
Abstract
NUT (midline) carcinoma is a rare, highly aggressive, poorly differentiated carcinoma that characteristically harbors a rearrangement of the NUTM1 gene. Most of these tumors occur in adolescents and young adults, arise from the midline structures of the thorax, head, and neck, and are associated with extremely poor outcomes. Rare cases originating from salivary glands have been reported with clinicopathologic features comparable to NUT carcinoma of other sites. Outcome studies regarding this subgroup are currently lacking. We report a case of NUT carcinoma arising in a submandibular gland of a 12-year-old boy. Diagnosis was confirmed by fluorescence in situ hybridization demonstrating fusion of the BRD4 (19p13.12) and NUTM1 (15q14) gene loci. A systematic review of all previously reported salivary gland NUT carcinomas (n = 15) showed exclusive occurrence of pediatric cases (n = 6) in males compared to adult patients (n = 9, male: female = 1:2; p < 0.05). The median survival was 24 and 4 months for pediatric and adult patients, respectively (95% confidence interval was 8–24 and 1–7 months, respectively; p < 0.01). The 1-year overall survival was 67% for pediatric and 11% for adult patients. Among all NUT carcinomas, pediatric salivary gland tumors may represent a distinct clinical subset associated with male predilection and comparatively prolonged survival.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
NUT (midline) carcinoma is a rare variant of poorly differentiated carcinoma defined by translocations involving the NUTM1 gene on chromosome 15q14 [1,2,3]. The NUTM1 gene is most commonly fused to the Bromodomain and Extra-Terminal (BET) family genes BRD4 gene on chromosome 19p13.12, and less frequently to the BRD3 gene on chromosome 9q34.2 or the NSD3 gene on chromosome 8p11.23 [1, 3, 4].
Originally considered to be restricted to the midline head and neck structures or mediastinal region in young individuals, NUT carcinomas now are identified in patients of all ages, affecting males and females equally, and can rarely arise outside of midline locations. Outcomes are almost universally dismal, with most patients presenting with advanced primary tumors and distant metastases [3,4,5]. Overall survival (OS) does not appear to correlate with gender, tumor site, histology, or lymph node involvement [5, 6]. Response to conventional chemotherapy is poor. However, new treatment strategies involving small-molecule BET inhibitors and histone deacetylase inhibitors are currently in development and hold promise [7,8,9]. Prompt enrollment of patients with early, treatable NUT carcinoma into clinical trials requires early diagnosis by recognition of the clinicopathologic features and a high degree of suspicion.
Salivary gland is an unusual site for NUT carcinoma. To the best of our knowledge, 14 cases of salivary gland NUT carcinoma (5 pediatric and 9 adult) have been reported in the literature [6, 10,11,12,13,14,15,16,17,18,19]. Here, we describe the clinicopathologic features and autopsy findings of a sixth pediatric case arising from the submandibular gland. A systematic review of the reported cases demonstrated male predilection and significantly prolonged OS in the pediatric group.
Case Report
An 11-year-old male presented with an enlarging and painful mass at the angle of the left mandible for 2 months. Physical examination showed a large, complex, firm mass extending downward from the angle of the left mandible without evidence of cellulitis of the overlying skin and soft tissue. Lymph nodes in the submental region and the left posterior triangle of the neck were enlarged. Computed tomography (CT) with contrast enhancement of the neck showed a partially necrotic lesion in the left submandibular space measuring 5.5 × 5.4 × 5.1 cm, as well as a separate 3.6 × 3.2 × 3 cm mass deep to the left sternocleidomastoid muscle. The clinical and radiologic findings were suggestive of a malignant neoplasm originating from the left submandibular gland with nodal involvement.
