
Abstract
Sinonasal tract (SNT) alveolar rhabdomyosarcoma (ARMS) are frequently misdiagnosed, especially in adults. Fifty-two adult (≥18 years) patients with SNT ARMS were reviewed and characterized by immunohistochemistry and molecular studies. Twenty-six females and 26 males (18–72 years; mean 43.2 years), presented after a short duration (mean 2.6 months) with a large (mean 5.5 cm) destructive nasal cavity mass, involving multiple contiguous paranasal sites (n = 46) and with cervical adenopathy (n = 41). The tumors showed an alveolar, nested to solid growth pattern below an intact, but often involved (n = 9) epithelium with frequent necrosis (n = 37), destructive bone invasion (n = 30), and lymphovascular invasion (n = 25). The neoplastic cells were dyshesive and dilapidated, with crush artifacts. Rhabdoid features (n = 36) and tumor cell multinucleation (n = 28) were common. Mitotic counts were high (mean 17/10 HPFs). The neoplastic cells showed the following immunohistochemical positive findings: desmin (100%), myogenin (100%), MYOD1 (100%), MSA (96%), SMA (52%), CAM5.2 (50%), AE1/AE3 (36%); other positive markers included S100 protein (27%), CD56 (100%), synaptophysin (35%), and chromogranin (13%). Overall, 54% show epithelial marker reactivity. Molecular studies showed FOXO1 translocations (81%) with PCR demonstrating PAX3 in 72.7% tested. Patients presented with high stage (IV 24; III 26) and metastatic disease (lymph nodes n = 41; distant metastases n = 25) (IRSG grouping). Surgery (n = 16), radiation (n = 41) and chemotherapy (n = 45) yielded an overall survival of 36.1 months (mean; range 2.4–286); 18 alive without disease (mean 69.6 months); 7 alive with disease (mean 11.0 months); 1 dead without disease (63.7 months); and 26 dead with disease (mean 18.5 months). SNT ARMS frequently present in adults as a large, destructive midline mass of short symptom duration, with high stage disease. The alveolar to solid pattern of growth of cells with rhabdoid-plasmacytoid features suggests the diagnosis, but epithelial immunohistochemistry markers are present in 54% of cases, leading to misdiagnosis as carcinomas if muscle markers are not also performed. Overall survival of 36.1 months is achieved with multimodality therapy, but 64% have incurable disease (16.9 months). Mixed anatomic site (p = 0.02) was a significant adverse prognostic indicator, while stage (0.06) and tumor size >5 cm (0.06) approached marginal significance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The “small blue round cell” differential diagnosis in the sinonasal tract can be quite daunting as the biopsies are frequently small, with only limited material for examination and crush artifacts are frequently present. The clinical symptoms and imaging findings overlap significantly between the diagnostic differential considerations, and thus careful histologic examination in combination with additional ancillary testing is required to reach a definitive diagnosis. The treatments for carcinoma, lymphoma, sarcoma and melanoma in this anatomic site are quite different from each other, requiring a definitive diagnosis before therapies are undertaken. Many of these treatments can be quite disfiguring and associated with significant side effects and/or complications and morbidities, emphasizing the need for an accurate classification. Knowing that squamous cell carcinoma is the most common malignancy of the upper aerodigestive tract, and sinonasal tract specifically in adult patients, may result in practitioners selecting a limited immunophenotypic panel to confirm the hematoxylin and eosin impression of a poorly differentiated carcinoma. Thus, a pancytokeratin (AE1/AE3, OSCAR or CAM5.2) positive tumor may be classified as a poorly differentiated squamous cell carcinoma, failing to appreciate the frequency with which other neoplasms in this anatomic site are keratin immunoreactive also, yet not representing epithelial derived neoplasms [1, 2]. This background framed the proposed study of alveolar rhabdomyosarcoma of the sinonasal tract in adult patients, with an emphasis on epithelial immunoreactivity, while also evaluating the clinical features and treatment outcomes for this uncommon sinonasal tract tumor. Alveolar rhabdomyosarcoma, a primitive malignant mesenchymal tumor with skeletal muscle differentiation, frequently displays a small blue round cell histologic appearance, potentially leading to a wrong diagnosis and thus inappropriate treatment [3, 4]. On the other hand, embryonal rhabdomyosarcoma usually show a spindled more heterogeneous morphology and is not generally considered in the epithelial-derived tumor differential diagnostic categories.
Materials and Methods
Fifty-two cases of sinonasal tract alveolar rhabdomyosarcomas were retrieved from the consultation files of the authors. An approximate incidence is suggested by 12 sinonasal tract tumors identified in adults out of a total of 145 rhabdomyosarcomas diagnosed between January 1, 2006 to January 1, 2016 (10 years) at one author’s institution (LDRT). There were overall 76 females and 69 males; ages ranged from 1 week to 88.4 years, with 86 patients older than or equal to 18 years, and 59 patients younger than 18 years of age. Thus, approximately 10% of adult alveolar RMS involve the head and neck with an approximate relative incidence of 0.4/1 million population (Southern California Permanente Medical Group mid-year average population for same 10 years). Cases were excluded if sufficient clinical, immunophenotypic or follow-up data was not available.
