
Abstract
Objective
To ascertain the impact of pituitary size as judged by Magnetic Resonance Imaging (MRI), on response to Growth Hormone (GH) therapy in GH deficient children.
Methods
Thirty nine children (9.1 ± 2.7 y, 22 boys) with non-acquired GH deficiency (21 Isolated GH deficiency and 18 Combined pituitary hormone deficiency) were consecutively recruited and followed up for one year. Clinical, radiological (bone age and MRI) and biochemical parameters were studied.
Results
Children with hypoplastic pituitary (pituitary height < 3 mm) had more severe height deficit (height for age Z-score −6.0 vs. −5.0) and retardation of skeletal maturation (bone age chronological age ratio of 0.59 vs. 0.48) at baseline as compared to children with normal pituitary heights (p < 0.05 for both). After one year of GH therapy, height for age Z scores and percentage change in height for age Z scores were significantly higher in children with hypoplastic pituitaries (13.8 ± 3.6 and 28.7 % vs. 11.2 ± 4.1 and 21.4 %). Significant co-relation was observed between pituitary height and height for age Z-scores at baseline (r = 0.39, p < 0.05). The predicted adult height using Tanner Whitehouse-2 equations improved from 140.8 to 152.3 cm in children with hypoplastic pituitary when compared to an increase from 145.8 to 153.5 cm observed in children with normal pituitary height (p < 0.05).
Conclusions
Indian growth hormone deficient children with hypoplastic pituitary respond better to therapy with GH in short term.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Growth hormone deficiency (GHD) is an important treatable cause of pathological short stature [1]. Growth of children with non-acquired growth hormone deficiency is believed to be influenced by many factors such as pre-treatment height velocity, age at onset of therapy with growth hormone, parental target height, etc. [2]. However, in developing countries such as India, treatment with recombinant Human Growth hormone (rhGH) is still very expensive and cost-benefit needs to be considered when putting patients on therapy with rhGH [3]. Hence, investigating factors which influence growth response of growth hormone deficient children to growth hormone therapy is crucial.
Magnetic resonance imaging (MRI) is an important diagnostic tool which complements hormonal assessment and anthropometric analysis in growth hormone deficient children. Morphological alterations in the pituitary such as ectopic posterior pituitary and pituitary stalk interruption provide vital clues to the etiology of hypopituitarism. Previous studies report that MRI abnormalities are linked to severity in GH deficiency [4, 5]. However, reports on pituitary abnormalities as judged by an MRI and their association with response to GH therapy are scarce.
Hence, the objectives of the present study were 1) to study the pituitary morphology in Indian patients with non-acquired growth hormone deficiency and 2) to assess the association of pituitary size with response to a year of recombinant human growth hormone therapy in them.
Material and Methods
The present study was conducted as a prospective study from January 2008 through February 2011. Thirty nine non-acquired GH-deficient children (22 boys, 17 girls) with a mean age of 9.1 ± 2.7 y, referred to a tertiary-level pediatric endocrine care unit in Pune, India were consecutively recruited. The diagnosis of GHD was based on severe short stature (<−3.0 SD below the mean height for age- and sex-matched children), low growth velocity (below 10th percentile for age), failure to show serum GH concentrations above 7 μg/L after two provocation tests using clonidine (0.15 mg/m2) and glucagon (0.03 mg/kg) as stimulating agents prior to enrolment in the study (priming, boys: depot testosterone IM 3–5 d before the test, girls: 50–100 μgm/d ethinyl estradiol for three consecutive days) and low baseline IGF-1 concentrations (IGF-I SDS ≤ −0.5) [6]. Of the enroled subjects, 21 children had isolated GHD (IGHD), while 18 had combined pituitary hormone deficiency (2 or >2 hormone deficiencies; CPHD). Subjects with CPHD were on stable substitution therapy [with thyroxin (100 μg/m2/d) and/or hydrocortisone (10 mg/m2/d)]. Previous research has shown that growth in children with IGHD and appropriately treated CPHD is comparable in terms of initial response to GH and final height achieved [7]. Further, there were no differences in study parameters in children with IGHD and CPHD at the onset of study (Height for age Z scores at baseline IGHD −5.4 ± 1.5, CPHD −5.6 ± 1.5, P > 0.05). Hence, children with IGHD and CPHD (on stable substitution therapy) have been considered as one group for analysis. Similarly, there were no differences in boys and girls, hence data were analysed together (Data not shown). Children with dysmorphic syndromes, chromosomal abnormalities and acquired causes for GHD like tumors and trauma were excluded from the study. Informed consent was obtained from parents and assent from children (both for patients and controls) before the study was commenced. The study was approved by Institutional ethics committee.