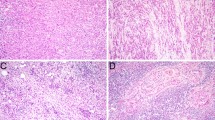
An incisional biopsy of the large mass was performed. Microscopic examination showed cohesive sheets of medium-sized round blue tumor cells intimately associated with a reactive lymphoid process in the background of extensive necrosis. The tumor cells exhibited scant to moderate cytoplasm, mild nuclear pleomorphism, evenly dispersed chromatin, inconspicuous nucleoli, and abundant mitotic figures of both typical and atypical forms (Fig. 1a–d). Squamous differentiation was lacking. The tumor cells exhibited strong but patchy immunoreactivity for AE1/AE3 (Fig. 1e), CK19, CK5/6 and CAM 5.2. The tumor cells were also positive for synaptophysin (Fig. 1f), p63 (patchy, Fig. 1g), CD99, INI-1 (retained nuclear expression), Bcl2 and S-100 (patchy). Negative stains included GFAP, epithelial membrane antigen (EMA), chromogranin A, CD56, NB84, CD34, smooth muscle actin, desmin, myogenin, Melan-A, HMB-45 and CD117. Epstein–Barr virus-encoded small RNAs (EBER) in situ hybridization was negative. Based on the histologic features and immunohistochemical profile, a preliminary diagnosis of poorly differentiated carcinoma was made, with NUT carcinoma and myoepithelial carcinoma being considered more likely on the differential diagnosis. Subsequent fluorescent in situ hybridisation (FISH) study for the BRD4 and NUTM1 spanning probe set was positive, confirming the diagnosis of NUT carcinoma (Fig. 1i). FISH studies for rearrangement of the EWSR1 locus seen in some myoepithelial tumors and the SS18 locus in synovial sarcomas were both negative. A retrospective NUT immunostain showed diffuse and strong nuclear positivity in tumor cells (Fig. 1h).

Histologic, immunohistochemical and molecular cytogenetic findings of the initial biopsy tissue. a Touch preparation showing a cluster of cohesive poorly differentiated cells. b Frequent areas of geographic tumor necrosis. c Tumor cells demonstrating a predominantly solid growth pattern. d Poorly differentiated tumor cells with scant to moderate, eosinophilic and occasionally dense cytoplasm, moderate pleomorphism, and brisk mitotic activities. e Pan cytokeratin (AE1/AE3) stain showing diffuse and variably strong immunoreactivity in the poorly differentiated cells. f A subset of tumor cells strongly positive for synaptophysin with granular cytoplasmic staining. G, p63 stain showing patchy but strong nuclear immunoreactivity. h NUT stain showing diffuse and strong expression in tumor cell nuclei. i Representative interphase nuclei illustrating tightly juxtaposed orange (BRD4) and green (NUTM1) signals (arrows) indicative of BRD4-NUTM1 fusion
Imaging studies demonstrated metastatic disease in the liver, brain and bone. A magnetic resonance imaging (MRI) of the brain revealed dural enhancement with a 0.6 × 1.8 cm nodule over the left frontal lobe. Approximately 2 weeks after the initial biopsy, chemotherapy was initiated with vorinostat (180 mg/m2/dose on days 1–5 and 8–12), paclitaxel (250 mg/m2/dose on day 1) and cisplatin (50 mg/m2/dose on days 1 and 2). Restaging after two cycles of chemotherapy demonstrated a mixed response. There was 50% reduction in size of the primary tumor, but new metastatic bony sites emerged in the spine associated with back pain. The chemotherapeutic regimen was then changed to ifosfamide (1800 mg/m2/dose on days 1–5), etoposide (100 mg/m2/dose on days 1–5) and vorinostat (180 mg/m2/dose on days 1–5 and 8–12). The patient received four cycles of this chemotherapy regimen with partial response at the primary and metastatic sites including resolution of PET avidity. He subsequently underwent resection of the primary tumor with a modified left radical neck dissection. Pathologic examination showed residual NUT carcinoma with extensive therapy effect (< 5% tumor viability), negative surgical margins, and 5 out of 32 lymph nodes positive for metastatic NUT carcinoma. Within 2–3 weeks after surgery, the patient experienced rapid growth of tumor at the primary site. During the following two months, the patient developed new and progressive tumor recurrence in the left submandibular region, left orbit, right parietal bone, and leptomeninges, most extensively in the posterior fossa. The patient did not demonstrate response to salvage chemotherapy with cyclophosphamide, topotecan and vorinostat. He received palliative radiation therapy and died 8 months after initial presentation from progressive disease.