Materials within the files were supplemented by a review of the patient demographics (sex, age, and race); symptoms and physical findings and duration at presentation including mass, obstructive or congestive symptoms, facial pain, ophthalmologic disturbances, headache, epistaxis, chronic sinusitis, central nervous system related symptoms and loss of smell. In addition, imaging studies, surgical pathology, and operative reports were reviewed when available, with follow-up information obtained by direct written or oral communication with the referring pathologist, patient’s physician, oncology data services and tumor registries, or the patient (patient’s family member[s]). Follow-up data included information regarding presence of recurrent disease, treatment modalities used, and the current patient status. Cases in general were submitted as consultations to several of the authors, who conducted this study as a retrospective review, without actually treating the patients. Submitted diagnoses by the primary pathologists included squamous cell carcinoma, carcinoma, not otherwise specified, neuroendocrine carcinoma, olfactory neuroblastoma, sinonasal undifferentiated carcinoma, melanoma and sarcoma. This clinical investigation was conducted in accordance and compliance with an Internal Review Board authorization (#5968) performed under the direction of Southern California Permanente Medical Group.
Specific information about the exact location, laterality, and tumor size (greatest dimension in cm) was documented (imaging reports and/or surgical pathology material). Hematoxylin and eosin-stained slides from all cases were reviewed to document specific histologic features, to include: surface epithelium (present or absent; ulcerated; involved by tumor: pagetoid spread); tumor invasion (destructive bone invasion, perineural invasion, lymphovascular invasion); architectural pattern of growth (alveolar, nested, solid); necrosis (absent or present; comedotype); cell type (polygonal, plasmacytoid, spindled, cuboidal); multinucleation (present, or absent); strap cells (present or absent); mitotic figures [number of mitotic figures per ten high power fields (magnification at ×40 with a ×10 objective lens using Olympus BX41 microscope)]; atypical mitotic figures (present or absent, and defined by abnormal chromosome spread, tripolar or quadripolar forms, circular forms, or indescribably bizarre); background fibrous connective tissue septa; and the presence of other notable microscopic pathologic findings.
Immunophenotypic analysis was performed in cases with sufficient suitable material by a standardized Envision™ method employing 4 µm-thick, formalin fixed, paraffin embedded sections. Table 1 documents the pertinent, commercially available immunohistochemical antibody panel used. The analysis was performed on a single representative block for each primary tumor. However, the biopsies were often small, yielding a limited amount of tissue for additional examination, thus prioritizing immunohistochemistry studies for diagnostic clarity was paramount. Epitope retrieval was performed, as required by the manufacturer guidelines. Standard positive controls were used throughout, with serum used as the negative control. The antibody reactions were graded as positive or negative; positives were separated into strong, moderate and weak; diffuse (>50%), patchy (10–50%) or focal (<10%); and by pattern (cytoplasmic, membrane, dot-like, nuclear, nuclear and cytoplasmic). Staining equivalent reactions would be absent to weak (0 to 1+), moderate (2+ to 3+) and strong (4+) staining.
Sanger sequencing was performed upon available material, with electropherograms created specifically for the transcripts for PAX3 exon 7 (2q35), PAX7 (1p36.2-p36.12) and FOXO1 exon 2 (FOXO1 is the forkhead box O1 gene, previously FKHR). Breakpoints were documented. Beta-globulin controls were positive.
A review of the English literature based on a MEDLINE search from 1966 to 2017 was performed and all cases of sinonasal tract rhabdomyosarcomas were reviewed, with specific attention to the clinical series and those which included immunohistochemistry information.
Statistical evaluation was performed using a standard statistics software package with categorical variables analyzed using Chi square tests and Fisher’s Exact tests to compare observed and expected frequency distributions. Comparison of means between groups was made with independent t-tests (including 1-tailed and 2-tailed tests with degrees of freedom) or one-way analysis of variance, depending on whether there were two groups or more than two groups, respectively. Confidence intervals of 95% were generated for all positive findings. The alpha level was set at p < 0.05.
Results
Clinical
The patients included 26 women and 26 men (Table 2), all adults, ranging from 18 to 72 years (mean 43.2 years). Patients presented with symptoms of a relatively short duration (mean 2.6 months), suggesting rapid clinical onset of the tumor. The symptoms were non-specific in nature, although all patients presented with a mass. The vast majority of tumors (n = 46) involved more than one anatomic site/subsite, with nasal cavity, maxillary sinus, frontal, sphenoid, and ethmoid sinuses affected, frequently showing extension into the skull base or adjacent soft tissue structures. Thus, tumors were large, with a mean of 5.5 cm, usually confined to a single side (bilateral, n = 2), but often showing midline involvement (Fig. 1). Imaging studies identified destructive masses that usually involved multiple sites and frequently highlighted cervical lymph node metastases (n = 41 cases), the latter meeting size and radiographic criteria for lymph node disease. The imaging findings were not unique or specific to the diagnosis, but showed a high SUV value when evaluated by PET studies (Fig. 1). No patients were part of a familial or syndrome association (specifically, Li-Fraumeni, Costello syndrome or Beckwith-Wiedemann).

