
Abstract
N-myristoylation is the attachment of a 14-carbon fatty acid, myristate, onto the N-terminal glycine residue of target proteins, catalysed by N-myristoyltransferase (NMT), a ubiquitous and essential enzyme in eukaryotes. Many of the target proteins of NMT are crucial components of signalling pathways, and myristoylation typically promotes membrane binding that is essential for proper protein localisation or biological function. NMT is a validated therapeutic target in opportunistic infections of humans by fungi or parasitic protozoa. Additionally, NMT is implicated in carcinogenesis, particularly colon cancer, where there is evidence for its upregulation in the early stages of tumour formation. However, the study of myristoylation in all organisms has until recently been hindered by a lack of techniques for detection and identification of myristoylated proteins. Here we introduce the chemistry and biology of N-myristoylation and NMT, and discuss new developments in chemical proteomic technologies that are meeting the challenge of studying this important co-translational modification in living systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Post-translational modification and myristoylation
The proteome is a larger and more dynamic entity than the genome, a result of both increased sequence diversity at the mRNA level due to alternative splicing of genes, and increased chemical and functional complexity at the protein level due to post-translational modification (PTM). This explains to some extent why larger genomes do not necessarily translate into more complex organisms. Post-translational modification allows the incorporation of chemistries and new molecular functions that cannot be directly encoded by the gene sequence. This includes internal protein changes (e.g. disulfide bond formation [1]), removal of amino acid residues (proteolysis [2], N-terminal initiator methionine removal [3]), attachment of other proteins (e.g. ubiquitination [4], sumoylation [5]), N-terminal modifications (acetylation [6], arginylation [7], lipidation by myristoylation [8] or palmitoylation [9]) and side-chain modifications (e.g. acetylation [6], methylation [10], phosphorylation [11], glycosylation [12], lipidations such as palmitoylation and prenylation [13, 14] and sulfenic acid attachment [15]). Modifications such as these have incredibly diverse biological functions and are involved in protein maturation, signalling, protein localisation, trafficking, extracellular communication, protein regulation and metabolism.
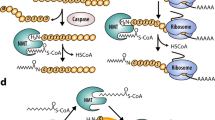
Myristoylation is the attachment of a 14-carbon saturated fatty acid, myristate, to the N-terminal glycine of a subset of eukaryotic proteins [8, 16, 17]. Although often referred to as a PTM, it usually occurs co-translationally [18], when fewer than 100 residues have been polymerised by the ribosome[19], and follows removal of the leader methionine residue by a methionine aminopeptidase to expose an N-terminal glycine [18] (Fig. 1). More rarely, post-translational myristoylation can occur following exposure of an internal glycine after cleavage of pro-apoptotic proteins by caspases in the apoptotic cascade [20]. Myristoylation of a substrate can have numerous effects, including influencing protein–protein interactions, enhancing interactions of the protein with either organelle or plasma membranes and changes in protein stability [13, 21].

a Co-translational and b post-translational myristoylation. MetAP methionine aminopeptidase
Myristoyl-CoA:protein N-myristoyltransferase
The enzyme that catalyses the transfer of myristate is myristoyl-CoA:protein N-myristoyltransferase (NMT). It appears to be ubiquitous in eukaryotes and has been isolated and characterised in yeast and fungi (Candida albicans, Saccharomyces cerevisiae [22], Cryptococcus neoformans [23] and Aspergillus nidulans [24]), parasitic protozoa (plasmodium species [25], Leishmania major [26] and, Trypanosoma brucei [26]), insects (Drosophila melanogaster [27]), plants (Arabidopsis thaliana [28]) and mammals (including mouse, rat, cow and human). NMT has been shown to be essential for the survival of S. cerevisiae [29], C. albicans [30], C. neoformans [31] and the bloodstream forms of the parasites L. major and T. brucei [32]. It is important in the development of Drosophila [27] and mice [33]. There is no precisely defined “myristoylation motif”, although substrate specificity can be rationalised to a certain extent and the enzyme has an absolute requirement for a peptide or protein substrate with glycine at the N terminus. NMT also appears to be very specific for transfer of myristate, at least in vivo [8].
Catalytic mechanism and enzyme structure
The catalytic cycle of NMT involves a sequential, ordered Bi-Bi mechanism [22, 34]. The myristoyl-CoA substrate binds first to the apo-enzyme, inducing a conformational change that allows binding of the peptide substrate. The chemical transformation proceeds by nucleophilic substitution via attack by the N-terminal glycine amine on the myristoyl-CoA (myr-CoA) thioester; CoA product is released first, followed by myristoylated peptide [34] (Fig. 2).

NMT catalytic cycle
This mechanism is supported by both biochemical and structural studies, including site-directed mutagenesis experiments [35–37]. An initial crystal structure was published for C. albicans NMT (CaNMT) and defined the enzyme as monomeric, with a compact globular structure and a large, saddle-shaped β-sheet flanked by several α-helices dominating the core [38]. The protein appears to have internal twofold symmetry, with two distinct but structurally similar regions corresponding to the N- and C-terminal halves. The “NMT fold” was novel at the time of its discovery [38]. A crystal structure of S. cerevisiae NMT (ScNMT) with substrate analogues of myristoyl-CoA (myr-CoA) and peptides bound confirmed these observations [39]. These and subsequent co-crystal structures have given insight into the binding of myr-CoA and peptide substrates as well as chemical transformation [40–42]. Structures of myr-CoA and analogues bound to CaNMT and ScNMT, and more recently also L. major and Leishmania donovani NMTs (LmNMT and LdNMT) indicate that myr-CoA binds in a bent “question mark” conformation (Fig. 3). The thioester carbonyl is positioned to interact with the main chain amides of two enzyme residues that create an oxy-anion hole. This hole polarises the carbonyl and activates it towards nucleophilic substitution, as well as stabilising the tetrahedral intermediate formed [39]. One of the bends directs the end of the fatty acyl chain into a deep, narrow pocket in the enzyme where the methyl group interacts with the pocket floor. This provides a way for the enzyme to “measure” the length of the acyl-CoA chain and position the CoA and thioester moieties appropriately, and goes some way to explaining the high specificity of NMT for myristoyl-CoA [39]. All NMTs characterised to date also have very high affinity for myristoyl-CoA; this is essential in vivo, since myristate is a very scarce intracellular acid [43]. The enzyme forms multiple interactions with the ADP moiety of Coenzyme A, and there is also evidence of an intramolecular hydrogen bond between the thioester sulphur and the N6 adenine amine of CoA [40]. This interaction could stabilise the thiolate leaving group and also facilitate diffusion of CoA product away from the enzyme as a compact, globular structure [40].

Crystal Structure of NMT and bound myristoyl-CoA (MYA) (PDB: 1IID) showing helical structure in purple, β-sheet in yellow, a peptide substrate in blue, myristate in red and CoA in green. Image generated with PyMol (2008, DeLano Scientific)
Peptide and protein substrates can only bind after complexation with myr-CoA [34]. One reason for this is that myr-CoA forms an integral part of the peptide binding site [39, 40]. By looking at the environment of each residue, it has been possible to rationalise some of the peptide substrate specificities [39, 40]. The peptide N-terminal glycine ammonium interacts electrostatically with the buried carboxylate of the C-terminal enzyme residue, in a very unusual example of direct catalytic involvement of the C-terminal carboxylate [40]. This carboxylate is probably responsible for deprotonation of the ammonium so that the newly generated nucleophilic amine can attack the thioester [40]. Gordon and co-workers have suggested that if the amine group is rotated around the backbone ψ angle, it can approach the polarised carbonyl of the thioester at the correct geometry for nucleophilic attack (Fig. 4) [40]. This mechanism could explain why the enzyme has such a strong requirement for an N-terminal glycine on the peptide substrate: such a rotation may be hindered in the confines of the enzyme for residues other than glycine, which all have substituents on the β carbon [40]. Pre-steady state kinetic analyses of NMT mutants support this proposed sequence of events [44].