Autopsy of the head and neck showed an 8 × 6 × 5 cm left submandibular recurrent tumor and a 7 × 6 × 2 cm tumor involving the left orbit and cranial bones (Fig. 2a). Intracranially, there was a 5 × 3 cm, hemorrhagic epidural metastasis extending from the superior sagittal sinus and eroding into the parietal bones. Tumor cells focally exhibited a pseudoalveolar and nested growth pattern (Fig. 2b) and prominent clear cell change (Fig. 2c) in the epidural location. The leptomeninges were grossly thickened with tan discoloration, and microscopically showed diffuse leptomeningeal carcinomatosis (Fig. 2d, e). Although the cerebral and cerebellar parenchyma appeared to be normal on gross examination, superficial cortical infiltration by carcinoma was evident microscopically, involving both frontal lobes, right temporal lobe, cerebellum and the brainstem (Fig. 2f).

Histologic features of metastatic disease. a Cribriform plate involvement by aggressive local invasion. b Sagittal suture metastasis showing a pseudoalveolar and nested pattern. c Tumor cells focally exhibiting prominent clear cytoplasm and vesicular chromatin. d Representative area showing extensive leptomeningeal carcinomatosis with superficial invasion into brain parenchyma. e Discohesive tumor cells in leptomeningeal spread. f Tumor cells breaking through the Virchow–Robin spaces and demonstrating an infiltrative growth pattern
Discussion
NUT carcinomas of probable or possible salivary gland origin are rare. Herein the clinicopathologic characteristics (including molecular alterations) of all reported cases (Table 1), and noted differences in sex and outcomes between the pediatric and adult groups are summarized. A systematic analysis comparing the parameters between these two groups (Table 2) showed all pediatric patients were male (6/6) while the male-to-female ratio was 1:2 in adults (p = 0.028). There was no statistically significant difference by all tumor characteristics, including size, type of salivary gland, primary tumor stage, lymph node involvement, and distant metastasis, although pediatric patients tended to have to smaller mean tumor size at presentation (3 cm in children versus 4.6 cm in adults). Molecular data was available in 3 of 6 pediatric and 8 of 9 adult cases, with 2 pediatric and 7 adult cases known to have BRD4‐NUTM1 translocation. A Kaplan–Meier analysis demonstrated significant prolonged OS in pediatric patients compared to adults (Fig. 3, p = 0.0014), with longer median OS time (24 versus 4 months) and higher 3-month (100% versus 67%), 7-month (83% versus 11%), and 1-year (67% versus 11%) OS rates (Table 2). Compared to the median OS time for NUT carcinomas of all ages and locations (4.7–6.7 months) [5, 6] and head and neck region only (9.7 months) [20], pediatric patients with salivary gland NUT carcinoma appears to have a better prognosis (Table 3). However, the results of this study must be interpreted cautiously due to the small sample size. Assembly of a much larger cohort of pediatric cases with molecular information will be necessary to draw more definitive conclusions.

