of changes is termed “sickness behaviors” and has been eloquently described by in reviews by Dantzer [64] and Raison et al. [65]. Briefly, acute infection induces increases in peripheral inflammatory cytokines that subsequently traverse through a complex web of pathways to the brain. These cytokines initiate a coordinated centrally mediated set of behavioral adjustments to deal with the infection, including but not limited to appetite changes, sleep changes, social withdrawal, and behavioral inactivity. These behavioral changes are hypothesized to be productive in the short run because they protect the individual from exposure to additional infection, protect the social network from contagion by sequestering the infected individual, and diverting energy resources to host defense.
In a well-functioning system, once the infection has cleared, the inflammatory responses are inhibited and the sickness behavior pattern remits. However, in cases where the organism does not adequately turn off inflammatory processes, chronic inflammation is thought to perturb the organism by prolonging sickness behavior symptoms, promoting depressive-like symptoms. In atherosclerosis, the chronic accumulation of low-density lipoproteins in atherosclerotic plaques continues to activate inflammatory processes, which could promote sickness behaviors and lead to chronic poor sleep as a consequence.
Depression, Insomnia, and Inflammation
One possible result of chronic elevations in inflammatory markers may be dysregulation of sickness behavior pathways resulting in long-lasting symptoms resembling depression, with poor sleep playing a role in connecting inflammation and depression. In a recent meta-analysis of over 100 studies, Howren and colleagues [66] found that depression was related to higher CRP, IL-6, and IL-1b levels and that the relationship was stronger in clinical samples and studies that used diagnostic assessment. A number of factors probably contribute to elevations in inflammatory markers in depressed patients including adiposity, smoking, alcohol use, physical inactivity, and poor sleep.
Poor sleep is prevalent in major depression, with as high as 90% of patients reporting frequent insomnia symptoms [67] with significantly more difficulty falling asleep, maintaining sleep, worse sleep-related daytime dysfunction, and poorer perceived sleep quality than nondepressed individuals [25]. Furthermore, insomnia symptoms lasting >2 weeks in the absence of other psychiatric symptoms prospectively predicts the development of major depression [67]. In depressed patients, insomnia symptoms may account for some of the relationship between depression and inflammatory markers.
In major depression, difficulty initiating sleep correlates with increased pre-sleep IL-6 and sICAM levels even after controlling for depressive status and other possible confounding variable such as age, body weight, education, and gender (all participants were men) [53]. Prather and colleagues [54] directly tested causal pathways through a longitudinal study with 95 nondepressed hepatitis C patients beginning interferon-alpha (IFNa) treatment. IFNa is a cytokine with proinflammatory effects, and pharmacologic treatment with IFNa induces depression in a subset of hepatitis patients. Prather and colleagues found that IFNa administration induced major depression in 22% of the sample within 3 months of treatment. Consistent with the findings from Motivala and colleagues [53], levels of IL-6 were higher in the subgroup of depressed patients than nondepressed patients, and sleep quality was significantly lower. In addition, time-lagged analyses showed that IL-6 levels predicted poorer sleep quality scores over the course of treatment, but the reverse was not true. In turn, poor sleep quality, measured by the Pittsburgh Sleep Quality Inventory at baseline, predicted subsequent Beck Depression Inventory scores during treatment. The reverse was not true: Higher depressive symptoms at baseline did not predict subsequent impairments in sleep quality. Thus, in hepatitis C patients undergoing IFN therapy, inflammation predicted poorer sleep quality which in turn predicted increased depressive symptoms. These findings suggest that inflammatory markers disrupt sleep and sleep disruption is important to the onset of subsequent depressive symptoms.
In the context of cardiovascular disease, a number of studies have demonstrated that depression prospectively predicts myocardial infarctions and CAD mortality (see Musselman et al. for review [68]). Depression involves a complex set of symptoms and associated behaviors that contribute to poor health outcomes, such as appetite change and weight gain, smoking and alcohol use, and poor self-care, but insomnia symptoms are also highly prevalent, with the majority of patients reporting poor sleep [25]. Furthermore, poor sleep could be an important “break-through” symptom that persists even during remission. In those with a history of depression but not currently depressed, 45% of individuals reported poor sleep, indexed via elevated scores on the Pittsburgh Sleep Quality Inventory (>5) [25]. Thus, insomnia may be a key symptom in depressed patients and those with a history of depression that propels chronic inflammation.