Part of the chemical transformation step. N-terminal peptide glycine (blue) can interact with Tyr3 of the substrate (blue), the enzyme C-terminal carboxylate (Leu455) and other enzyme residues (black). The angle of attack at the myristoyl-CoA thioester (red) is marked as a red line
Pre-steady state experiments have also revealed that the rate-determining step in the full enzyme mechanism is not the chemical transformation but a step subsequent to it [45]. This is likely to be a conformational change associated with release of myristoylated peptide product [45], although exactly which conformational changes occur upon myr-CoA binding and after the chemical reaction remains the subject of debate. Initial crystal structures implied that a particular loop was involved in closing over to form part of the peptide binding site [39–41] but a more recent study, which included the N terminus of NMT in the structure—a feature that had either been removed or was disordered in previous studies—indicated that regions of the N terminus could be involved instead [42]. However, this study was carried out with non-peptidic inhibitors of NMT, which bind in a slightly different way to peptide substrates. A structure of the full enzyme with a myr-CoA analogue and genuine peptide substrate would perhaps be more conclusive.
NMT substrate specificity
Whilst less is known about the specificity of NMT for CoA, binding of the acyl chain has been investigated thoroughly using myristic acid analogues, generally as a tool for investigating the myr-CoA binding site or as potential enzyme inhibitors [46–51]. These have included compounds with chains of varying length, containing ethers, thioethers, double and triple bonds, aromatic components, carbonyls and nitrogen functional groups. Generally, these studies have been performed using ScNMT, and are in good agreement with the structure of the binding pocket as determined by X-ray crystallography. A number of observations have been made, including:
-
Chain length is more important for binding than hydrophobicity [46];
-
The myristoyl-CoA binding site can accommodate a wide variety of polar and protruding functional groups in the chain, although activity generally decreases with increasing polarity [47, 49];
-
The fatty acyl-CoA binding pocket has structural features that “measure” the distance from the C-5/C-6 chain bend to the ω-terminus [50], the distance from the thioether carbonyl to the ω-chain end [52] and the steric bulk of the chain end [47, 48, 50];
-
Peptide binding is affected by the acyl-CoA analogue bound [46];
-
The myristoyl-CoA binding site is highly conserved across those species studied (for example, between yeast and plant NMT [54] and between yeast and human NMT) [53].
Chain length is an important consideration in vivo. Acyl chains one methylene shorter or longer than myristate are well tolerated, perhaps not unexpectedly given that 13- and 15-carbon fatty acids are very rare in eukaryotes [55]. However, palmitoyl-CoA, a 16-C fatty acyl-CoA, is found in cells in much higher concentration than myristate and can bind to NMT [55]. Although it is a poor substrate, palmitoyl-CoA is a competitive inhibitor for the enzyme [52, 56]. These findings have led to speculation that palmitoyl-CoA pools must in some way be made unavailable to NMT in vivo, perhaps by sequestration by acyl carrier proteins [52]. Although a wide range of myristate analogues can be incorporated into peptides by NMT, these analogues can have very different properties and would correspondingly affect the properties of the acylated peptide [46].
The protein substrate specificity of ScNMT has been extensively probed using shorter peptide analogues [57, 58], summarised in Fig. 5.

Summary of the peptide substrate specificity of ScNMT. Residues are numbered from the leader methionine, which must be removed by methionine aminopeptidase prior to myristoylation
The general consensus is that active peptide substrate recognition occurs with around the first 10 residues. By considering information from a number of sources, including the sequences of known myristoylated proteins, crystal structures and biochemical data from the literature, Maurer-Stroh and co-workers refined the N-terminal myristoylation motif and used it to predict myristoylated proteins from amino acid sequence [59, 60]. They propose that as many as 17 N-terminal residues are involved in determining whether the protein or peptide will be myristoylated, and that this section is split into three regions: region 1 (residue positions 2–7) which is bound in the active site pocket of the enzyme, region 2 (residues 8–11) which interacts with the surface of NMT around the catalytic site pocket opening and region 3 (residues 12–18) which is a hydrophilic linker section of protein. Region 2 tends to contain small, quite polar residues and region 3 increasingly hydrophilic residues and sequences without any particular secondary structure-forming propensity, presumably preventing the N-terminal section from partially folding and inhibiting recognition by NMT.
In contrast to the acyl-CoA binding site, the peptide binding site is not highly conserved across species. As a result of numerous anti-fungal NMT drug discovery programmes, most work has focussed on the differences in substrate specificity of ScNMT or CaNMT and HsNMT. An initial study co-expressed ScNMT in E. coli along with the mammalian Gα subunit proteins (known to be myristoylated by human NMT [61]) and found that ScNMT was unable to transfer myristate to G2α [62]. The differing substrate specificities of these enzymes have also been investigated using a series of octapeptides [35]. Starting from a ScNMT crystal structure, Maurer-Stroh and co-workers used sequence alignment and modelling to predict substrate specificities for NMTs from a variety of species. They propose that HsNMT has a requirement for glycine at position 2 (i.e. the first amino acid after removal of leader met), prefers polar residues at positions 3 and 4, large hydrophobic residues at position 5, small, polar residues such as serine at position 6 and lysine at position 7 [59]. Acidic residues at positions 8 and 9 are not as disfavoured as in the yeast case due to the presence of some positively charged residues in the enzyme loop above this section [59].
Mammalian NMT isozymes
All mammalian species studied to date, including humans, appear to have two NMT enzymes encoded by distinct genes [63]. The human isozymes, HsNMT1 and HsNMT2, share 77% identity and have homologues in mouse and rat [63, 64]. Each isozyme is highly conserved amongst mammals: for example, HsNMT1 and mouse NMT1 share 97% identity and HsNMT2 and mouse NMT2 share 96% of their amino acid sequence [63], suggesting a specific role for each isozyme in mammalian physiology or development. In addition, there appear to be multiple forms of NMT1 protein in mammals. Prior to the identification of the second mammalian gene several forms of NMT, differing in molecular weight or subcellular distribution, had been observed in several tissues from different mammals [61, 65, 66]. However, it was unclear whether these isoforms arose from differential post-translational processing of the enzymes, nor whether these differences were tissue-specific. There is now some evidence that additional complexity could result from alternative splicing of the mRNA [67]. McIlhinney and co-workers identified two potential Met start sites which would give rise to long and medium forms of NMT. Splicing of the mRNA could shift the reading frame, allowing translation initiation to commence at an internal methionine (Fig. 6) giving rise to a short NMT (NMTS). These authors and others have observed differential tissue distribution of the various NMT isoforms, but it remains unclear whether NMT1 and NMT2 themselves are expressed differently in diverse tissues of the mature organism [64].

NMT splice variants
Interestingly, in all the isoforms of NMT isolated, the main differences occur at the N-termini of the enzymes. NMT1 and NMT2 have the most sequence divergence at their N-termini [63] and any splice variants or isoforms arising from initiation of translation at different start codons will have truncations in the N-terminal domain. This region is not required for catalytic activity, and it may be involved in targeting the enzyme to various subcellular locations such as ribosomes, allowing efficient co-translational myristoylation to occur [68]. It is possible, therefore, that these isoforms have different functions based primarily on their localisation rather than differences in inherent substrate specificity. Little is known about distinct functions of the NMT1 isoforms, but recent work investigating NMT1 and NMT2 has established that they have overlapping but distinct substrate specificities in vivo and in vitro [63], potentially translating into different roles in development and cellular processes such as apoptosis and proliferation. Young and co-workers investigated the functional redundancy of NMT isozymes in vivo by knocking out the NMT1 gene in mice via an insertional mutation process termed gene trapping [33]. They found that in normal mice NMT1 and NMT2 were expressed to a similar extent in a wide variety of tissues but that in embryos NMT1 was expressed more than NMT2. NMT1 −/− (homozygous deletion mutant) mice were not viable and NMT1 +/− heterozygous mice were born less frequently and died earlier. NMT2 could not rescue the development of NMT1 −/− mice, which had very low NMT2 activity, suggesting that NMT1 is the main NMT in embryonic development of mice.
Differential activity of NMT isozymes may also help explain the process of heterogeneous acylation that occurs in the retina and results in N-terminal acylation of retinal proteins with myristate but also with other fatty acids, notably unsaturated C14 and saturated C12 acids [69]. While it seems that no one NMT isoform is responsible for this phenomenon, each isoform has different kinetic parameters for each fatty acyl-CoA/peptide pair, a reflection of the inter-dependence of fatty acyl-CoA and peptide binding. This may contribute to the regulation of heterogeneous acylation, but further insights into this fundamental process await the application of advanced analytical techniques in retinal cell lines [70, 71]. The effect of the NMT isozymes on cellular processes such as apoptosis and proliferation is discussed later, in the context of the myristoylated proteins involved (Sections Myristoylation and apoptosis and NMT in cancer).