Imaging studies of sinonasal tract rhabdomyosarcoma. a A coronal computed tomography scan shows a large destructive mass in the maxillary sinus, expanding into the orbit and nasal cavity. b A MRI (sagittal) T1 SE image shows a large destructive mass within the sinonasal tract. c An axial MRI T2 axial image shows a destructive mass breaking though the medial wall of the maxilla. d A fused PET/CT image shows high avidity in the nasal cavity and maxillary sinus tumor
Pathologic Features
Microscopic
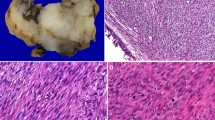
The fragments of tissue were commonly polypoid, fleshy, and pale, demonstrating fibrous connective tissue stroma and bands dissecting between the tumor islands (Fig. 2). The tumors were contained within fragments of tissue, as most samples were curetted rather than an “en bloc” resection. The neoplastic cells were separated by fibrous septae into nested, alveolar and pseudoalveolar patterns, while a more solid pattern was seen in 26 cases (Table 3). Destructive bone or cartilage involvement was present in 30 cases (Fig. 2), although bone was not included in several samples. Lymphovascular invasion was seen in 25 cases, but due to fragmentation and retraction artifacts, it was difficult to determine with certainty. While nerves were identified in the samples, perineural invasion was only identified in one case. None of the cases showed a botryoid pattern. The majority of tumors demonstrated overlying intact respiratory surface epithelium, with ulceration seen in only 4 cases. Surface involvement by the neoplasm was present in nine, showing a pagetoid spread into the surface respiratory or squamous epithelium (Fig. 3). Alveolar spaces were defined by a space that showed a clinging or dilapidated periphery, with the cells dropping off into the center, while a nested pattern was defined by sheets of cells without peripheral degeneration or dilapidation (n = 10; Fig. 3). The neoplastic cells were associated with a desmoplastic stromal reaction, with neoplastic cells seen infiltrating into fibrous connective tissue (Fig. 4) while focal myxoid background stroma was noted. Central comedonecrosis was common (n = 37). Many cases showed crush artifact (n = 44) of the neoplastic cells (Fig. 4). The cells were generally 2–3 times as large as lymphocytes, with rhabdoid, eccentric cytoplasm in 36 cases (Fig. 3). It is important to note that the rhabdoid appearance may have been focal, in isolated cells, showing eosinophilic cytoplasm pulled off to one side of the nucleus. True strap cells (tadpole, elongated cytoplasmic extension) with cross striations were only seen focally in 11 cases. The neoplastic cells were round, even though they were dyshesive, but were still more associated with one another than would be seen in a lymphoma or melanoma. Typical tumor multinucleation was common (n = 28). The nuclear chromatin was slightly open to vesicular, usually with prominent nucleoli. Importantly, the nuclei did not show salt-and-pepper nuclear chromatin, as would be seen in a neuroendocrine tumor. Mitoses were easy to identify, with a mean of 17/10 high power fields, while atypical forms were seen in 25 cases. An inflammatory infiltrate was not prominent; a well developed pseudoepitheliomatous hyperplasia was not seen; cytoplasmic pigment was not identified, and significant tumor cell spindling was not appreciated.

The tumors were usually polypoid masses (a), showing an intact squamous or respiratory epithelial lining (b). There was a nested appearance. Crush artifacts with fibrosis were common (c). Bone invasion by the neoplastic cells was frequently noted (d)

The tumor was arranged in a nested or alveolar pattern (a), while in other areas a more solid appearance could be seen (b). An intraepithelial growth was present in a few cases (c)

The neoplastic cells were separated into nests by a heavy stromal fibrosis (a), sometimes desmoplastic (b), which partially obscured the neoplastic cells. Crush artifacts created a carcinoma appearance (c). A peritheliomatous pattern was noted in some tumors, with adjacent necrosis (d)
Immunohistochemical Results
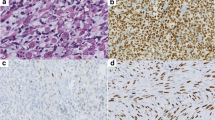
The immunohistochemistry studies performed confirmed the skeletal muscle differentiation (Table 1), with all tumors tested showing a strong and diffuse reaction with desmin, myogenin and MYOD1 (Fig. 5), while myoglobin was seen in 37% (7/19). Muscle specific actin (96%) was more strongly reactive and in a greater number of cases than smooth muscle actin (52%). The pancytokeratin (36%), CAM5.2 (50%), and CK5/6 (5%) were positive in a sufficient number of cases that the differential with a carcinoma would certainly be raised during initial evaluation, if they had been performed (not all antibodies were tested on the cases during initial clinical evaluation, but only during the research protocol). Overall, 54% of cases showed reactivity with one of the epithelial markers tested (Fig. 6). There was no difference in expression of epithelial markers between the cases that were confirmed by molecular evaluation versus those that were not. Further, neuroendocrine markers, such as synaptophysin (35%) and chromogranin (13%) were also positive, while CD56 demonstrated a strong, diffuse reactivity in all cases tested (32/32). S100 protein showed a focal, but still positive reaction in 27% of cases, but SOX10 was negative in all cases tested (0/16). It is important to note that the epithelial marker immunoreactivity was frequently in a dot-like or Golgi distribution (Fig. 6), a feature that can mimic a neuroendocrine tumor.