Standing height was measured using a stadiometer (Leicester Height Meter, Child Growth Foundation, UK, range 60–207 cm). The child stood on the flat base of the stadiometer with the back of the head, shoulder blades, buttocks and heels touching the vertical rod, and head in the Frankfurt plane. Gentle upward traction was applied to the mandibular process and the headboard lowered. Height reading was taken to the last completed millimetre, avoiding parallax, and three such readings were averaged for analysis. Weight was measured using electronic weighing scales (Salter, India) to the last 100 g. Measurements were recorded by the same pediatrician at both time points. Height and weight were measured at baseline and then after 1 y of therapy with rhGH. Body mass index (BMI) was calculated using the formula weight in kg/height in square meters. The anthropometric measures were converted into Z-scores based on ethnic reference data [8]. Pubertal status was assessed by a pediatric endocrinologist [9]. Growth velocity was converted into Z-scores using available published references [10, 11].
Serum GH concentrations were assessed by a solid-phase, two-site chemiluminescent immunometric assay with an intraassay coefficient of variation (CV) of 5.3 % and interassay CV of 5.5 %. The serum IGF-1 concentrations were analyzed by a solid-phase, enzyme-labeled chemiluminescent immunometric assay with an intraassay CV of 5.8 % and interassay CV of 3.1 %. The IGF-1 concentrations were then converted into SDS using available normative data [12].
Magnetic resonance imaging (MRI) scans were performed in a 1.5 T unit (Signa, GE, Milwaukee, WI) using T1-weighted sagittal and coronal scans with TR:350 ms and TE:20 ms. The coronal images were obtained using 3.0-mm slice thickness with 10 % gap before and after administration of 0.1 mmol/L of a contrast (gadolinium). None of the subjects required sedation.
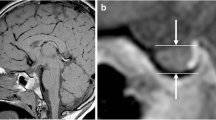
MRI were then separately analyzed according to the same protocol by two experienced neuroradiologists, who were blinded to the clinical and biochemical details of the subjects. Maximal height of the pituitary gland was measured perpendicular to the sella turcica floor. Height of the pituitary gland increases with age, thus, the measurements were compared with available age matched normative data [13]. Pituitary volume was calculated using the formula for the volume of an ellipsoid (0.5 × length × width × height) and compared with published reference values using the same measurement and calculation technique, previously published [14]. Ectopia of the neurohypophysis was diagnosed on the basis of lack of visualization of the high signal on T1 image in the sella turcica, and the presence of this signal in the infundibular recess of the third ventricle. Pituitary stalk with a normal diameter from the level of the optic chiasm to its insertion on the pituitary gland was considered normal. The stalk was considered thin when it had a continuous but extremely thin appearance and its proximal and distal diameter size were below normal. MRI scans were assessed for midline defects such as agenesis of corpus callosum, optic nerve hypoplasia, Chiari I malformation and any other abnormality of the brain. Partial empty sella syndrome was defined when less than half of the sella was filled with CSF, while, total empty sella syndrome was diagnosed when more than half of the sella was filled with CSF [15]. The cerebellar tonsils were judged to be low lying if they were displaced below the foramen magnum. Medial deviation of the carotid arteries to a dimension less than the transverse diameter of the pituitary gland indicated an abnormal course of the carotid arteries [16]. Pituitary height ≤ 3 mm prepubertally was considered hypoplastic [17].
Bone age was estimated at initiation of treatment and 12 mo later by the same radiologist using the RUS score of Tanner Whitehouse 2 method (TW2 method) [18]. Final height was predicted by the Tanner Whitehouse 2 equations [18].