NMT regulation
As mentioned previously, it appears that palmitoyl-CoA can competitively inhibit the enzyme in vitro, implying that for efficient myristoylation to take place there must be segregation of the enzyme from the palmitoyl-CoA pools in vivo [52]. This has importance for all eukaryotes, since myristoylation is essential in most of these organisms and involved in crucial cellular processes such as signalling [13]. The enzyme appears to be constitutively active the majority of the time in most tissues, although the issue of isozymes and isoforms and their subcellular or tissue distribution has yet to be clearly elucidated, and may play a role in regulation.
The regulation of myristoylation in the yeast S. cerevisiae is relatively well understood. The (single) enzyme appears to be continuously active in the cell and largely cytosolic [72], and the issue of access to fatty acyl-CoA pools is therefore of critical importance. It has been shown that myristoyl peptides bind membranes with a Gibbs free binding energy of around 8 kcal/mol [73], whereas the more hydrophobic palmitoyl-CoA binds more strongly, resulting in around 15-fold greater partitioning of palmitoylated substrates to the membrane [72]. Palmitoyl acyltransferases have only recently been characterised, but in most cases studied they also localise to membranes [74, 75]. This could be one mechanism by which certain fatty acyl-CoA substrates are made unavailable to NMT in the cell. A summary of the regulation of myristoylation in vivo in S. cerevisiae is shown in Fig. 7. Both fatty acid synthetase and acyl-CoA synthetase (FAA) genes are involved in regulating myr-CoA pools.

Regulation of myristoylation in S. cerevisiae. FAA1 is an acyl-CoA synthetase; MetAP methionine aminopeptidase, FA fatty acid
Several other regulatory mechanisms have been suggested. Sharma and King isolated a protein from the particulate fraction of bovine brain that can inhibit myristoylation in vitro [76]. They showed that this protein, named NIP71, appears to show mixed inhibition with respect to peptide and myristoyl-CoA substrates [77] and is homologous to heat shock cognate protein 70 (a chaperone molecule in the cell) [78]. However, the regulation of NMT activity by this protein in vivo has yet to be proven. Although myristoylation is generally regarded as an irreversible modification, pools of non-myristoylated protein substrates have been shown to exist in cells, for example, non-myristoylated myristoylated alanine-Rich C Kinase Substrate (MARCKS) in synaptosomes [79]. It was subsequently demonstrated that an activity capable of apparently demyristoylating MARCKS exists in the cytoplasmic fraction of the cells, and is dependent on calcium [80]. Vergères and co-workers more recently investigated the N-terminal cleavage of MARCKS with macrophage cytosolic and calf brain extracts which effectively demyristoylated the protein [81]. It is not clear, however, whether this demyristoylation activity is the same as that observed in synaptosomes, nor whether it is due to a specific enzymatic activity. Tentative evidence suggests that NMT1 activity can be modulated by degradation by calcium-dependent proteases (calpains) [82], and that NMT2 may be regulated by caspases [83], though these hypotheses have not yet been rigorously tested.
Sharma and Raju have suggested that coenzyme A can regulate myristoylation versus demyristoylation by NMT but the relevance of this in vivo has yet to be demonstrated [84]: NMT can catalyse the reverse demyristoylation reaction in vitro but only at greatly reduced rates, and this reaction is highly dependent on coenzyme A concentrations [85]. NMT can also be phosphorylated by some of its tyrosine kinase substrates, and in a recent review Selvakumar and Sharma suggested that this could be a regulatory mechanism for myristoylation [86]. However, any functional differences of this phosphorylated NMT have yet to be reported, and so the significance of this process in vivo is unclear. In summary, there is relatively little conclusive data on the regulation of NMT and myristoylation in vivo. Access to fatty acyl-CoA pools is likely to be important and should help explain both why NMT is not significantly inhibited by palmitoyl-CoA in the cell and the pattern of heterogeneous acylation in the retina (Section Mammalian NMT isozymes).
Myristoylated proteins
The prevalence of N-terminally myristoylated proteins in eukaryotes has been estimated at between 0.5% and 3% of the cellular proteome, depending on species and the predictive model used [60, 87]. These proteins have a broad range of functions and include protein kinases and phosphatases, Gα proteins, nitric oxide synthase [88], ADP-ribosylation factors (ARFs), calcium binding proteins and membrane or cytoskeleton-associated structural proteins such as MARCKS [21, 61]. Many of these proteins are involved in signalling cascades. In addition, several viral proteins and, more recently, some bacterial proteins have been shown to be myristoylated in vivo, usually by the eukaryotic host cell NMT [89].
The myristoyl moiety can perform one or a combination of several roles in the cell:
-
promote reversible membrane binding which, in conjunction with other interactions, localises a protein to a membrane,
-
form an integral part of the protein tertiary structure to stabilise the conformation,
-
form part of a protein–protein interaction site.
An important example of a protein whose 3D structure is stabilised by a myristoyl group is cAMP-dependent protein kinase, in which the myristoyl moiety binds in a deep hydrophobic cleft [90]. In the picornavirus protein VP4, myristate moieties on different proteins interact and this is thought to contribute to protein–protein interactions driving assembly of the viral capsid [91, 92]. Mammalian NADH-cytochrome b(5) reductase provides an unusual example of myristate competing for a protein binding site. A recent report suggests that myristoylation interferes with recognition by the signal recognition particle, resulting in a proportion of this protein remaining outside the ER where it may relocate to the mitochondrial outer membrane [93].
The promotion of reversible membrane binding by myristate is well established and likely to occur by simple insertion of the hydrophobic myristate moiety into the lipid bilayer. Studies by Peitzsch and McLaughlin on the association of myristoylated peptides with lipid membranes indicates that the Gibbs free energy of binding of such peptides is around 8 kcal/mol, corresponding to an effective dissociation constant of 10−4 M, which is barely sufficient to localise the peptide to the membrane [73]. As a result, myristate is generally necessary but not sufficient to confer membrane localisation to myristoylated proteins.
The two-signal membrane binding model
It has been proposed that a second signal is required to establish strong localisation of a myristoyl protein to a membrane. This signal could be another fatty acyl chain such as palmitate, a polybasic cluster of amino acids that interacts with the head groups of acidic phospholipids, or a domain that will interact with another membrane-bound protein (Fig. 8) [13, 21]. Proteins that use the myristate plus basic motif include pp60src [94, 95], the HIV-1 viral protein Gag [96] and MARCKS [97]. Other Src family kinases and Gα proteins use dual fatty acylation with palmitate and myristate to generate strong membrane binding [21]. Membrane binding is sometimes essential for the biological function of the protein; for example, non-myristoylated mutant of pp60v-src (a viral oncogene) does not localise to the plasma membrane and is not able to transform cells [94].

Two-signal hypothesis of myristate-mediated membrane binding. Myristate (red), palmitate (pink), protein (blue) and membrane-bound protein (green). See text for detail
This two-signal model provides mechanisms whereby membrane association may be modulated, via so-called myristoyl switches. In one scenario, ligand binding induces a conformational change in the protein and the myristoyl group flips into a hydrophobic binding pocket (Fig. 9a). For example, binding of ARF GTPase (which mediates vesicle trafficking in cells) to the plasma membrane is thought to be regulated by such a switch (Fig. 9d) [98].

Myristoyl switches, A, B and C. Ligands (green); acyl chain (red); protein (blue). See text for detail. D ARF myristoyl switch
A variation on this theme is an electrostatic switch, where either ligand binding, binding of ions such as calcium, or phosphorylation attenuates the positive charge on a polybasic domain contributing to membrane binding, causing dissociation from the membrane (Fig. 9b and c) [99]. An example of a protein regulated in this manner is MARCKS, which is involved in processes such as cell spreading and exocytosis. Phosphorylation of a polybasic effector region reduces the positive charge in the polybasic domain and hence the binding affinity, causing release of MARCKS from the membrane (as in Fig. 9c) [99].
Myristoylation and apoptosis
There is recent evidence that post-translational myristoylation of proteins may be an important mechanism in the regulation of programmed cell death, or apoptosis [100]. Apoptosis is a tightly controlled process whereby cells undergo a series of sequential and broadly conserved morphological changes in response stimuli such as injury to cellular membranes, DNA or mitochondria, cytotoxic T-cell killing and infection by some viruses [101]. At the molecular level, structural components such as the nuclear membrane and cytoskeleton are dismantled, DNA is degraded and DNA repair enzymes deactivated. Mitochondrial dysfunction may also be initiated. The “executioners” of this process are proteases called caspases (cysteine-containing Asp-ases) which can cleave upwards of 100 target proteins in the cell. The development of technology to map these proteolytic cleavage events is ongoing and two novel mass spectrometry-based proteomic methods were recently compared and reviewed [102].