Pathways Connecting Poor Sleep and Inflammation
The Autonomic Nervous System and Catecholamines
Primary insomnia can be conceptualized as a psychophysiologic disorder involving heightened sympathetic arousal. Bonnet and Arand [69] have shown that through the night, chronic insomnia patients have autonomic balance shifted toward sympathetic dominance indexed via heart rate variability assessment when compared to healthy sleepers. Similarly, Irwin and colleagues [70] have demonstrated that nighttime levels of catecholamines are increased in male insomnia patients as compared to healthy sleeping men.
Sleep deprivation studies indicate that nighttime wakefulness is associated with increased sympathetic activation. Partial sleep deprivation and brief total sleep deprivation (e.g., for one night) are associated with increased systolic blood pressure [71, 72]. Irwin and colleagues [73] found that late night sleep deprivation from 4 to 8 am led to increased norepinephrine and epinephrine levels in 17 healthy male participants. More recent work has corroborated these findings using noninvasive methods; Zhong and colleagues [71] found that sympathetic activity was increased relative to parasympathetic activity, indexed via assessment of heart rate variability, during 36 h of total sleep deprivation in a sample of mostly male participants. The findings by Irwin and colleagues [73] are particularly significant because all participants remained supine while they were awake; other studies that have shown increased autonomic activity and blood pressure levels have not always controlled for posture and movement during nighttime wakefulness [71]. Thus, acute wakefulness through the night is associated with increased sympathetic arousal, and observational studies with chronic insomnia patients indicate that even during sleep, sympathetic activity is elevated.
Heightened sympathetic, relative to parasympathetic autonomic input, may play an important role in spurring inflammation via NFkB. The latter is a ubiquitous transcription factor residing in the cytoplasm that when activated translocates to the cell nucleus and promotes gene expression of a range of inflammatory cytokines. NFkB signaling can be engaged by a range of extracellular factors including oxidized LDL, LPS, and TNFa. Norepinephrine also appears to upregulate NFkB signaling [74]. In a series of studies, Bierhaus and colleagues found that in vitro incubation of norepinephrine at physiological dosage levels increased NFkB signaling in human-derived PBMCs, and furthermore, pre-incubation with alpha1, beta1, and beta2 adrenergic antagonists (prazosin, metoprolol, and butoxamine, respectively) each reduced NFkB activation.
Alternatively, poor sleep may lead to inflammation by promoting increased blood pressure and thus increased shearing stress at sites of major bifurcations in arteries. In these areas, increased blood pressure damages endothelial cells in blood vessels; these cells then recruit leukocytes to the sites of stress, via the release of chemokines and adhesion molecules. Typically, nighttime is characterized by low circulating levels of these molecules [75]. Consistent with this, experimental sleep deprivation produces increases in blood pressure [8] along with selectin and intracellular adhesion molecule [76]. Likewise, for chronic sleep impairment, normotensive primary insomnia patients have increased nighttime blood pressure when compared to healthy sleepers [77]. And in individuals with major depression, difficulty falling asleep positively correlates with higher intracellular adhesion molecules [53].
These studies suggest a model in which sleep loss promotes sympathetic activation which triggers inflammatory processes by activating NFkB, increased gene expression of inflammatory cytokines in PBMCs, and increased production of inflammatory cytokines by immune cells, which in turn likely cause an increase in circulating levels of inflammatory markers and trigger the liver to produce CRP. Similarly, sleep loss also promotes increased blood pressure promoting endothelial-mediated increases in inflammation.
Cortisol
The steroid hormone cortisol exerts powerful modulatory effects on the immune system; whether it plays a major role in the relationship between poor sleep and inflammation is less clear. The hypothalamic–pituitary–adrenocortical (HPA) axis is a multi-organ circuit resulting in release of cortisol from the adrenal cortex. Cortisol is a multifunctional hormone that affects energy metabolism and immune function. Cortisol affects insulin and glucose metabolism, promoting energy release and uptake. Cortisol levels can affect immune cells and inflammatory processes, but the relationship is complex, with cortisol evidencing both stimulatory and inhibitory functions [78].