The neoplastic cells shows reactivity with a number of markers, including a myogenin; b MYOD1; c CD56; d synaptophysin

In many cases, the neoplastic cells were positive with pancytokeratin (AE1/AE3) (a) or CAM5.2 (b). c A FISH break apart probe shows a positive result for FOXO1. d An electropherogram shows the breakpoint in the PAX3 exon 7, fused to FOXO1 exon 2 in this Sanger sequence
Molecular Results
Eighteen of 21 cases evaluated by FISH showed a FOXO1 break apart positive result. Additional reverse transcriptase polymerase chain reaction studies were performed on 22 cases for PAX3 and PAX7, with results obtained in 16 cases, all classified as PAX3/FOXO1 rearrangements. No PAX7/FOXO1 rearrangements were identified in this series. Non-informative results were seen in 6 cases (perhaps due to the age of the paraffin samples tested).
Treatment and Follow-Up
All patients had a diagnostic biopsy as their initial evaluation, but only 16 were managed by any sort of resection or wide excision. Thus, while observed, a positive margin status was not separately evaluated since most of the samples were biopsy only, and received as multiple fragments of tissue. Radiation (n = 41) and/or chemotherapy (n = 45) was the main treatment, with 38 patients receiving both; seven patients had chemotherapy only, while four patients were treated by radiation only, and in 3 patients the information about radiation and chemotherapy was unknown. Of the patients managed with chemotherapy alone, 3 are dead with disease (mean 8.8 months), one is alive with disease (24 months), and 3 are alive with no evidence of disease (mean 9.2 months). All patients managed by radiation alone, were dead of disease (mean 14.5 months).
Follow-up was available in all patients, with those who died in <12 months considered to have died of disease if the specific information was not available. The overall average follow-up was 36.1 months (Table 4). Forty-three (83%) patients had metastatic disease at the time of initial presentation, with the majority showing lymph node metastases (n = 40, 77%), while three additional patients showed distant metastases without cervical lymph node disease. Eight patients developed recurrent or showed persistent local disease. Overall, 18 patients were alive without evidence of disease (mean 69.6 months; range 2.5–285.9 months), including 10 patients who had presented with metastatic disease. Seven patients are alive but with disease (mean 11 months; range 2.5–24 months), 6 of these presenting with metastatic disease. One patient died with no evidence of disease (63.7 months), while 26 patients died with disease (mean 18.5 months; range 2.4–75.2 months). There was no difference in patient outcome based on age, sex, tumor size, keratin reactivity, stage, or treatment, with only multiple anatomic subsites of tumor involvement conferring a worse outcome. However, both size and tumor stage approached significance (p = 0.06). It may be that there are too few patients in each group to achieve statistical significance.
Staging was difficult to perform, as most of the staging systems are based on pediatric rhabdomyosarcomas, staging is not performed for non-epithelial tumors of the sinonasal tract, or the soft tissue staging systems do not include head and neck sites. Thus, staging was based on the Intergroup Rhabdomyosarcoma Study Group (IRSG) surgical pathology grouping system and staging system [5]. By definition, a biopsy only procedure places the tumor in a group III category; distant metastases places the tumor in group IV; any tumors of the sinonasal tract are considered parameningeal for staging purposes, and automatically at least stage II; if lymph node metastases were present, irrespective of the tumor size, the tumor was placed in stage III. Thus, by definition, for sinonasal tract disease, there is no stage I category, with all tumors in stage II to IV. Overall, using this staging, 2 patients were in stage II; 26 in stage III; and 24 in stage IV. As expected, even though not statistically significant, the patients with stage II disease were both alive (mean 55.2 months) with no deaths from disease. However, 10 of 26 patients (38%) showed disease at last following for stage III patients, while 17 of 24 (71%) showed disease at last follow-up for stage IV patients. Thus, a trend is noted towards a worse outcome for higher stage (p = 0.068).