One of the proteins cleaved by caspase-8 is the pro-apoptotic protein Bid. In 2000 it was shown by Zha and co-workers that the p15 fragment of Bid contains an N-terminal glycine exposed by caspase cleavage that is myristoylated post-translationally by NMT during apoptosis [20]. The two fragments were known to relocalise to the mitochondrial membrane as a non-covalent complex, where the p15 fragment causes oligomerisation of another protein Bak, which initiates mitochondrial dysfunction and release of cytochrome c into the cytosol (Fig. 10). Post-translational myristoylation is required for the translocation of Bid to the mitochondrial outer membrane. Subsequent studies have suggested a role for post-translational myristoylation of caspase-cleaved actin (a cytoskeleton structural element) and gelsolin (an actin regulatory protein) [103, 104]. The biological functions of these myristoylation processes are not yet well understood.

Post-translational myristoylation of Bid during apoptosis
Another caspase substrate post-translationally myristoylated in apoptotic cells is PAK-2 (p21-activated protein kinase 2), a serine/threonine kinase activated by small GTPases and cleaved by caspases-3 during apoptosis [105]. The C-terminal cleaved portion of PAK-2 is myristoylated on the exposed glycine and then localises to internal membranes and regions of the plasma membrane termed membrane ruffles. Here, PAK-2 signals through the stress-activated signalling pathway to promote further events in apoptosis, although it does not directly promote mitochondrial dysfunction. Recently, in vivo metabolic labelling with an azido-myristate analogue (see Section Chemical proteomics) has been used to study post-translational myristoylation in apoptosis [106]. The authors showed that 15 proteins undergo myristoylation upon caspases cleavage in Jurkat cells and identified five of these.
As previously alluded to in Section Mammalian NMT isozymes, there is evidence to suggest that the NMT isozymes may have distinct roles in apoptosis [107]. Small interfering RNA (siRNA) was used to knock down the levels of each isozyme in two cancer cell lines. The authors reported cross-talk between the two isozymes such that when NMT1 is knocked down, NMT2 expression levels increase; however, the inverse does not apply. Apoptosis increased when either enzyme was ablated but the effect was more significant with NMT2. In addition, expression of anti-apoptotic proteins such as Bcl-XL and Mcl-1 decreased in response to NMT2 knock down, but the effects of losing NMT1 were not as significant.
NMT as a potential drug target
Myristoylation is clearly an important cellular process. Many myristoylated proteins are involved in signalling networks, including proto-oncogenes like pp60c-src, and myristoylation may be important in apoptosis. Given the involvement of these proteins in controlling aspects of cell growth, proliferation and survival, one might expect myristoylation to be misregulated in cancer [108]. In addition, several eukaryotic human pathogens such as pathogenic fungi and parasites require NMT for their survival and some viruses and bacteria use the host NMT enzyme to myristoylate their own proteins.
To avoid toxicity, one might expect that it will be important that a drug targets only a single NMT; whether that means exploiting substrate specificity differences of a fungal NMT compared to human NMT to inhibit the fungal enzyme, or targeting an NMT isozyme specifically responsible for myristoylating a viral protein or in promoting proliferation of cancer cells. Interestingly, there has been no study reported to date on the toxicity of on-target NMT inhibition in vivo, and it remains to be seen how much of a challenge this may present in taking NMT inhibitors to the clinic. In general, it has proven relatively straightforward to differentiate between the single NMT from single-celled organisms and mammalian NMTs; however, there has not yet been conclusive evidence for inhibitors that can target a single mammalian NMT isoform in live cells. Methionine aminopeptidase (MetAP) lies upstream of NMT in the co-translational modification cascade; however, studies using MetAP inhibitors have not been very informative with regard to myristoylation due to myriad other effects associated with inhibition of MetAP, particularly in the cell cycle [109].
NMT in infectious disease
NMT is essential for the vegetative growth of the opportunistic human pathogen C. albicans [30]. An immunosuppressed mouse model was used to show that C. albicans producing mutant NMT with greatly reduced activity did not kill the mice, unlike the wild-type NMT strain, suggesting a role in virulence. Similarly, it was shown that C. neoformans (which can cause chronic meningitis in immunosuppressed patients) possessing a less active mutant version of NMT cannot survive at 37 °C without the addition of myristic acid to the medium [31]. The search for species-specific inhibitors has focused on the peptide binding pocket of NMT since this is not strictly conserved across species, unlike the myristoyl-CoA binding site (see Section NMT substrate specificity). Initial work identified a dipeptide inhibitor with an IC50 of around 50 nM and 250-fold selectivity for CaNMT over HsNMT [110] and subsequent investigations have focused on peptomimetic inhibitors of the enzyme, which are likely to have increased half-life in vivo and bioavailability [111]. Inhibitors of C. neoformans are also under investigation [112]. Prominent anti-fungal NMT inhibitor programmes were launched by both Pfizer and Roche in the 1990s, following the discovery and characterisation of NMT in pathogenic yeasts by Gordon et al. [29, 31].
Several parasitic protozoa also possess NMTs essential for their survival in the host [113]. NMT enzymes have been characterised in L. major [32] (which causes leishmaniasis), T. brucei (responsible for African sleeping sickness) [32] and P. falciparum (the parasite causing most cases of malaria in humans) [25]. Several studies have sought to identify selective inhibitors of the NMTs in these organisms [26, 113, 114].
Another potential target for NMT inhibitors is HIV-1 infection. Both the Gag and Nef viral proteins require myristoylation by the host cell NMT to carry out their functions properly. Gag is the polyprotein precursor for the four major structural components of the viral capsid and is targeted to the plasma membrane by myristoylation where it interacts through a myristate plus basic motif (see Section The two-signal membrane binding model) [96]. Myristoylation is required for the assembly and budding of normal virions; NMT inhibitors block processing of the gag precursor at the cell membrane, and greatly reduce the infectivity of released virus particles [115]. Accumulation of non-myristoylated gag in the cytoplasm also has negative downstream effects on other viral proteins such as env.
Any anti-HIV inhibitor of myristoylation would have to target human NMT, raising the possibility of toxicity in uninfected cells. One recent report suggests that a drug may be able to exploit the distinct substrate profiles of human NMT isozymes [116]. The authors reported that both isozymes prefer Nef over Gag, with NMT2 showing increased affinity in vitro and in the cell. Nef enhances the infectivity and replication potential of HIV in an infected cell by altering cell surface moieties such as those involved in immune recognition. However, subsequent work [117] has suggested that NMT1 myristoylates Gag in vivo and that reducing activity of a specific NMT1 isoform, but not NMT2, negatively affects HIV-1 production. A previous study had suggested that Gag binds to NMT2 only whereas Nef prefers NMT1 over NMT2 [118]. As these conflicting observations make clear, current methods for predicting and understanding NMT substrate specificity in vivo are lacking in precision and reliability, and the potential of NMT inhibitors as anti-HIV agents with acceptable toxicity profiles remains an open question pending the development of improved techniques (see Section Chemical proteomics).
NMT in cancer
Given that several myristoylated proteins are involved in signalling processes, including those that regulate cell proliferation and growth, Felsted, Glover and Hartman first proposed in 1995 that myristoylation should be considered as a chemotherapeutic target for cancer [108]. They suggested that further research would be required to (1) establish that abnormal myristoylation contributes to or results from carcinogenesis, (2) understand how myristoylation is regulated in mammalian cells, and (3) establish ways of selectively interfering with myristoylation in cancer cells to avoid general toxicity.
There is now substantial evidence to suggest that NMT is involved in cancer. Sharma and co-workers in particular have amassed considerable data suggesting that NMT is over-expressed in human colorectal tumours and adenocarcinomas [119, 120]. Over-expression also occurs relatively early in carcinogenesis, in colonic polyps that are not yet malignant [119]. There is also evidence for the over-expression of NMT in gallbladder and oral squamous cell carcinomas [121, 122] and brain tumours [123]. Clegg and co-workers have looked at NMT in mammary epithelial cells and found that proliferative capacity correlated with NMT activity [124]. They also reported that there may be a redistribution of NMT from the membrane to the cytosol in cancer cells.