Cortisol is inversely correlated with IL-6 levels and inhibits IL-6 production in vitro [79]. IL-6 stimulates cortisol release, suggesting that increases in IL-6 trigger a compensatory HPA response, increasing cortisol levels, which then work to decrease IL-6 levels. Peak cortisol levels occur in the morning, usually some time between 7 and 9 am, and then decrease through the day reaching a nadir in the late evening. Levels remain low in the first half of the night, slowly increasing through the latter half of the night.
In clinical samples, the extent of HPA dysregulation is unclear, with variable findings and small sample sizes, but there is some suggestion of cortisol dysregulation with greater insomnia severity. Riemann and colleagues [80] did not find any differences in cortisol levels when comparing ten insomnia patients (men and women) with healthy controls; however, insomnia patients slept just as well as controls in their sleep laboratory. From the same research group, Rodenbeck and colleagues [81] found that cortisol levels were higher in seven male severe insomnia patients compared to controls and that cortisol levels before sleep correlated with number of nighttime awakenings. Vgontzas and colleagues [82] also found that 11 insomnia patients (both men and women) had higher levels of cortisol than gender-matched controls, primarily in the evening and early nighttime and that more severe insomnia symptoms was associated with higher cortisol. Thus, there is some evidence of heightened cortisol secretion in chronic insomnia patients, most likely in those with more severe symptoms.
Experimental sleep loss findings have been complex and contrast with clinical findings. Single night partial sleep deprivation does not appear to affect nighttime cortisol expression during deprivation [83, 84]. The effects on cortisol levels during recovery sleep are conflicting. Leproult and colleagues [84] found that cortisol levels increased in the evening immediately before recovery sleep. Similar findings occur when partial sleep deprivation is extended for six nights [85]. Rising cortisol levels along with rising IL-6 levels would suggest that the typical negative feedback loop between the HPA and IL-6 levels is temporarily weakened following deprivation. These findings contrast with those of Vgontzas et al. [86] who found that one full night of sleep deprivation led to decreases in 24-h AUC and peak amplitude levels of cortisol. Vgontzas conducted a follow-up study involving 1 week of partial sleep deprivation and found that peak cortisol levels were lower during recovery sleep [45]. Findings from Vgontzas’s studies suggest that sleep deprivation reduces HPA activity evidenced by reduced cortisol levels and that this could play a role in enhancing or disinhibiting IL-6 levels. The verdict on cortisol is that there appears to be some alteration in HPA inflammatory functioning, but the exact mechanisms and the nature of the alterations remain somewhat unclear.
If a clearer picture emerges confirming that chronic insomnia is associated with progressive increases in cortisol levels, then poor sleep may contribute to cardiovascular disease pathogenesis through the HPA axis as well. Increased morning levels of cortisol are associated with atherosclerosis in men, and steroid medications can lead to increased triglyceride and cholesterol levels [68].
Conclusions and Caveats with an Eye to the Future
In contemplating findings from cross-sectional and prospective studies with community populations, naturalistic studies with clinical populations, and experimental sleep deprivation studies, four major themes become clear. First, clearly differentiating insomnia from other sleep difficulties linked to poor health and inflammation are vitally important to determine whether inadequacies in the ability to fall asleep and maintain sleep are specifically related to poor health outcomes. In this regard, there is evidence that those with insomnia symptoms do not typically experience sleep apnea. However, those with apnea may have upward of 50% experience co-occurring insomnia symptoms [60].
Second, the directionality and clinical significance of the relationships between sleep and inflammation need to be determined. Currently, acute sleep deprivation studies indicate that sleep loss can lead to brief elevations in inflammatory markers [10], and two clinical studies suggest that primary insomnia is associated with elevations as well. But longitudinal data are needed linking insomnia, inflammation, and clinical outcomes. Clinical trials to reduce insomnia would provide a test of whether improvements in sleep lead to subsequent reductions in inflammatory markers. As of yet, such studies have not been done, and so it is unclear whether sleep improvement has a salutary effect on health. Behavioral treatment of insomnia appears to have durable effects lasting at least 6 months after intervention completion [87]. Whether these long-lasting improvements are important in producing meaningful reductions in inflammatory cytokines is unknown. In the sickness behavior model, chronic inflammation disrupts sleep; clinical trials examining administration of inflammatory agents such as IFNa are important to demonstrate that cytokines can disrupt sleep. Additionally, examination of changes in sleep when inflammatory cytokines are blocked, typically with biologic medications that specifically target cytokines such as TNFa, IL-1b, and IL-6, will provide clearer evidence for the role of cytokines in affecting sleep in humans. Considerable evidence indicates that cytokines affect sleep in other species [88], but the nature and magnitude of these relationships are less clear in humans.