Discussion
Alveolar rhabdomyosarcoma is a relatively uncommon malignancy of the sinonasal tract, encountered in the setting of a primitive small round blue cell neoplasm [3, 6,7,8,9,10,11,12,13,14,15,16,17]. The tumor is usually considered in pediatric patients in contrast to adults, where the diagnosis is more common [2, 18]. The identification of eosinophilic, eccentric cytoplasm in the neoplastic population helps to suggest rhabdomyosarcoma, but is often a focal and limited histologic feature. In this setting, the diagnosis is usually confirmed with the application of a pertinent immunohistochemistry panel and additionally perhaps, genetic (FISH) studies. In an attempt at tissue conservation in samples that have very limited material, an initial immunohistochemistry panel generally includes a pancytokeratin, CD45RB and S100 protein, an initial screen for carcinoma, lymphoma, and melanoma. Squamous cell carcinoma, sinonasal undifferentiated carcinoma, NUT carcinoma, SMARCB1-deficient carcinoma, and neuroendocrine carcinoma will virtually all be cytokeratin positive and negative with the other two markers. Desmoplastic small round cell tumor is exceedingly exceptional in the sinonasal tract, and usually shows a more polyphenotypic expression with multiple lines of differentiation identified [19, 20]. As “epithelial” neoplasms are more common, additional markers, such as CK5/6, p63, p40, EMA, CAM5.2, CK7 and K903 may be applied to more fully elucidate the nature of the carcinoma. But, when some or all these additional markers are negative, the more generic poorly differentiated carcinoma diagnosis may be applied, without further immunohistochemistry evaluation. Alternatively, depending on the pattern of epithelial immunoreactivity, neuroendocrine markers may be obtained, including synaptophysin, chromogranin, neuron specific enolase and/or CD56. If these markers show any reactivity, then either a carcinoma with neuroendocrine differentiation, neuroendocrine carcinoma, pituitary adenoma or even olfactory neuroblastoma may be the diagnosis rendered [1, 3, 10, 21,22,23,24,25,26,27,28,29,30]. Thus, it is critical in primitive appearing and non-keratinizing squamous cell carcinomas of the sinonasal tract, that a muscle marker, such as desmin, myogenin or MYOD1, be performed in order to exclude the possibility of a cytokeratin positive rhabdomyosarcoma [1,2,3, 18]. Myogenin and MYOD1 are considered sensitive and specific markers of rhabdomyoblastic differentiation, two of the myogenic transcriptional regulatory proteins, expressed in a higher percentage of cells in alveolar rhabdomyosarcoma than in embryonal rhabdomyosarcoma [31]. As shown in this series, up to 54% of rhabdomyosarcoma show epithelial immunoreactivity, with CAM5.2 showing the highest reactivity (50%), but pancytokeratin (AE1/AE3) showing positivity in 38% of cases. In this series, the AE1/AE3 reactivity was strong in 12 of the 16 cases tested, although the reaction was focal in 15 of 16 cases. This finding is significantly higher than reported in earlier, more limited series [2]. Importantly, the keratin expression was often in a dot-like or Golgi distribution, a finding similar to what is seen in neuroendocrine tumors, and thus a potential pitfall in interpretation of the final diagnosis. CAM5.2 was also strong in 17 of 22 cases tested, most showing a focal distribution. The dot-like pattern was only seen in 12 cases, the remaining cases showing a diffuse cytoplasm reaction. The differential of rhabdomyosarcoma with neuroendocrine carcinomas in the sinonasal tract is further complicated by the relatively high expression of neuroendocrine markers in rhabdomyosarcoma, 35% in this study, but up to 43% in other studies [1, 9]. Synaptophysin is more commonly expressed in this study than in others [1, 9]. CD56 was present in all cases tested in this series, similar to previously reported results [1], but in general is not considered to be a specific neuroendocrine marker and should not be used without other neuroendocrine markers. It is important to consider that immunohistochemistry findings are seldom specific for a particular tumor or diagnosis, with well documented immunohistochemical cross-reactivity between tumor types and putative myogenic and epithelial markers. Thus, anomalous expression of markers can be seen in a wide variety of tumor types, with lineage infidelity quite common [2]. Overall, in this series, 10 of 48 cases (21%) tested for epithelial and neuroendocrine markers, showed coexpression of epithelial and neuroendocrine markers (synaptophysin and chromogranin), a finding which may suggest true neuroepithelial differentiation in a significant subset of SNT ARMS.
It is important to note that the classical rhabdomyoblastic differentiation with strap cells or cross striations is seldom seen in ARMS, and when present is an isolated finding. Thus, it is imperative in sinonasal tract tumors that show a primitive, undifferentiated or round blue cell pattern, to obtain an expanded immunohistochemistry panel that includes epithelial, mesenchymal, melanocytic, lymphoid and neuroendocrine markers, whether in pediatric or adult patients. Such a panel might include a pancytokeratin, S100 protein, CD45RB, desmin and synaptophysin as the possible initial studies, with additional confirmatory immunohistochemistry or molecular studies performed to confirm the diagnostic interpretation [1,2,3]. It is probably even more significant in adult populations to include such a panel, as ARMS is less common in the SNT, and thus may be potentially overlooked. ARMS frequently show specific fusion translocations involving the FOXO1 gene on chromosome 13q14 with either PAX3 (chromosome 2q35) or PAX7 (chromosome 1p36) [32], with a majority showing PAX3 fusion (59%). In this series, only PAX3 was identified specifically (100%), but only 16 of 22 cases studied had an informative result. FISH break apart demonstrated a FOXO1 result in 18 of 21 cases tested, suggesting fusion negative cases. Thus, a difference in outcome based on the differences in PAX3 versus PAX7 could not be validated in this study. No other studies of just adult sinonasal tract ARMS have shown similar molecular results, which will need further clarification.