Myristoylation of an oncogene: Src
It is well established that the NMT substrate Src has elevated activity in some human cancers and that this contributes to pathogenicity [125]. Src is a tyrosine kinase and the cellular homologue of v-src (the viral oncogene from Rous sarcoma virus), whose transformation potential is dependent on localisation to the plasma membrane, mediated by myristoylation at the N terminus [94]. Myristoylation has been shown to be required for transformation by Src and for its activation during mitosis [126]. Src is activated by autophosphorylation at Tyr416 and by dephosphorylation of Tyr527, which when phosphorylated causes an inter-domain interaction that folds the protein into a closed, inactive state [127]. A non-myristoylated mutant has a decreased rate of phosphorylation and suppressed kinase activity, and so is not as efficient at inducing events in proliferation [128]. The accessibility of Src to regulatory phosphatases and kinases may depend on subcellular localisation and hence on myristoylation.
In particular, Src shows increased activity in tumours of the colon and breast. In one investigation of Src activity in tissues ranging from benign colonic polyps to metastatic lesions, enzyme activity and level was elevated relative to normal tissue and increased with progressively more advanced cancer [129], indicating a role for Src in colon cancer, both in the early stages of carcinogenesis and in cancer progression. Src also shows increased activity in human breast cancers, where it may couple with the growth factor receptor pathway which is often constitutively activated in this disease [130]. The membrane association of Src is important in this case, and Src activation during mitosis may occur specifically in so-called lipid rafts (cholesterol-rich domains of cell membranes), as demonstrated in breast cancer cell lines [131]. This study also implied that inhibiting Src family tyrosine kinase activity in lipid rafts suppressed the phosphorylation of Cyclin D2 (a key player in cell cycle progression) in tumour but not in normal cells.
It is possible that increased NMT activity is required for localisation and activation of over-expressed myristoyl-proteins involved in cellular proliferation (like Src). Enhanced NMT activity could also potentially lead to abnormal myristoylation patterns in cancer cells and contribute to pathogenesis [119].
The two NMT isozymes have been shown to have distinct roles in apoptosis, as previously discussed (Section Myristoylation and apoptosis), and in proliferation [107]. NMT1 knockdown reduced proliferation by around 27%, whereas NMT2 knockdown did not inhibit cell division. Ablating NMT1 also had an effect on several signalling proteins involved in proliferation, including Src: generally Src levels were increased in knockdown cells but activated, phosphorylated Src levels were decreased. One of the substrates of Src, FAK, was found to be less abundant in its activated phosphorylated form when NMT1 levels were reduced. These data are consistent with the idea that Src must be myristoylated in order to carry out its biological functions. Inhibiting NMT1 also affected the levels of MARCKS and reduced signalling through the c-Raf/MEK/ERK/Elk pathway, which is also implicated in cell cycle progression and may be activated by Src.
Myristoylation of a potential tumour suppressor: Fus1
Whilst targeting the myristoylation of oncogenes such as Src may be a potential avenue for novel cancer therapy, there are also potential negative consequences for broad-spectrum inhibition of myristoylation in carcinogenesis. Tumour suppressor genes are often mutated or their protein products deregulated in human cancers. Fus1 is a myristoylated candidate tumour suppressor implicated in lung cancers and, although much of the mechanism of action of Fus1 remains unknown, it appears to promote apoptosis through the intrinsic apoptotic signalling pathway, as summarised in Fig. 11 [132]. Uno and co-workers have reported evidence that non-myristoylated Fus1 loses its tumour suppressor activity and so myristoylation could play an important role in retarding lung cancer pathogenesis [133]. Whilst in normal cells only myristoylated Fus1 could be detected, in cancer cells non-myristoylated protein was also present in significant amounts. It had a much reduced half-life compared to wild-type Fus1 and lost its characteristic localisation at cellular membranes. Experiments with protease inhibitors suggested that non-myristoylated Fus1 may be degraded by proteases [133]. The reason for defective myristoylation of Fus1 is unknown. Any mutations in the N-terminal region of the Fus1 gene have yet to be identified and it is possible that the myristoylation process itself is deregulated. Further work is needed to establish why Fus1 in cancer cells is not myristoylated as efficiently.

The involvement of Fus1 in apoptosis
In keeping with the diverse nature of both myristoylation and cancer, inhibiting NMT in cancer cells has the potential to impact negatively or positively on proliferation. From this point of view, if antineoplastic therapies targeting NMT are developed they are likely to be useful in a specific subset of cancers, and the possibility of provoking further carcinogenesis cannot be ruled out.
Approaches for the analysis of myristoylation
Despite the importance of myristoylation (and the chemically related palmitoylation), there are few methods for the biochemical analysis of these modifications. Traditionally, the main options for detection have been metabolic labelling with radiolabelled fatty acids or direct mass spectrometry of fatty-acylated proteins; both have significant disadvantages and limitations [134].
Traditional approaches
Radiolabelling with [3H]myristate and detection by fluorography is a laborious process due to the very long (weeks or even months) exposure times required. Metabolic labelling with iodo-fatty acid analogues containing [125I] shows greatly improved sensitivity with much faster detection times but necessitates working with relatively large quantities of hazardous material, since the isotope is a potent γ-emitter. Furthermore, it is not always straightforward to obtain radiolabelled substrates, and there is the potential for non-native processing of unnatural iodinated fatty acids.
Mass spectrometry (usually MALDI-TOF) can also be used to detect fatty acylation of proteins but generally the protein of interest must first be purified to some extent and lipophilic fatty-acylated proteins can be lost during preparation. The identification of modified proteins in a complex cell lysate can be highly problematic due to current limitations in proteomic technology [135]; indeed, the efficient detection of post-translational modifications has become something of a ‘holy grail’ in current proteomic research. Most current efforts are focussed on developing methods for the selective enrichment of a proteome in the PTM of interest, which greatly increases the utility of standard proteomic approaches for the detection of PTMs. For example, an in vitro chemical labelling strategy termed acyl–biotin exchange has been developed for palmitoylated proteins by Drisdel and Green [136]. This method has been used in conjunction with mass spectrometric analysis to detect global protein palmitoylation in total yeast membrane lysates [137] and in rat neurons [138]. It is not, however, applicable to myristoylation because myristate is attached via a chemically stable amide bond that cannot be selectively cleaved in the presence of all the amide linkages in a protein. For both myristoylation and a wide range of other important PTMs, chemical proteomics is emerging as the technology of choice.
Chemical proteomics
Chemical proteomics [135, 139] is a multidisciplinary field at the interface between chemistry, proteomics and the life sciences, and promises to transform our understanding of PTM by providing a qualitatively new way to identify and quantify modified proteins. The key enabling technology underlying this approach is the development of ‘bioorthogonal ligation’ chemistry (Fig. 12), which permits the highly selective modification of biologically inert chemical tags.

The three key bioorthogonal ligation reactions in common use in chemical proteomics and protein labelling. A Staudinger-Bertozzi ligation between a phosphine and an organic azide; B Cu(I) catalysed 3+2 cycloaddition reaction between an alkyne and an azide; C strain-promoted 3+2 cycloaddition reaction between a cyclooctyne and an azide. R and R1 can be any set of labels, proteins, DNA or other biomolecules
In the case of myristoylation, these tags can be appended to myristate analogues to enable downstream addition of a panel of labels for detection (e.g. dyes) and purification (e.g. affinity tags), without influencing the upstream biosynthetic labelling process. The field of chemical proteomics applied to PTMs has been reviewed recently [135, 140] and here we consider the subsequent developments most relevant to myristoylation.
In 2007, Ploegh and co-workers reported the use of azido-fatty acid analogues of varying chain length (AzC(11, 13, 14, 15), Fig. 13) for metabolic labelling of fatty-acylated proteins in cells [141]. Tagged proteins were captured via Staudinger Ligation [142, 143] with a biotinylated phosphine, separated by gel electrophoresis and detected by streptavidin blotting (Fig. 14). The different analogues showed distinct protein profiles, with the longer chains (AzC14-16) attached via a hydroxylamine-labile linkage, indicating that these analogues were being incorporated as thioesters in the same manner as palmitate [141]. Cycloheximide (an inhibitor of protein synthesis) reduced incorporation of the shortest analogue tested (AzC12), consistent with the analogue of this chain length acting as a myristate analogue and being incorporated co-translationally [141].