Third, the mechanisms that link sleep loss to cytokine expression need to be identified. There is consistent evidence that primary insomnia is associated with heightened sympathetic autonomic arousal, and heightened sympathetic drive can promote increases in inflammatory cytokines. Yet no study has linked the two together in a clinical sample.
Lastly, a better understanding of the course of insomnia is needed. Acute insomnia is incredibly common in healthy individuals, but at what point does insomnia begin to meaningfully negatively impact health? How severe and how long-lasting does it need to be? These questions have been largely unexplored and are important.
As voiced by Shakespeare’s character Macbeth, who describes sleep as the “balm of hurt minds” and “chief nourisher in life’s feast,” sleep has long been considered important for health. Psychoneuroimmunology research is providing a map of interactions between behavior, the brain, and immune-inflammatory processes. These pathways provide a promising set of mechanisms linking poor sleep to deleterious cardiovascular outcomes.
References
Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143
Mohamed-Ali V, Goodrick S, Rawesh A, et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab. 1997;82(12):4196–4200.
Blake GJ, Ridker PM. C-reactive protein and other inflammatory risk markers in acute coronary syndromes. J Am Coll Cardiol. 2003;41(4 Suppl S):37S-42S
Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101(15):1767–1772.
Besedovsky HO, del Rey A, Sorkin E, DaPrada M, Burri R, Honegger C. The immune response evokes changes in brain noradrenergic neurons. Science. 1983;221:564–566.
Tracey KJ. The inflammatory reflex. Nature. 2002;420(6917):853–859
Dantzer R. Cytokines and sickness behavior. In: Kronfol Z, ed. Cytokines and mental health. Boston: Kluwer Academic; 2003:129–146.
Meier-Ewert HK, Ridker PM, Rifai N, et al. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol. 2004;43(4):678–683
Irwin MR, Rinetti G. Disordered sleep, nocturnal cytokines, and immunity: Interactions between alcohol dependence and African-American ethnicity. Alcohol. 2004;32(1):53–61.
Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med. 2006;166(16):1756–1762
Opp MR. Cytokines and sleep. Sleep Med Rev. 2005;9(5):355–364.
Ohayon MM. Epidemiology of insomnia: What we know and what we still need to learn. Sleep Med Rev. 2002;6(2):97–111.
Morin CM, Hauri PJ, Espie CA, Spielman AJ, Buysse DJ, Bootzin RR. Nonpharmacologic treatment of chronic insomnia. An American Academy of Sleep Medicine review. Sleep. 1999;22(8):1134–1156
Wingard DL, Berkman LF. Mortality risk associated with sleeping patterns among adults. Sleep. 1983;6(2):102–107.
Kripke DF, Garfinkel L, Wingard DL, Klauber MR, Marler MR. Mortality associated with sleep duration and insomnia. Arch Gen Psychiatry. 2002;59(2):131–136.
Dew MA, Hoch CC, Buysse DJ, et al. Healthy older adults’ sleep predicts all-cause mortality at 4 to 19 years of follow-up. Psychosom Med. 2003;65(1):63–73.
Taylor DJ, Mallory LJ, Lichstein KL, Durrence HH, Riedel BW, Bush AJ. Comorbidity of chronic insomnia with medical problems. Sleep. 2007;30(2):213–218
Mallon L, Broman JE, Hetta J. Sleep complaints predict coronary artery disease mortality in males: A 12-year follow-up study of a middle-aged Swedish population. J Intern Med. 2002;251(3):207–216.
Eaker ED, Pinsky J, Castelli WP. Myocardial infarction and coronary death among women: Psychosocial predictors from a 20-year follow-up of women in the Framingham Study. Am J Epidemiol. 1992;135(8):854–864
Schwartz S, McDowell Anderson W, Cole SR, Cornoni-Huntley J, Hays JC, Blazer D. Insomnia and heart disease: A review of epidemiologic studies. J Psychosom Res. 1999;47(4):313–333
Schwartz SW, Cornoni-Huntley J, Cole SR, Hays JC, Blazer DG, Schocken DD. Are sleep complaints an independent risk factor for myocardial infarction? Ann Epidemiol. 1998;8(6):384–392.