Kaplan–Meier survival analysis of patients with salivary gland NUT carcinoma. The probability of overall survival is estimated for adult (age ≥ 21; n = 9) and pediatric patients (age < 21; n = 6). Log-rank test was performed to compare the survival distributions of these two groups
Classifying a small round blue cell neoplasm in the head and neck region can be challenging. Due to its rarity and non-midline location, salivary gland NUT carcinoma may be an underrecognized diagnostic consideration in the differential diagnosis of any poorly differentiated salivary gland tumor. In fact, NUT carcinoma in general has been confused with squamous cell carcinoma, NOS, poorly differentiated carcinoma, NOS, thymic carcinoma, sinonasal undifferentiated carcinoma, Ewing sarcoma, or classified as undifferentiated epithelioid/round cell malignant neoplasms, creating significant diagnostic challenge [21,22,23,24,25,26,27]. If present, foci of squamous differentiation, often abrupt, can be helpful in distinguishing NUT carcinoma from most small round blue cell tumors. However, it is not uncommon for NUT carcinoma to lack any morphologic evidence of squamous differentiation (such as the current case), especially in biopsies or specimens of small size. Some tumors may exhibit features mimicking myoepithelial carcinoma with myxoid matrix, and rarely mesenchymal differentiation as described in one pediatric salivary gland NUT carcinoma [11]. Immunohistochemically, expression of cytokeratins, EMA, myoepithelial markers, and CD99 in NUT carcinoma overlaps with myoepithelial carcinoma and Ewing sarcoma. Fortunately, NUT IHC has been proven to be a relatively sensitive (87%) and highly specific (nearly 100%) tool for the diagnosis of NUT carcinoma, demonstrating diffuse and strong nuclear reactivity with a speckled pattern [22]. Recently, NUT immunoexpression and NUTM1 rearrangements have also been described in tumors with a favored diagnosis of sarcoma [28,29,30]. An alternative to NUT IHC is molecular analysis to detect rearrangement of NUTM1 gene using FISH, reverse‐transcriptase polymerase chain reaction (RT-PCR), cytogenetics, or next‐generation sequencing‐based approaches. A reciprocal translocation between NUTM1 and BRD4 genes can be demonstrated in up to 70% of NUT carcinomas by molecular analysis. NUTM1 has also been shown to be fused to BRD3 or NSD3, and rarely to ZNF532, ZNF592 or CIC [3, 30, 31]. In the current study, BRD4 was the only identified NUTM1 fusion gene partner for the subset of cases reviewed (9/15) for which molecular assessment capable of identifying partnerships was performed. Interestingly, it has been suggested that NSD3- or BRD3‐NUTM1‐positive tumors may be associated with significantly better overall survival than those with BRD4‐NUTM1 [3]. Molecular analysis to identify specific fusion partner to NUTM1 therefore may be of potential clinical relevance.
In summary, salivary gland NUT carcinomas are rare, diagnostically challenging, and probably underrecognized due to overlapping histopathologic features with other poorly differentiated neoplasms. Among all patients with NUT carcinoma, children with salivary gland origin may represent a distinct subset with male predilection and better overall survival.
References
French C. NUT midline carcinoma. Nat Rev Cancer. 2014;14(3):149–50.
French CA. Pathogenesis of NUT midline carcinoma. Annu Rev Pathol. 2012;7:247–65.
French CA. NUT Carcinoma: clinicopathologic features, pathogenesis, and treatment. Pathol Int. 2018;68(11):583–95. https://doi.org/10.1111/pin.12727.
French CA. The importance of diagnosing NUT midline carcinoma. Head Neck Pathol. 2013;7(1):11–6.
Bauer DE, Mitchell CM, Strait KM, Lathan CS, Stelow EB, Luer SC, et al. Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin Cancer Res. 2012;18(20):5773–9.
Lemelle L, Pierron G, Freneaux P, Huybrechts S, Spiegel A, Plantaz D, et al. NUT carcinoma in children and adults: a multicenter retrospective study. Pediatr Blood Cancer. 2017. https://doi.org/10.1002/pbc.26693.
Stathis A, Zucca E, Bekradda M, Gomez-Roca C, Delord JP, de La Motte RT, et al. Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov. 2016;6(5):492–500. https://doi.org/10.1158/2159-8290.CD-15-1335.
Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. Phase Ib trial with Birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J Clin Oncol. 2018;36(30):3007–144. https://doi.org/10.1200/JCO.2018.78.2292.
Maher OM, Christensen AM, Yedururi S, Bell D, Tarek N. Histone deacetylase inhibitor for NUT midline carcinoma. Pediatr Blood Cancer. 2015;62(4):715–7. https://doi.org/10.1002/pbc.25350.
Park HS, Bae YS, Yoon SO, Lim BJ, Hong HJ, Ro JY, et al. Usefulness of Nuclear Protein in Testis (NUT) Immunohistochemistry in the cytodiagnosis of NUT midline carcinoma: a brief case report. Korean J Pathol. 2014;48(4):335–8. https://doi.org/10.4132/KoreanJPathol.2014.48.4.335.
den Bakker MA, Beverloo BH, van den Heuvel-Eibrink MM, Meeuwis CA, Tan LM, Johnson LA, et al. NUT midline carcinoma of the parotid gland with mesenchymal differentiation. Am J Surg Pathol. 2009;33(8):1253–8.
Ziai J, French CA, Zambrano E. NUT gene rearrangement in a poorly-differentiated carcinoma of the submandibular gland. Head Neck Pathol. 2010;4(2):163–8.
Storck S, Kennedy AL, Marcus KJ, Teot L, Vaughn J, Gnekow AK, et al. Pediatric NUT-midline carcinoma: therapeutic success employing a sarcoma based multimodal approach. Pediatr Hematol Oncol. 2017;34(4):231–7. https://doi.org/10.1080/08880018.2017.1363839.
Vulsteke C, Lurquin E, Debiec-Rychter M, Gheysens O, Nuyts S, Schoenaers J, et al. First evidence of treatment efficacy in metastatic carcinoma of the parotid gland with BRD4/NUT translocation. J Chemother. 2016;28(3):242–6. https://doi.org/10.1179/1973947815Y.0000000046.
Andreasen S, French CA, Josiassen M, Hahn CH, Kiss K. NUT carcinoma of the sublingual gland. Head Neck Pathol. 2016;10(3):362–6. https://doi.org/10.1007/s12105-015-0672-7.
Klijanienko J, Le Tourneau C, Rodriguez J, Caly M, Theocharis S. Cytological features of NUT midline carcinoma arising in sino-nasal tract and parotid gland: Report of two new cases and review of the literature. Diagn Cytopathol. 2016;44(9):753–6. https://doi.org/10.1002/dc.23506.
Seim NB, Philips RHW, Schoenfield L, Teknos TN, Rocco JW, Agrawal A, et al. NUT midline carcinoma of the sublingual gland: clinical presentation and review. Head Neck Pathol. 2017;11(4):460–8. https://doi.org/10.1007/s12105-017-0809-y.
Agaimy A, Fonseca I, Martins C, Thway K, Barrette R, Harrington KJ, et al. NUT carcinoma of the salivary glands: clinicopathologic and molecular analysis of 3 cases and a survey of NUT expression in salivary gland carcinomas. Am J Surg Pathol. 2018;42(7):877–84. https://doi.org/10.1097/PAS.0000000000001046.
Cho Y, Keam BS, Jung KC, Kim BH. A case of nuclear protein in testis midline carcinoma arising from the submandibular gland duct in a pregnant patient. J Oral Maxillofac Surg. 2017;75(9):2020–4. https://doi.org/10.1016/j.joms.2017.02.002.
Chau NG, Hurwitz S, Mitchell CM, Aserlind A, Grunfeld N, Kaplan L, et al. Intensive treatment and survival outcomes in NUT midline carcinoma of the head and neck. Cancer. 2016;122(23):3632–40. https://doi.org/10.1002/cncr.30242.
Evans AG, French CA, Cameron MJ, Fletcher CD, Jackman DM, Lathan CS, et al. Pathologic characteristics of NUT midline carcinoma arising in the mediastinum. Am J Surg Pathol. 2012;36(8):1222–7. https://doi.org/10.1097/PAS.0b013e318258f03b.