Myristic acid, myristoyl-CoA and analogues

Example of a tagging by substrate experiment for myristoylation, such as that used by Ploegh and co-workers [141]
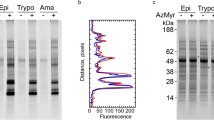
A flurry of subsequent papers demonstrated the robustness and versatility of this approach. Work by Kostiuk and co-workers looked at the detection of palmitoylated mitochondrial proteins using in vitro incorporation of an azido-palmitate analogue (AzC14) and Staudinger Ligation with a variety of labelled phosphines (including biotinylated and fluorescein-conjugated reagents) [144]. Proteins were separated first by charge and then on the basis of size by SDS-PAGE and identified by Tandem-MS. Incorporation via thioester bonds was demonstrated by hydroxylamine and alkali sensitivity. In comparison with in vitro radiolabelling, the technique was found to be rapid, sensitive and specific [144]. A similar method was applied to the detection of proteins post-translationally myristoylated in apoptotic Jurkat T cells [106]. In this study, proteins were metabolically tagged in vivo using an azido-myristate analogue (AzC12) and then captured with a biotin-conjugated phosphine for analysis by streptavidin blotting. The authors also directly compared their method with radioactive labelling using [3H]myristate and found a million-fold amplification of the signal. By combining prediction analysis to determine which proteins cleaved by caspases could be subsequently myristoylated, and the chemical tagging technique, the authors were able to identify several new post-translational myristoylation events.
Parallel studies in our own groups set out to assess the robustness and sensitivity of this tagging-by-substrate approach and the efficiency of the ligation reaction. In vitro experiments were carried out to assess the incorporation of 1AzC11-CoA (Fig. 13) into a test peptide substrate consisting of the N-terminal region of the P. falciparum protein ARF1 (PfARF1), which is known to be myristoylated in vivo [145]. Good incorporation was observed and AzC11-CoA was also readily transferred to the full PfARF1 protein in vitro. Staudinger Ligation with a biotinylated phosphine followed by anti-biotin HRP blot allowed detection of analogue incorporation and capture. A further handle was provided by the use of a C-terminal His6 affinity tag engineered into the recombinant PfARF1 used in the experiments, which can be detected via an anti-His HRP blot. The authors were able to establish that the efficiency of the ligation was >99% by looking for protein in the anti-His blot following pull-down of biotinylated protein by NeutrAvidin™-agarose beads.
In an in vivo approach, E. coli engineered to express CaNMT and PfARF1 protein substrate were fed with AzC11, lysed and probed for azide incorporation using the Staudinger Ligation with a biotinylated phosphine [145]. Again, efficiency of labelling was shown to be >99%.
This work was further extended to metabolic labelling via the alkynyl-myristate YnC12 [146]. In vitro, both analogues AzC11-CoA and YnC12-CoA were incorporated by CaNMT into the model peptide and protein at rates comparable to that of the natural substrate, although alkyne-tagged YnC12-CoA was incorporated twice as fast as azide-tagged AzC11-CoA, perhaps reflecting slightly different enzyme–substrate affinities [146]. Both analogues enabled tagging of protein in the bacterial co-expression system. Capture was carried out with a biotinylated phosphine or by copper-catalysed [3+2] click chemistry [137–139], depending on which tag was used. Both bioorthogonal ligations were highly selective, with the click method being more efficient [146]. This observation is in agreement with a 2006 study comparing the Staudinger Ligation, copper-catalysed cycloaddition and strain-promoted [3+2] cycloaddition as bioorthogonal ligations of azides [147].
The fatty acid probes were designed as isosteric analogues of myristate, in order to mimic the chain length known to be important for recognition by NMT (see Section NMT substrate specificity) [46]. In AzC11 the three terminal carbon atoms have been replaced by nitrogen atoms and in YnC12 there is a triple bond in place of the terminal carbon–carbon single bond [146]. Recently, Hang and co-workers reported the use of both alkynyl- and azido-fatty acids of varying lengths (AzC12, AzC14, YnC12, YnC14 and YnC16), combined with fluorescent or biotinylated capture reagents, to profile fatty acylation in mammalian cells [148]. Fluorescent probes allow captured proteins separated by SDS-PAGE to be detected by in-gel fluorescence, which is both rapid and sensitive. Consistent with the previous studies discussed above, the authors noted superior detection with the copper-catalysed click reaction compared to the Staudinger Ligation. They also observed similar profiles with alkynyl- and azido-fatty acids, providing further evidence for the use of these functional groups as “silent” tags, although they did note improved sensitivity with the combination alkyne-tagged protein plus azide-capture. The study also reports the imaging of tagged fatty-acylated proteins in fixed cells.
Concurrent with the publication of this work, Cravatt and Martin reported their study on protein palmitoylation and took the approach to the next level of refinement by enriching tagged and captured proteins for proteomic analysis via mass spectrometry [149]. Together with the previous studies on the identification of palmitoylated mitochondrial proteins [144] and post-translationally myristoylated proteins associated with apoptosis [106] (both discussed above), this work demonstrates that this chemical proteomics approach can be used to profile fatty-acylated proteomes. The study also employed fluorescent probes to detect alkynyl-tagged proteins, using YnC16, and copper-catalysed click reaction as the bioorthogonal ligation. A third recent study sought to investigate the subcellular distribution of fatty-acylated proteins via a similar approach [150]. Alkynyl-fatty acids YnC(8, 9, 11, 12, 14 and 16) were metabolically incorporated into proteins and then captured via copper-catalysed click chemistry with fluorescent probes. Distribution of fatty-acylated proteins within cells was visualised by carrying out the bioorthogonal ligation step on fixed cells.
The studies described above suggest that AzC11, AzC12, YnC11 and YnC12 generally act as myristic acid analogues, whereas AzC14, AzC15 and AzC16, along with YnC14 and YnC16, target proteins normally palmitoylated.
Conclusion
Post-translational modification may have dramatic or exquisitely subtle effects on protein function. N-Myristoylation affects only a few percent of the eukaryotic proteome, yet is essential for the survival or development of these organisms and has implications in diseases as diverse as cancer and malaria. The subtleties of NMT regulation and the differing roles of the NMT isozymes in mammalian cells remain largely unresolved. The study of global myristoylation has, until recently, been hindered by the low abundance of myristoylated proteins in the cell, the complex and (currently) difficult to predict substrate specificity of NMT and the issues of separating myristoylated proteins from complex cell lysates.
The solution to such a complex problem often comes about through a multidisciplinary approach. Chemical proteomics is emerging as a technology with the potential to successfully tackle these issues and complement the latest mass spectrometry-based proteomics techniques. Tools such as these will be essential in furthering our understanding of this complex biological process and in the development of therapeutics targeting NMT.