Siegrist J, Peter R, Motz W, Strauer B. The role of hypertension, left ventricular hypertrophy and psychosocial risks in cardiovascular disease: Prospective evidence from blue collar men. Eur Heart J. 1992;13(Suppl D):89–95
Appels A, de Vos Y, van Diest R, Hoppner P, Mulder P, de Groen J. Are sleep complaints predictive of future myocardial infarction? Act Nerv Super (Praha). 1987;29(2):147–151.
Koskenvuo M, Kaprio J, Partinen M. Poor sleep quality, emotional stress and morbidity: A six year followup of 10778 persons aged 35–59 years. In: Achete K, Pakaslahti A, eds. Stress and Psychosomatics. Vol . Helsinki: Pyschiatric Fenica; 1986:115–120.
Motivala SJ, Levin MJ, Oxman MN, Irwin MR. Impairments in health functioning and sleep quality in older adults with a history of depression. J Am Geriatr Soc. 2006;54(8):1184–1191.
Vgontzas AN, Tan TL, Bixler EO, Martin LF, Shubert D, Kales A. Sleep apnea and sleep disruption in obese patients. Arch Intern Med. 1994;154(15):1705–1711
Frey WC, Pilcher J. Obstructive sleep-related breathing disorders in patients evaluated for bariatric surgery. Obes Surg. 2003;13(5):676–683.
Patel SR, Hu FB. Short sleep duration and weight gain: A systematic review. Obesity (Silver Spring). 2008;16(3):643–653.
Vgontzas AN, Bixler EO, Tan TL, Kantner D, Martin LF, Kales A. Obesity without sleep apnea is associated with daytime sleepiness. Archives of Internal Medicine. 1998;158(12):1333–1337.
Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet. 2004;364(9438):937–952
O’Keefe JH, Bybee KA, Lavie CJ. Alcohol and cardiovascular health: The razor-sharp double-edged sword. J Am Coll Cardiol. 2007;50(11):1009–1014
Roehrs T, Roth T. Sleep, sleepiness, and alcohol use. Alcohol Res Health. 2001;25(2):101–109.
Irwin M, Miller C, Gillin JC, Demodena A, Ehlers CL. Polysomnographic and spectral sleep EEG in primary alcoholics: An interaction between alcohol dependence and African-American ethnicity. Alcohol Clin Exp Res. 2000;24(9):1376–1384.
Breslau N, Roth T, Rosenthal L, Andreski P. Sleep disturbance and psychiatric disorders: A longitudinal epidemiological study of young adults. Biol Psychiatry. 1996;39(6):411–418
Jefferson CD, Drake CL, Scofield HM, et al. Sleep hygiene practices in a population-based sample of insomniacs. Sleep. 2005;28(5):611–615
Frohlich M, Sund M, Lowel H, Imhof A, Hoffmeister A, Koenig W. Independent association of various smoking characteristics with markers of systemic inflammation in men. Results from a representative sample of the general population (MONICA Augsburg Survey 1994/95). Eur Heart J. 2003;24(14):1365–1372.
Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874
Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282(21):2035–2042
Pearson TA, Mensah GA, Hong Y, Smith SC, Jr. CDC/AHA workshop on markers of inflammation and cardiovascular disease: Application to clinical and public health practice: Overview. Circulation. 2004;110(25):e543–544
O’Connor MF, Bower JE, Cho HJ, et al. To assess, to control, to exclude: Effects of biobehavioral factors on circulating inflammatory markers. Brain Behav Immun. 2009;23(7):887–897.
Mohamed-Ali V, Pinkney JH, Coppack SW. Adipose tissue as an endocrine and paracrine organ. Int J Obes Relat Metab Disord. 1998;22(12):1145–1158.
Oliveira A, Rodriguez-Artalejo F, Lopes C. Alcohol intake and systemic markers of inflammation—shape of the association according to sex and body mass index. Alcohol Alcohol. 2010;45(2):119–125
van Leeuwen W, Lehto M, Karisola P, et al. Sleep restriction increases the risk of developing cardiovascular diseases by augmenting proinflammatory responses through IL-17 and CRP. PLOS One. 2009;4(2):1–7.