All subjects were treated with recombinant human growth hormone (rhGH) [Novo Nordisk A/S Pharma, Banglore, Maharashtra, India or Eli Lilly and Company Gurgaon (India) Private Limited, India] for a period of 12 mo (dose 10 mg/m2/wk, subcutaneously at night) with an injection frequency of 7 injections/wk. The GH dose was then adjusted in accordance with the body surface area of the children every 3 mo. The criteria for discontinuation of GH treatment was growth velocity less than 2 cm/y in the previous 6 mo with a bone age greater than 14 y for girls and 16 y for boys [19].
Results are expressed as the Mean ± SD. Student’s t test was used for comparison between the groups. Chi-square test was applied to compare proportions. Pearson correlation coefficient (r) was calculated to measure the association between pituitary height, body height for age and IGF levels. p < 0.05 was considered significant. Statistical tests were performed with the SPSS 9.0 statistical package (SPSS, Inc., Chicago, IL).
Results
Children were divided as per their pituitary heights, as greater (normal) or lesser (hypoplastic) than 3 mm [17]. Anthropometric characteristics, bone age, IGF1 and IGFBP3 concentrations of study children (as per pituitary height) at baseline and at 1 y of rhGH therapy are presented in Table 1. Mean age of the study children was 9.1 ± 2.7 y. Children with hypoplastic pituitary (nine children had IGHD, nine CPHD) had more severe height deficit (height for age Z-score −6.0 vs. −5.0) and retardation of skeletal maturation (bone age to chronological age ratio of 0.59 vs. 0.48) compared to children with normal pituitary heights (p < 0.05 for both).
In the study subjects, six children had ectopic posterior lobe, 18 children had empty or partially empty sella. The number of children with vascular and cranio vertebral anomalies was 9 and 6 respectively. The pituitary stalk was noted to be thin, thick and interrupted in 20, 1 and 1 case respectively.
The height as well as height for age Z scores were significantly lower in the group with a hypoplastic pituitary at baseline. The percentage change in height and height for age Z-scores at the end of the year of therapy in subjects with hypoplastic pituitary were significantly higher than in children with pituitary height >3 mm (p < 0.05). The growth velocity and growth velocity Z-scores were also marginally higher in children with hypoplastic pituitary (p < 0.1). The predicted adult height using Tanner Whitehouse-2 equations was significantly higher in children with a pituitary height of >3 mm at baseline; it improved significantly from 140.8 to 152.3 cm in children with hypoplastic pituitary when compared to an increase from 145.8 to 153.5 cm observed in children with normal pituitary height (p < 0.05) (Fig. 1).

Change in Predicted Adult height (PAH) in children with normal and hypoplastic pituitary
A statistically significant correlation was observed between pituitary height and height for age Z-scores at baseline (r = 0.39, p < 0.05) (Fig. 2). Pituitary height also significantly correlated with IGF-I Z-score at baseline (r = 0.33 and p < 0.05).

Correlation of baseline HAZ with the pituitary height
All the children tolerated GH therapy well; none developed any therapy related complications. All the enroled subjects completed the study and were prepubertal at baseline and at the end of 1 y of therapy.
Discussion
To the best of authors’ knowledge, this is the first Indian study that describes the association of pituitary size with response to rhGH therapy in GH deficient children. Children with pituitary height of < 3 mm as judged by an MRI were significantly shorter at baseline. They also had a lower height velocity SDS and skeletal age as compared to children with a pituitary height of ≥ 3 mm. Children who had a hypoplastic pituitary (pituitary height < 3 mm) responded better to rhGH therapy in terms of percentage change in height for age Z-scores, absolute height (in cm), growth velocity Z-scores and improvement in predicted adult height based on bone age. The authors have also described the morphological features of the pituitary in Indian GHD children.
In the present study, nine children (23 %) had vascular abnormalities and six children (15.4 %) had cranio-vertebral abnormalities. Scotti et al. [20] and Marwaha et al. [21] made similar observations in 13.5 % and 22.7 % children respectively. These abnormalities may be a result of birth trauma or a part of midline syndrome. The occurrence of ectopic posterior lobe in authors’ series, 6/39 (15.4 %) is much lower than other published series by Duttah et al. [4] (12/31) and Bordallo et al. [22] (14/37). This may be related to the mode of delivery—wherein traumatic delivery leads to transaction of the stalk and distal regeneration leading to an ectopic posterior pituitary. However, the authors were not able to retrieve the mode of delivery from the records of their study cohort. Also, they did not find any significant difference between IGHD and CPHD children with respect to their height for age Z-scores, possibly because the CPHD children were on appropriate substitution therapy.