References
Woycechowsky KJ, Raines RT (2000) Curr Opin Chem Biol 4:533–539
Ehrmann M, Clausen T (2004) Annu Rev Genet 38:709–724
Giglione C, Boularot A, Meinnel T (2004) Cell Mol Life Sci 61:1455–1474
Hershko A, Ciechanover A (1998) Annu Rev Biochem 67:425–479
Zhao J (2007) Cell Mol Life Sci 64:3017–3033
Cheung WL, Briggs SD, Allis CD (2000) Curr Opin Cell Biol 12:326–333
Karakozova M, Kozak M, Wong CC, Bailey AO, Yates JR 3rd, Mogilner A, Zebroski H, Kashina A (2006) Science 313:192–196
Farazi TA, Waksman G, Gordon JI (2001) J Biol Chem 276:39501–39504
Buglino JA, Resh MD (2008) J Biol Chem 283:22076–22088
Paik WK, Paik DC, Kim S (2007) Trends Biochem Sci 32:146–152
Cohen P (2000) Trends Biochem Sci 25:596–601
Spiro RG (2002) Glycobiology 12:43R–56R
Resh MD (2006) Nat Chem Biol 2:584–590
Resh MD (2004) Subcell Biochem 37:217–232
Poole LB, Karplus PA, Claiborne A (2004) Annu Rev Pharmacol Toxicol 44:325–347
Gordon JI, Duronio RJ, Rudnick DA, Adams SP, Gokel GW (1991) J Biol Chem 266:8647–8650
Boutin JA (1997) Cell Signal 9:15–35
Wilcox C, Hu JS, Olson EN (1987) Science 238:1275–1278
Deichaite I, Casson LP, Ling HP, Resh MD (1988) Mol Cell Biol 8:4295–4301
Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ (2000) Science 290:1761–1765
Resh MD (1999) Biochim Biophys Acta-Mol Cell Res 1451:1–16
Towler DA, Adams SP, Eubanks SR, Towery DS, Jackson-Machelski E, Glaser L, Gordon JI (1987) Proc Natl Acad Sci U S A 84:2708–2712
Lodge JK, Johnson RL, Weinberg RA, Gordon JI (1994) J Biol Chem 269:2996–3009
Shaw BD, Momany C, Momany M (2002) Eukaryot Cell 1:241–248
Gunaratne RS, Sajid M, Ling IT, Tripathi R, Pachebat JA, Holder AA (2000) Biochem J 348(Pt 2):459–463
Panethymitaki C, Bowyer PW, Price HP, Leatherbarrow RJ, Brown KA, Smith DF (2006) Biochem J 396:277–285
Ntwasa M, Aapies S, Schiffmann DA, Gay NJ (2001) Exp Cell Res 262:134–144
Boisson B, Giglione C, Meinnel T (2003) J Biol Chem 278:43418–43429
Duronio RJ, Towler DA, Heuckeroth RO, Gordon JI (1989) Science 243:796–800
Weinberg RA, McWherter CA, Freeman SK, Wood DC, Gordon JI, Lee SC (1995) Mol Microbiol 16:241–250
Lodge JK, Jackson-Machelski E, Toffaletti DL, Perfect JR, Gordon JI (1994) Proc Natl Acad Sci U S A 91:12008–12012
Price HP, Menon MR, Panethymitaki C, Goulding D, McKean PG, Smith DF (2003) J Biol Chem 278:7206–7214
Yang SH, Shrivastav A, Kosinski C, Sharma RK, Chen M-H, Berthiaume LG, Peters LL, Chuang P-T, Young SG, Bergo MO (2005) J Biol Chem 280:18990–18995
Rudnick DA, McWherter CA, Rocque WJ, Lennon PJ, Getman DP, Gordon JI (1991) J Biol Chem 266:9732–9739
Rocque WJ, Mcwherter CA, Wood DC, Gordon JI (1993) J Biol Chem 268:9964–9971
Peseckis SM, Resh MD (1994) J Biol Chem 269:30888–30892
Zhang L, Jackson-Machelski E, Gordon JI (1996) J Biol Chem 271:33131–33140
Weston SA, Camble R, Colls J, Rosenbrock G, Taylor I, Egerton M, Tucker AD, Tunnicliffe A, Mistry A, Mancia F, de la Fortelle E, Irwin J, Bricogne G, Pauptit RA (1998) Nat Struct Biol 5:213–221
Bhatnagar RS, Futterer K, Farazi TA, Korolev S, Murray CL, Jackson-Machelski E, Gokel GW, Gordon JI, Waksman G (1998) Nat Struct Mol Biol 5:1091–1097
Farazi TA, Waksman G, Gordon JI (2001) Biochem 40:6335–6343
Sogabe S, Masubuchi M, Sakata K, Fukami TA, Morikami K, Shiratori Y, Ebiike H, Kawasaki K, Aoki Y, Shimma N, D’Arcy A, Winkler FK, Banner DW, Ohtsuka T (2002) Chem Biol 9:1119–1128
Wu J, Tao Y, Zhang M, Howard MH, Gutteridge S, Ding J (2007) J Biol Chem 282:22185–22194
Rioux V, Legrand P (2007) Curr Opin Clin Nutr Metab Care 10:752–758
Farazi TA, Manchester JK, Waksman G, Gordon JI (2001) Biochem 40:9177–9186
Farazi TA, Manchester JK, Gordon JI (2000) Biochem 39:15807–15816
Heuckeroth RO, Glaser L, Gordon JI (1988) Proc Natl Acad Sci U S A 85:8795–8799
Heuckeroth RO, Jackson-Machelski E, Adams SP, Kishore NS, Huhn M, Katoh A, Lu T, Gokel GW, Gordon JI (1990) J Lipid Res 31:1121–1129
Kishore NS, Lu TB, Knoll LJ, Katoh A, Rudnick DA, Mehta PP, Devadas B, Huhn M, Atwood JL, Adams SP et al (1991) J Biol Chem 266:8835–8855
Devadas B, Lu T, Katoh A, Kishore NS, Wade AC, Mehta PP, Rudnick DA, Bryant ML, Adams SP, Li Q et al (1992) J Biol Chem 267:7224–7239
Lu T, Li Q, Katoh A, Hernandez J, Duffin K, Jackson-Machelski E, Knoll LJ, Gokel GW, Gordon JI (1994) J Biol Chem 269:5346–5357
Pasha MK, Selvakumar P, Ashakumary L, Qureshi M, Guziec FS Jr, Dimmock JR, Felsted RL, Glover CJ, Sharma RK (2004) Int J Mol Med 13:557–563
Bhatnagar RS, Schall OF, Jackson-Machelski E, Sikorski JA, Devadas B, Gokel GW, Gordon JI (1997) Biochem 36:6700–6708
Kishore NS, Wood DC, Mehta PP, Wade AC, Lu T, Gokel GW, Gordon JI (1993) J Biol Chem 268:4889–4902
Heuckeroth RO, Towler DA, Adams SP, Glaser L, Gordon JI (1988) J Biol Chem 263:2127–2133
Rudnick DA, Lu TB, Jacksonmachelski E, Hernandez JC, Li Q, Gokel GW, Gordon JI (1992) Proc Natl Acad Sci U S A 89:10507–10511
Bhatnagar RS, Jackson-Machelski E, McWherter CA, Gordon JI (1994) J Biol Chem 269:11045–11053
Towler DA, Eubanks SR, Towery DS, Adams SP, Glaser L (1987) J Biol Chem 262:1030–1036
Towler DA, Gordon JI, Adams SP, Glaser L (1988) Annu Rev Biochem 57:69–97
Maurer-Stroh S, Eisenhaber B, Eisenhaber F (2002) J Mol Biol 317:523–540
Maurer-Stroh S, Eisenhaber B, Eisenhaber F (2002) J Mol Biol 317:541–557
Raju RV, Magnuson BA, Sharma RK (1995) Mol Cell Biochem 149–150:191–202
Duronio RJ, Rudnick DA, Adams SP, Towler DA, Gordon JI (1991) J Biol Chem 266:10498–10504
Giang DK, Cravatt BF (1998) J Biol Chem 273:6595–6598
Rioux V, Beauchamp E, Pedrono F, Daval S, Molle D, Catheline D, Legrand P (2006) Mol Cell Biochem 286:161–170
King MJ, Sharma RK (1992) Mol Cell Biochem 113:77–81
Glover CJ, Felsted RL (1995) J Biol Chem 270:23226–23233
McIlhinney RA, Young K, Egerton M, Camble R, White A, Soloviev M (1998) Biochem J 333(Pt 3):491–495
Glover CJ, Hartman KD, Felsted RL (1997) J Biol Chem 272:28680–28689
DeMar JC, Rundle DR, Wensel TG, Anderson RE (1999) Prog Lipid Res 38:49–89
Rundle DR, Rajala RVS, Anderson RE (2002) Exp Eye Res 75:87–97
Rundle DR, Rajala RV, Alvarez RA, Anderson RE (2004) Mol Vis 10:177–185
Johnson DR, Bhatnagar RS, Knoll LJ, Gordon JI (1994) Annu Rev Biochem 63:869–914
Peitzsch RM, McLaughlin S (1993) Biochem 32:10436–10443
Berthiaume L, Resh MD (1995) J Biol Chem 270:22399–22405
Das AK, Dasgupta B, Bhattacharya R, Basu J (1997) J Biol Chem 272:11021–11025
King MJ, Sharma RK (1993) Biochem J 291:635–639
King MJ, Sharma RK (1994) Mol Cell Biochem 141:79–86
Selvakumar P, Lakshmikuttyamma A, Pasha MK, King MJ, Olson DJH, Mori S, Ross ARS, Hayashi K, Dimmock JR, Sharma RK (2004) J Cell Biochem 92:573–578