Children with rhabdomyosarcoma have an overall 5-year survival of 34–48% [33, 34] versus 31–40% for adults [35, 36], suggesting there is not a significant difference in outcome in general for rhabdomyosarcoma. When all head and neck rhabdomyosarcomas are included, children have a reported 5-year overall survival of 75% [37] versus adults with 36% [38] in general, although when age is not evaluated, there is an overall 62.8% 5-year survival for head and neck RMS [39, 40]. When sinonasal tract RMS are reviewed, children are reported to have a 53–58% 5-year overall survival versus 32% for adults [41, 42].
Further, all SNT RMS are reported to have a 47% 5-year overall survival and a 46% 10-year survival, suggesting that those who survive the first 5 years are likely to be considered cured [43]. The cohort of 14 SNT RMS cases reported by Fu et al. showed 11 patients dead of disease within 2 years of diagnosis (79%) [44], although they only had a single adult patient with alveolar histology. In this cohort of adult patients with ARMS specifically, 63.5% of patients had disease at last follow-up, with an average of 16.9 months (range 2.4–75.2 months), suggesting a worse prognosis for this adult population with ARMS (36.5% alive or dead without evidence of disease), a finding similar to 44% 5-year overall survival reported by Callender, et al. [41] Further, there is a much lower overall 5-year survival of 28–38% in patients with SNT ARMS versus 49–57% for SNT embryonal type, although not stratified based on age [40, 41, 43]. These findings are similar to the findings in this report on a cohort of adult patients with SNT ARMS. Thus, it seems that SNT ARMS have a worse overall outcome than other anatomic sites, a worse prognosis than SNT embryonal RMS, and adults a worse prognosis than children, making the SNT, ARMS type, and adult patients all unfavorable factors in RMS [40, 41, 43, 45].
There is no accepted staging for SNT RMS, with modifications trying to establish a TNM type reporting. However, in reported cases, there was a better outcome for stage III (57% 5-year) and stage II patients (49%) compared to stage I patients (40%) [41], which does not support generalized principles of higher stage tumors usually associated with a worse prognosis. The Intergroup Rhabdomyosarcoma Study Group (IRSG) surgical pathology grouping system and staging system [5] has been applied to sinonasal tract, using the parameningeal criteria. However, in this case series a statistically significant difference in outcome based on IRSG staging was not identified. In general, however, there is a high rate of cervical lymph node involvement for head and neck RMS in general (28–50%) [36, 38, 46, 47] further supported by this study showing a 78.8% lymph node metastasis rate and a 48% M1 rate at diagnosis. Thus, in practical terms, SNT ARMS in adults must be managed much more aggressively with a vigorous systemic therapy if a better prognosis is to be expected [36].
Conclusion
In conclusion, the findings in this series of adult SNT ARMS have shown a high aberrant coexpression of epithelial and neuroendocrine immunohistochemistry markers, highlighting the necessity of obtaining a panel of immunohistochemistry markers that includes skeletal muscle markers (desmin, myogenin, myoD1) when primitive, undifferentiated, small cell or rhabdoid tumors are identified histologically. Mistakes in interpretation of epithelial and neuroendocrine markers can be avoided by implementing this approach. Multimodality therapies, specifically to include chemotherapy regimens, should be considered in SNT ARMS in adults, as there is a very high likelihood of cervical lymph node metastases and distant metastases. Overall, adult patients with SNT ARMS experience a worse outcome than children with SNT ARMS, than patients with SNT embryonal RMS, and than patients with RMS in other head and neck or extremity sites.
References
Bahrami A, Gown AM, Baird GS, Hicks MJ, Folpe AL. Aberrant expression of epithelial and neuroendocrine markers in alveolar rhabdomyosarcoma: a potentially serious diagnostic pitfall. Mod Pathol. 2008;21(7):795–806.
Miettinen M, Rapola J. Immunohistochemical spectrum of rhabdomyosarcoma and rhabdomyosarcoma-like tumors. Expression of cytokeratin and the 68-kD neurofilament protein. Am J Surg Pathol. 1989;13(2):120–32.
Thompson LD. Small round blue cell tumors of the sinonasal tract: a differential diagnosis approach. Mod Pathol. 2017;30(s1):S1–26.
Thompson LD. Update from the 4th edition of the World Health Organization classification of head and neck tumours: tumours of the ear. Head Neck Pathol. 2017;11(1):78–87.
Raney RB, Maurer HM, Anderson JR, Andrassy RJ, Donaldson SS, Qualman SJ, et al. The intergroup rhabdomyosarcoma study group (IRSG): major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma. 2001;5(1):9–15.
Bishop JA, Thompson LD, Cardesa A, Barnes L, Lewis JS Jr, Triantafyllou A, et al. Rhabdomyoblastic differentiation in head and neck malignancies other than rhabdomyosarcoma. Head Neck Pathol. 2015;9(4):507 – 18.
Bishop JA, Antonescu CR, Westra WH. SMARCB1 (INI-1)-deficient carcinomas of the sinonasal tract. Am J Surg Pathol. 2014;38(9):1282–9.
Bishop JA, Alaggio R, Zhang L, Seethala RR, Antonescu CR. Adamantinoma-like Ewing family tumors of the head and neck: a pitfall in the differential diagnosis of basaloid and myoepithelial carcinomas. Am J Surg Pathol. 2015;39(9):1267–74.