Haack M, Sanchez E, Mullington JM. Elevated inflammatory markers in response to prolonged sleep restriction are associated with increased pain experience in healthy volunteers. Sleep. 2007;30(9):1145–1152
Vgontzas AN, Zoumakis E, Bixler EO, et al. Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J Clin Endocrinol Metab. 2004;89(5):2119–2126.
Dinges DF. The state of sleep deprivation: From functional biology to functional consequences. Sleep Med Rev. 2006;10(5):303–305.
Born J, Lange T, Hansen K, Molle M, Fehm HL. Effects of sleep and circadian rhythm on human circulating immune cells. J Immunol. 1997;158:4454–4464.
Frey DJ, Fleshner M, Wright KP, Jr. The effects of 40 hours of total sleep deprivation on inflammatory markers in healthy young adults. Brain Behav Immun. 2007;21(8):1050–1057.
Irwin MR, Carrillo C, Olmstead R. Sleep loss activates cellular markers of inflammation: Sex differences. Brain Behav Immun. 2010;24(1):54–57
Shearer WT, Reuben JM, Mullington JM, et al. Soluble TNF-alpha receptor 1 and IL-6 plasma levels in humans subjected to the sleep deprivation model of spaceflight. J Allergy Clin Immunol. 2001;107(1):165–170.
Burgos I, Richter L, Klein T, et al. Increased nocturnal interleukin-6 excretion in patients with primary insomnia: A pilot study. Brain Behav Immun. 2005;20:246–253
Vgontzas AN, Zoumakis M, Papanicolaou DA, et al. Chronic insomnia is associated with a shift of interleukin-6 and tumor necrosis factor secretion from nighttime to daytime. Metabolism. 2002;51(7):887–892.
Motivala SJ, Sarfatti A, Olmos L, Irwin MR. Inflammatory markers and sleep disturbance in major depression. Psychosom Med. 2005;67(2):187–194.
Prather AA, Rabinovitz M, Pollock BG, Lotrich FE. Cytokine-induced depression during IFN-alpha treatment: The role of IL-6 and sleep quality. Brain Behav Immun. 2009;23(8):1109–1116.
Suarez EC. Self-reported symptoms of sleep disturbance and inflammation, coagulation, insulin resistance and psychosocial distress: Evidence for gender disparity. Brain Behav Immun. 2008;22(6):960–968.
Ciftci TU, Kokturk O, Bukan N, Bilgihan A. The relationship between serum cytokine levels with obesity and obstructive sleep apnea syndrome. Cytokine. 2004;28(2):87–91
Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: A population health perspective. Am J Respir Crit Care Med. 2002;165(9):1217–1239
Mant A, Eyland EA, Hewitt H, et al. Sleep-disordered breathing in elderly people and subjective sleep-wake disturbance. Age Ageing. 1992;21(4):262–268.
Kripke DF, Ancoli-Israel S, Klauber MR, Wingard DL, Mason WJ, Mullaney DJ. Prevalence of sleep-disordered breathing in ages 40–64 years: A population-based survey. Sleep. 1997;20(1):65–76.
Hagen C, Patel A, McCall WV. Prevalence of insomnia symptoms in sleep laboratory patients with and without sleep apnea. Psychiatry Res. 2009;170(2–3):276–277
Krell SB, Kapur VK. Insomnia complaints in patients evaluated for obstructive sleep apnea. Sleep Breath. 2005;9(3):104–110.
Chung KF. Insomnia subtypes and their relationships to daytime sleepiness in patients with obstructive sleep apnea. Respiration. 2005;72(5):460–465.
Coleman RM, Roffwarg HP, Kennedy SJ, et al. Sleep–wake disorders based on a polysomnographic diagnosis. A national cooperative study. JAMA. 1982;247(7):997–1003
Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am. 2009;29(2):247–264.
Raison CL, Capuron L, Miller AH. Cytokines sing the blues: Inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24–31.
Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom Med. 2009;71(2):171–186.
Buysse DJ, Angst J, Gamma A, Ajdacic V, Eich D, Rossler W. Prevalence, course, and comorbidity of insomnia and depression in young adults. Sleep. 2008;31(4):473–480
Musselman DL, Evans DL, Nemeroff CB. The relationship of depression to cardiovascular disease: Epidemiology, biology, and treatment. Archives of General Psychiatry. 1998;55(7):580–592.