Growth hormone deficient children with a hypoplastic pituitary gland have been shown to be significantly shorter than children with a normal MRI [23]. Further, studies also suggest that the severity of growth hormone deficiency is related to MRI findings [24]. This may be a reflection of the fact that morphological alterations on MRI of the hypothalamic-pituitary area are an expression of the severity of hypopituitarism. Pituitary height is thought to be directly related to GH levels, as GH-secreting cells are the most abundant cell population in the pituitary gland. In the index cohort of children, those with smaller pituitaries were shorter and had more bone age delay. Thus, the present results are in line with previously published data [23, 24].
In a study on 216 short Polish children with GHD, which aimed to assess the relationship between growth hormone secretion and pituitary size, Hilczer et al. have reported that response to rhGH therapy was best in children with severely hypoplastic pituitary glands [25]. The index results also suggest that the response to rhGH therapy in children with hypoplastic pituitary vs. children with normal pituitary height is greater in terms of improvement of absolute height (in cm), height for age Z-scores and growth velocity Z-scores. Further, percentage improvement in height for age Z-scores in the present cohort was 38 % in children with short pituitary height vs. 28 % in children with normal pituitary height. This is comparable to an improvement in height for age Z-score of +2.7 vs. +1.3 in children with abnormal and hypoplastic pituitary reported by others [23].
Improvement in predicted adult height, either by using the Tanner Whitehouse equations or Bayley Pinneau method (if Greulich and pyle atlas is used to compute bone age), is an important parameter to assess response to GH therapy. Height predictions, based on current chronological age, bone age, height and menarchal status. The authors have demonstrated a better improvement in predicted adult height based on bone age after 1 y of GH therapy in children with a hypoplastic pituitary vs. in children with a normal pituitary height. This has not been reported earlier. However, the final height achieved would be close to the predicted height only if GH therapy is continued beyond the study period.
The index study is not without limitations. Details pertaining to the mode of delivery could not be retrieved from the case records. Further, it would be preferable that the study subjects be followed till they reach adult height, even so, the present results suggest that in the short term, GHD children with hypoplastic pituitaries respond better to rhGH therapy.
To conclude, Indian growth hormone deficient children with hypoplastic pituitary respond better to therapy with rhGH. Thus, pituitary height, as judged by MRI provides valuable information concerning response to GH therapy in Indian growth hormone deficient children.
References
Desai MP, Colaco P. Growth hormone deficiency and insensitivity. In: Desai MP, Menon PSN, Bhatia V, eds. Pediatric endocrine disorders. Hyderabad: Universities Press (India) Pvt. Ltd; 2008. pp. 121–52.
Zenaty D, Garel C, Limoni C, Czernichow P, Léger J. Presence of magnetic resonance imaging abnormalities of the hypothalamic-pituitary axis is a significant determinant of the first 3 years growth response to human growth hormone treatment in prepubertal children with nonacquired growth hormone deficiency. Clin Endocrinol (Oxf). 2003;58:647–52.
Bajpai A, Menon PS. Growth hormone therapy. Indian J Pediatr. 2005;72:139–44.
Dutta P, Bhansali A, Singh P, Rajput R, Bhadada S. Clinico-radiological correlation in childhood hypopituitarism. Indian Pediatr. 2010;47:615–8.
Acharya SV, Gopal RA, Lila A, Sanghvi DS, Menon PS, Bandgar TR, et al. Phenotype and radiological correlation in patients with growth hormone deficiency. Indian Pediatr. 2011;78:49–54.
Wales JK. Evaluation of growth disorders. In: Brook C, Clayton P, Brown R, eds. Clinical pediatric endocrinology. Oxford: Wiley-Blackwell; 2009. pp. 124–54.
Lee PA, Sävendahl L, Oliver I, Tauber M, Blankenstein O, Ross J, et al. Comparison of response to 2-years’ growth hormone treatment in children with isolated growth hormone deficiency, born small for gestational age, idiopathic short stature, or multiple pituitary hormone deficiency: Combined results from two large observational studies. Int J Pediatr Endocrinol. 2012;2012:22.