Haack H, Johnson LA, Fry CJ, Crosby K, Polakiewicz RD, Stelow EB, et al. Diagnosis of NUT midline carcinoma using a NUT-specific monoclonal antibody. Am J Surg Pathol. 2009;33(7):984–91. https://doi.org/10.1097/PAS.0b013e318198d666.
Kubonishi I, Takehara N, Iwata J, Sonobe H, Ohtsuki Y, Abe T, et al. Novel t(15;19)(q15;p13) chromosome abnormality in a thymic carcinoma. Cancer Res. 1991;51(12):3327–8.
Mertens F, Wiebe T, Adlercreutz C, Mandahl N, French CA. Successful treatment of a child with t(15;19)-positive tumor. Pediatr Blood Cancer. 2007;49(7):1015–7. https://doi.org/10.1002/pbc.20755.
Petrini P, French CA, Rajan A, Cameron MJ, Jaffe ES, Zucali PA, et al. NUT rearrangement is uncommon in human thymic epithelial tumors. J Thorac Oncol. 2012;7(4):744–50. https://doi.org/10.1097/JTO.0b013e3182460f8f.
Stelow EB, Bellizzi AM, Taneja K, Mills SE, Legallo RD, Kutok JL, et al. NUT rearrangement in undifferentiated carcinomas of the upper aerodigestive tract. Am J Surg Pathol. 2008;32(6):828–34. https://doi.org/10.1097/PAS.0b013e31815a3900.
Toretsky JA, Jenson J, Sun CC, Eskenazi AE, Campbell A, Hunger SP, et al. Translocation (11;15;19): a highly specific chromosome rearrangement associated with poorly differentiated thymic carcinoma in young patients. Am J Clin Oncol. 2003;26(3):300–6. https://doi.org/10.1097/01.COC.0000020960.98562.84.
Tamura R, Nakaoka H, Yoshihara K, Mori Y, Yachida N, Nishikawa N, et al. Novel MXD4-NUTM1 fusion transcript identified in primary ovarian undifferentiated small round cell sarcoma. Genes Chromosomes Cancer. 2018;57(11):557–63. https://doi.org/10.1002/gcc.22668.
Stevens TM, Morlote D, Xiu J, Swensen J, Brandwein-Weber M, Miettinen MM, et al. NUTM1-rearranged neoplasia: a multi-institution experience yields novel fusion partners and expands the histologic spectrum. Modern Pathol. 2019;32(6):764–73. https://doi.org/10.1038/s41379-019-0206-z.
Le Loarer F, Pissaloux D, Watson S, Godfraind C, Galmiche-Rolland L, Silva K, et al. Clinicopathologic features of CIC-NUTM1 sarcomas, a new molecular variant of the family of CIC-fused sarcomas. Am J Surg Pathol. 2019;43(2):268–76. https://doi.org/10.1097/PAS.0000000000001187.
Schaefer IM, Dal Cin P, Landry LM, Fletcher CDM, Hanna GJ, French CA. CIC-NUTM1 fusion: a case which expands the spectrum of NUT-rearranged epithelioid malignancies. Genes Chromosomes Cancer. 2018;57(9):446–51. https://doi.org/10.1002/gcc.3.
Author information
Authors and Affiliations
Contributions
All authors contributed to material preparation, data collection and analysis of the case report. Literature review and analysis were performed by Huiying Wang and Jiancong Liang. The first draft of the manuscript was written by Huiying Wang and Jiancong Liang. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, H., Weiss, V.L., Hoffman, R.D. et al. Salivary Gland NUT Carcinoma with Prolonged Survival in Children: Case Illustration and Systematic Review of Literature. Head and Neck Pathol 15, 236–243 (2021). https://doi.org/10.1007/s12105-020-01141-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-020-01141-3