Bonnet MH, Arand DL. Heart rate variability in insomniacs and matched normal sleepers. Psychosom Med. 1998;60(5):610–615.
Irwin M, Clark C, Kennedy B, Gillin JC, Ziegler M. Nocturnal catecholamines and immune function in insomniacs, depressed patients, and control subjects. Brain Beh Immun. 2003;17:365–372.
Zhong X, Hilton HJ, Gates GJ, et al. Increased sympathetic and decreased parasympathetic cardiovascular modulation in normal humans with acute sleep deprivation. J Appl Physiol. 2005;98(6):2024–2032.
Lusardi P, Zoppi A, Preti P, Pesce RM, Piazza E, Fogari R. Effects of insufficient sleep on blood pressure in hypertensive patients: A 24-h study. Am J Hypertens. 1999;12(1 Pt 1):63–68.
Irwin M, Thompson J, Miller C, Gillin JC, Ziegler M. Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: Clinical implications. J Clin Endocrinol Metab. 1999;84(6):1979–1985.
Bierhaus A, Wolf J, Andrassy M, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A. 2003;100(4):1920–1925
Mullington JM, Haack M, Toth M, Serrador JM, Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog Cardiovasc Dis. 2009;51(4):294–302.
Sanchez A, Haack M, Toth LA. Effects of sleep deprivation on blood pressure and vascular cellular adhesion molecules. Sleep. 2008;31(Supplement):A137
Lanfranchi PA, Pennestri MH, Fradette L, Dumont M, Morin CM, Montplaisir J. Nighttime blood pressure in normotensive subjects with chronic insomnia: Implications for cardiovascular risk. Sleep. 2009;32(6):760–766.
Dhabhar FS. Stress-induced augmentation of immune function—the role of stress hormones, leukocyte trafficking, and cytokines. Brain Behav Immun. 2002;16(6):785–798.
Wilder RL. Neuroendocrine–immune system interactions and autoimmunity. Annu Rev Immunol. 1995;13:307–338.
Riemann D, Klein T, Rodenbeck A, et al. Nocturnal cortisol and melatonin secretion in primary insomnia. Psychiatry Res. 2002;113(1–2):17–27
Rodenbeck A, Huether G, Ruther E, Hajak G. Interactions between evening and nocturnal cortisol secretion and sleep parameters in patients with severe chronic primary insomnia. Neurosci Lett. 2002;324(2):159–163
Vgontzas AN, Bixler EO, Lin HM, et al. Chronic insomnia is associated with nyctohemeral activation of the hypothalamic–pituitary–adrenal axis: Clinical implications. J Clin Endocrinol Metab. 2001;86(8):3787–3794.
Redwine L, Hauger RL, Gillin JC, Irwin M. Effects of sleep and sleep deprivation on interleukin-6, growth hormone, cortisol, and melatonin levels in humans. J Clin Endocrinol Metab. 2000;85(10):3597–3603.
Leproult R, Copinschi G, Buxton O, Van Cauter E. Sleep loss results in an elevation of cortisol levels the next evening. Sleep. 1997;20(10):865–870.
Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354(9188):1435–1439
Vgontzas AN, Bixler EO, Wittman AM, et al. Middle-aged men show higher sensitivity of sleep to the arousing effects of corticotropin-releasing hormone than young men: Clinical implications. J Clin Endocrinol Metab. 2001;86(4):1489–1495.
Morgenthaler T, Kramer M, Alessi C, et al. Practice parameters for the psychological and behavioral treatment of insomnia: An update. An American Academy of Sleep Medicine report. Sleep. 2006;29(11):1415–1419
Krueger JM, Toth LA. Cytokines as regulators of sleep. Ann NY Acad Sci. 1994;739:299–310.
Acknowledgments
This work was supported by an NIH career development grant (K23 AG027860) and the UCLA Cousins Center for Psychoneuroimmunology. I would like to thank Michael Irwin, M.D., Mary-Frances O’Connor, Ph.D., and Neil Schneiderman, Ph.D., for their valuable comments in the process of putting this paper together.
Conflict of Interest Statement
The author has no conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Motivala, S.J. Sleep and Inflammation: Psychoneuroimmunology in the Context of Cardiovascular Disease. ann. behav. med. 42, 141–152 (2011). https://doi.org/10.1007/s12160-011-9280-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12160-011-9280-2