McIlhinney RA, McGlone K (1990) Biochem J 271:681–685
Manenti S, Sorokine O, Van Dorsselaer A, Taniguchi H (1994) J Biol Chem 269:8309–8313
Braun T, McIlhinney RA, Vergeres G (2000) Biochimie 82:705–715
Raju RV, Kakkar R, Datla RS, Radhi J, Sharma RK (1998) Exp Cell Res 241:23–35
Selvakumar P, Smith-Windsor E, Bonham K, Sharma RK (2006) FEBS Lett 580:2021–2026
Raju RVS, Sharma RK (1996) Mol Cell Biochem 158:107–113
McIlhinney RA, Patel PB, McGlone K (1994) Eur J Biochem 222:137–146
Selvakumar P, Sharma RK (2006) Can J Physiol Pharmacol 84:707–712
Martinez A, Traverso JA, Valot B, Ferro M, Espagne C, Ephritikhine G, Zivy M, Giglione C, Meinnel T (2008) Proteomics 8:2809–2831
Braam B, Verhaar MC (2007) Curr Pharm Des 13:1727–1740
Maurer-Stroh S, Eisenhaber F (2004) Trends Microbiol 12:178–185
Zheng J, Knighton DR, Xuong NH, Taylor SS, Sowadski JM, Ten Eyck LF (1993) Protein Sci 2:1559–1573
Chow M, Newman JF, Filman D, Hogle JM, Rowlands DJ, Brown F (1987) Nature 327:482–486
Simons J, Rogove A, Moscufo N, Reynolds C, Chow M (1993) J Virol 67:1734–1738
Colombo S, Longhi R, Alcaro S, Ortuso F, Sprocati T, Flora A, Borgese N (2005) J Cell Biol 168:735–745
Cross FR, Garber EA, Pellman D, Hanafusa H (1984) Mol Cell Biol 4:1834–1842
Sigal CT, Zhou W, Buser CA, McLaughlin S, Resh MD (1994) Proc Natl Acad Sci U S A 91:12253–12257
Zhou W, Parent LJ, Wills JW, Resh MD (1994) J Virol 68:2556–2569
Swierczynski SL, Blackshear PJ (1996) J Biol Chem 271:23424–23430
Goldberg J (1998) Cell 95:237–248
McLaughlin S, Aderem A (1995) Trends Biochem Sci 20:272–276
Mishkind M (2001) Trends Cell Biol 11:191
Wyllie AH (1997) Br Med Bull 53:451–465
Simon GM, Dix MM, Cravatt BF (2009) ACS Chem Biol 4:401–408
Utsumi T, Sakurai N, Nakano K, Ishisaka R (2003) FEBS Lett 539:37–44
Sakurai N, Utsumi T (2006) J Biol Chem 281:14288–14295
Vilas GL, Corvi MM, Plummer GJ, Seime AM, Lambkin GR, Berthiaume LG (2006) Proc Natl Acad Sci U S A 103:6542–6547
Martin DD, Vilas GL, Prescher JA, Rajaiah G, Falck JR, Bertozzi CR, Berthiaume LG (2008) FASEB J 22:797–806
Ducker CE, Upson JJ, French KJ, Smith CD (2005) Mol Cancer Res 3:463–476
Felsted RL, Glover CJ, Hartman K (1995) J Natl Cancer Inst 87:1571–1573
Owa T, Yoshino H, Yoshimatsu K, Nagasu T (2001) Curr Med Chem 8:1487–1503
Devadas B, Zupec ME, Freeman SK, Brown DL, Nagarajan S, Sikorski JA, McWherter CA, Getman DP, Gordon JI (1995) J Med Chem 38:1837–1840
Nagarajan SR, Devadas B, Zupec ME, Freeman SK, Brown DL, Lu HF, Mehta PP, Kishore NS, McWherter CA, Getman DP, Gordon JI, Sikorski JA (1997) J Med Chem 40:1422–1438
Lodge JK, Jackson-Machelski E, Higgins M, McWherter CA, Sikorski JA, Devadas B, Gordon JI (1998) J Biol Chem 273:12482–12491
Bowyer PW, Tate EW, Leatherbarrow RJ, Holder AA, Smith DF, Brown KA (2008) Chem Med Chem 3:402–408
Bowyer PW, Gunaratne RS, Grainger M, Withers-Martinez C, Wickramsinghe SR, Tate EW, Leatherbarrow RJ, Brown KA, Holder AA, Smith DF (2007) Biochem J 408:173–180
Furuishi K, Matsuoka H, Takama M, Takahashi I, Misumi S, Shoji S (1997) Biochem Biophys Res Commun 237:504–511
Seaton KE, Smith CD (2008) J Gen Virol 89:288–296
Takamune N, Gota K, Misumi S, Tanaka K, Okinaka S, Shoji S (2008) Microbes Infect 10:143–150
Hill BT, Skowronski J (2005) J Virol 79:1133–1141
Magnuson BA, Raju RVS, Moyana TN, Sharma RK (1995) J Natl Cancer Inst 87:1630–1635
Raju RVS, Moyana TN, Sharma RK (1997) Exp Cell Res 235:145–154
Rajala RV, Radhi JM, Kakkar R, Datla RS, Sharma RK (2000) Cancer 88:1992–1999
Shrivastav A, Sharma AR, Bajaj G, Charavaryamath C, Ezzat W, Spafford P, Gore-Hickman R, Singh B, Copete MA, Sharma RK (2007) Oncol Rep 18:93–97
Lu Y, Selvakumar P, Ali K, Shrivastav A, Bajaj G, Resch L, Griebel R, Fourney D, Meguro K, Sharma RK (2005) Neurochem Res 30:9–13
Clegg RA, Gordge PC, Miller WR (1999) Adv Enzyme Regul 39:175–203
Frame MC (2002) Biochim Biophys Acta 1602:114–130
Wilson LK, Luttrell DK, Parsons JT, Parsons SJ (1989) Mol Cell Biol 9:1536–1544
Engen JR, Wales TE, Hochrein JM, Meyn MA, Ozkan SB, Bahar I, Smithgall TE (2008) Cell Mol Life Sci 65:3058–3073
Bagrodia S, Taylor SJ, Shalloway D (1993) Mol Cell Biol 13:1464–1470
Talamonti MS, Roh MS, Curley SA, Gallick GE (1993) J Clin Invest 91:53–60
Luttrell DK, Lee A, Lansing TJ, Crosby RM, Jung KD, Willard D, Luther M, Rodriguez M, Berman J, Gilmer TM (1994) Proc Natl Acad Sci U S A 91:83–87
Hitosugi T, Sato M, Sasaki K, Umezawa Y (2007) Cancer Res 67:8139–8148
Ji L, Roth JA (2008) J ThoracOncol 3:327–330
Uno F, Sasaki J, Nishizaki M, Carboni G, Xu K, Atkinson EN, Kondo M, Minna JD, Roth JA, Ji L (2004) Cancer Res 64:2969–2976
Resh MD (2006) Methods 40:191–197
Tate EW (2008) J Chem Biol 1:17–26
Drisdel RC, Green WN (2004) Biotechniques 36:276–285
Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates Iii JR, Davis NG (2006) Cell 125:1003–1013
Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates Iii JR, Davis NG, El-Husseini A (2008) Nature 456:904–909
Heal WP, Wickramasinghe SR, Tate EW (2008) Curr Drug Discov Technol 5:200–212
Gamblin DP, van Kasteren SI, Chalker JM, Davis BG (2008) Febs J 275:1949–1959
Hang HC, Geutjes EJ, Grotenbreg G, Pollington AM, Bijlmakers MJ, Ploegh HL (2007) J Am Chem Soc 129:2744–2745
Saxon E, Bertozzi CR (2000) Science 287:2007–2010
Maja Köhn RB (2004) Angew Chem Int Ed 43:3106–3116
Kostiuk MA, Corvi MM, Keller BO, Plummer G, Prescher JA, Hangauer MJ, Bertozzi CR, Rajaiah G, Falck JR, Berthiaume LG (2007) FASEB J 22:721–732
Heal WP, Wickramasinghe SR, Bowyer PW, Holder AA, Smith DF, Leatherbarrow RJ, Tate EW (2008) Chem Commun 4:480–482
Heal WP, Wickramasinghe SR, Leatherbarrow RJ, Tate EW (2008) Org Biomol Chem 6:2308–2315
Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR (2006) ACS Chem Biol 1:644–648
Charron G, Zhang MZM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC (2009) J Am Chem Soc 131:4967–4975
Martin BR, Cravatt BF (2009) Nature Methods 6:135–138
Hannoush RN, Arenas-Ramirez N (2009) ACS Chem Biol 4:581–587
Acknowledgments
MHW thanks the Chemical Biology Centre, Imperial College London, for the award of a studentship. WPH is supported by a Research Project Grant from Cancer Research UK to EWT and DJM (grant C29637/A9913). EWT thanks the Biotechnology and Biological Sciences Research Council (BBSRC), UK, for the award of a David Phillips Research Fellowship (grant BB/D02014X/1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wright, M.H., Heal, W.P., Mann, D.J. et al. Protein myristoylation in health and disease. J Chem Biol 3, 19–35 (2010). https://doi.org/10.1007/s12154-009-0032-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12154-009-0032-8