Houreih MA, Lin AY, Eyden B, Menasce LP, Harrison J, Jones D, et al. Alveolar rhabdomyosarcoma with neuroendocrine/neuronal differentiation: report of 3 cases. Int J Surg Pathol. 2009;17(2):135–41.
Iezzoni JC, Mills SE. “Undifferentiated” small round cell tumors of the sinonasal tract: differential diagnosis update. Am J Clin Pathol. 2005;124(Suppl):S110–21.
Smith SM, Schmitt AC, Carrau RL, Iwenofu OH. Primary sinonasal mucosal melanoma with aberrant diffuse and strong desmin reactivity: a potential diagnostic pitfall! Head Neck Pathol. 2015;9(1):165–71.
Thompson LDR. What is a small round blue cell tumour anyway? Pathology. 2009;41:8.
Win KT, Lee MY, Tan TD, Tsai MP, Bahrami A, Raimondi SC, et al. Nasopharyngeal alveolar rhabdomyosarcoma expressing CD56: a mimicker of extranodal natural killer/T cell lymphoma. Int J Clin Exp Pathol. 2014;7(1):451–5.
Wooff JC, Weinreb I, Perez-Ordonez B, Magee JF, Bullock MJ. Calretinin staining facilitates differentiation of olfactory neuroblastoma from other small round blue cell tumors in the sinonasal tract. Am J Surg Pathol. 2011;35(12):1786–93.
Agaimy A, Hartmann A, Antonescu CR, Chiosea SI, El-Mofty SK, Geddert H, et al. SMARCB1 (INI-1)-deficient sinonasal carcinoma: a series of 39 cases expanding the morphologic and clinicopathologic spectrum of a recently described entity. Am J Surg Pathol. 2017;41(4):458–71.
Bishop JA. Newly described tumor entities in sinonasal tract pathology. Head Neck Pathol. 2016;10(1):23–31.
Stelow EB, Bishop JA. Update from the 4th edition of the World Health Organization classification of head and neck tumours: tumors of the nasal cavity, paranasal sinuses and skull base. Head Neck Pathol. 2017;11(1):3–15.
Miettinen M, Lehto VP, Badley RA, Virtanen I. Alveolar. rhabdomyosarcoma. Demonstration of the muscle type of intermediate filament protein, desmin, as a diagnostic aid. Am J Pathol. 1982;108(2):246–51.
Downs-Kelly E, Shehata BM, Lopez-Terrada D, Weaver J, Patel RM, Hartke M, et al. The utility of FOXO1 fluorescence in situ hybridization (FISH) in formalin-fixed paraffin-embedded specimens in the diagnosis of alveolar rhabdomyosarcoma. Diagn Mol Pathol. 2009;18(3):138–43.
Sebire NJ, Gibson S, Rampling D, Williams S, Malone M, Ramsay AD. Immunohistochemical findings in embryonal small round cell tumors with molecular diagnostic confirmation. Appl Immunohistochem Mol Morphol. 2005;13(1):1–5.
Babin E, Rouleau V, Vedrine PO, Toussaint B, de Raucourt D, Malard O, et al. Small cell neuroendocrine carcinoma of the nasal cavity and paranasal sinuses. J Laryngol Otol. 2006;120(4):289–97.
Chapman-Fredricks J, Jorda M, Gomez-Fernandez C. A limited immunohistochemical panel helps differentiate small cell epithelial malignancies of the sinonasal cavity and nasopharynx. Appl Immunohistochem Mol Morphol. 2009;17(3):207–10.
Cordes B, Williams MD, Tirado Y, Bell D, Rosenthal DI, Al-Dhahri SF, et al. Molecular and phenotypic analysis of poorly differentiated sinonasal neoplasms: an integrated approach for early diagnosis and classification. Hum Pathol. 2009;40(3):283–92.
Ejaz A, Wenig BM. Sinonasal undifferentiated carcinoma: clinical and pathologic features and a discussion on classification, cellular differentiation, and differential diagnosis. Adv Anat Pathol. 2005;12(3):134–43.
Mills SE. Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinomas. Mod Pathol. 2002;15(3):264–78.
Patel TD, Vazquez A, Dubal PM, Baredes S, Liu JK, Eloy JA. Sinonasal neuroendocrine carcinoma: a population-based analysis of incidence and survival. Int Forum Allergy Rhinol. 2015;5(5):448–53.
Su SY, Bell D, Hanna EY. Esthesioneuroblastoma, neuroendocrine carcinoma, and sinonasal undifferentiated carcinoma: differentiation in diagnosis and treatment. Int Arch Otorhinolaryngol. 2014;18(Suppl 2):S149–56.
Weinreb I, Perez-Ordonez B. Non-small cell neuroendocrine carcinoma of the sinonasal tract and nasopharynx. Report of 2 cases and review of the literature. Head Neck Pathol. 2007;1(1):21–6.
Wenig BM. Undifferentiated malignant neoplasms of the sinonasal tract. Arch Pathol Lab Med. 2009;133(5):699–712.
Thompson LDR, Seethala RR, Müller S. Ectopic sphenoid sinus pituitary adenoma (ESSPA) with normal anterior pituitary gland: a clinicopathologic and immunophenotypic study of 32 cases with a comprehensive review of the English literature. Head Neck Pathol. 2012;6(1):75–100.
Dias P, Chen B, Dilday B, Palmer H, Hosoi H, Singh S, et al. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol. 2000;156(2):399–408.
Barr FG, Smith LM, Lynch JC, Strzelecki D, Parham DM, Qualman SJ, et al. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Intergroup Rhabdomyosarcoma Study-III trial: a report from the Children’s Oncology Group. J Mol Diagn. 2006;8(2):202–8.
Akyuz C, Sari N, Yalcin B, Varan A, Kutluk T, Buyukpamukcu M. Long-term survival results of pediatric rhabdomyosarcoma patients: a single-center experience from Turkey. Pediatr Hematol Oncol. 2012;29(1):38–49.
Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer 2009;115(18):4218–26.
Ferrari A, Dileo P, Casanova M, Bertulli R, Meazza C, Gandola L, et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer 2003;98(3):571–80.
Esnaola NF, Rubin BP, Baldini EH, Vasudevan N, Demetri GD, Fletcher CD, et al. Response to chemotherapy and predictors of survival in adult rhabdomyosarcoma. Ann Surg. 2001;234(2):215–23.
Raney RB, Chintagumpala M, Anderson J, Pappo A, Qualman S, Wharam M, et al. Results of treatment of patients with superficial facial rhabdomyosarcomas on protocols of the Intergroup Rhabdomyosarcoma Study Group (IRSG), 1984–1997. Pediatr Blood Cancer. 2008;50(5):958 – 64.
Wu Y, Li C, Zhong Y, Guo W, Ren G. Head and neck rhabdomyosarcoma in adults. J Craniofac Surg. 2014;25(3):922–5.
Turner JH, Richmon JD. Head and neck rhabdomyosarcoma: a critical analysis of population-based incidence and survival data. Otolaryngol Head Neck Surg. 2011;145(6):967–73.
Wurm J, Constantinidis J, Grabenbauer GG, Iro H. Rhabdomyosarcomas of the nose and paranasal sinuses: treatment results in 15 cases. Otolaryngol Head Neck Surg. 2005;133(1):42–50.
Callender TA, Weber RS, Janjan N, Benjamin R, Zaher M, Wolf P, et al. Rhabdomyosarcoma of the nose and paranasal sinuses in adults and children. Otolaryngol Head Neck Surg. 1995;112(2):252–7.
Raney RB, Asmar L, Vassilopoulou-Sellin R, Klein MJ, Donaldson SS, Green J, et al. Late complications of therapy in 213 children with localized, nonorbital soft-tissue sarcoma of the head and neck: a descriptive report from the Intergroup Rhabdomyosarcoma Studies (IRS)-II and -III. IRS Group of the Children’s Cancer Group and the Pediatric Oncology Group. Med Pediatr Oncol. 1999;33(4):362–71.
Sanghvi S, Misra P, Patel NR, Kalyoussef E, Baredes S, Eloy JA. Incidence trends and long-term survival analysis of sinonasal rhabdomyosarcoma. Am J Otolaryngol. 2013;34(6):682–9.
Fu YS, Perzin KH. Nonepithelial tumors of the nasal cavity paranasal sinuses, and nasopharynx: a clinicopathologic study. V. Skeletal muscle tumors (rhabdomyoma and rhabdomyosarcoma). Cancer 1976;37(1):364–76.
Szablewski V, Neuville A, Terrier P, Lae M, Schaub R, Garrel R, et al. Adult sinonasal soft tissue sarcoma: analysis of 48 cases from the French Sarcoma Group database. Laryngoscope 2015;125(3):615–23.
Luna-Ortiz K, Hurtado-Lopez LM, Valderrama-Landaeta JL, Ruiz-Vega A. Thyroglossal duct cyst with papillary carcinoma: what must be done? Thyroid 2004;14(5):363–6.
Bisogno G, Compostella A, Ferrari A, Pastore G, Cecchetto G, Garaventa A, et al. Rhabdomyosarcoma in adolescents: a report from the AIEOP Soft Tissue Sarcoma Committee. Cancer 2012;118(3):821–7.
Acknowledgements
A special thanks to Ms. Hannah B. Herrera for her research assistance. The views expressed are those of the authors solely and do not represent endorsement from Southern California Permanente Medical Group.
Funding
No external funding was obtained for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that he/she has not conflict of interest as it relates to this research project.
Ethical Approval
All procedures performed in this retrospective data analysis involving human participants were in accordance with the ethical standards of the institutional review board (IRB #5968), which did not require informed consent.
Rights and permissions
About this article

Cite this article
Thompson, L.D.R., Jo, V.Y., Agaimy, A. et al. Sinonasal Tract Alveolar Rhabdomyosarcoma in Adults: A Clinicopathologic and Immunophenotypic Study of Fifty-Two Cases with Emphasis on Epithelial Immunoreactivity. Head and Neck Pathol 12, 181–192 (2018). https://doi.org/10.1007/s12105-017-0851-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-017-0851-9