Khadilkar VV, Khadilkar AV, Cole TJ, Sayyad MG. Cross sectional growth curves for height, weight and body mass index for affluent Indian children, 2007. Indian Pediatr. 2009;46:477–89.
Tanner JM. Growth and Adolescence. 2nd ed. Oxford, England: Blackwell Scientific Publications; 1962.
Tanner JM, Whitehouse RH, Takaishi M. Standards from birth to maturity for height, weight, height velocity, and weight velocity: British children, I965. Arch Dis Child. 1966;41:613.
Rikken B, Wit JM. Prepubertal height velocity references over a wide age range. Arch Dis Child. 1992;67:1277–80.
Juul A, Bang P, Hertel NT, Main K, Dalgaard P, Jørgensen K, et al. Serum insulin-like growth factor-I in 1030 healthy children, adolescents, and adults: Relation to age, sex, stage of puberty, testicular size, and body mass index. J Clin Endocrinol Metab. 1994;78:744–52.
Argyropoulou M, Perignon F, Brunelle F, Brauner R, Rappaport R. Height of normal pituitary gland as a function of age evaluated by magnetic resonance imaging in children. Pediatr Radiol. 1991;21:247–9.
Takano K, Utsunomiya H, Ono H, Ohfu M, Okazaki M. Normal development of the pituitary gland: Assessment with three-dimensional MR volumetry. AJNR Am J Neuroradiol. 1999;20:312–5.
Marinis L, Bonadonna S, Bianchi A, Maira G, Giustina A. Primary empty sella. J Clin Endocrinol Metab. 2005;91:3329–36.
Hamilton J, Blaser S, Daneman D. MR imaging in idiopathic growth hormone deficiency. AJNR Am J Neuroradiol. 1998;19:1609–15.
Nagel BH, Palmbach M, Petersen D, Ranke MB. Magnetic resonance images of 91 children with different causes of short stature: pituitary size reflects growth hormone secretion. Eur J Pediatr. 1997;156:758–63.
Tanner JM, Whitehouse RH, Cameron N, Marshall WA, Healy MJR, Goldstein H. Assessment of skeletal maturity and prediction of adult height (TW2 method). London: Academic Press; 1983.
Ranke MB. Growth hormone therapy in children: when to stop? Horm Res. 1995;43:122–5.
Scotti G, Triulzi F, Chiumello G, di Natale B. New imaging techniques in endocrinology: magnetic resonance of the pituitary gland and sella turcica. Acta Paediatr Stand. 1989;356S:5–14.
Marwaha R, Menon PS, Jena A, Pant C, Sethi AK, Sapra ML. Hypothalamo-pituitary axis by magnetic resonance imaging in isolated growth hormone deficiency patients born by normal delivery. J Clin Endocrinol Metab. 1992;74:654–9.
Bordallo MA, Tellerman LD, Bosignoli R, Oliveira FF, Gazolla FM, Madeira IR, et al. Neuroradiological investigation in patients with idiopathic growth hormone deficiency. J Pediatr (Rio J). 2004;80:223–8.
Coutant R, Rouleau S, Despert F, Magontier N, Loisel D, Limal J-M. Growth and adult height in GH-treated children with nonacquired GH deficiency and idiopathic short stature: the influence of pituitary magnetic resonance imaging findings. J Clin Endocrinol Metab. 2001;86:4649–54.
Maghnie M, Strigazzi C, Tinelli C, Autelli M, Cisternino M, Loche S, et al. Growth hormone (GH) deficiency (GHD) of childhood onset: Reassessment of GH status and evaluation of the predictive criteria for permanent GHD in young adults. J Clin Endocrinol Metab. 1999;84:1324–8.
Hilczer M, Szalecki M, Smyczynska J, Stawerska R, Kaniewska D, Lewinski A. Growth hormone (GH) secretion and pituitary size in children with short stature. Efficacy of GH therapy in GH-deficient children, depending on the pituitary size. Neuro Endocrinol Lett. 2005;26:447–52.
Acknowledgements
The authors would like to thank the children and their parents who participated in the study.
Contributions
JS is the neuroradiologist involved in the radiological evaluation of the MRI scans. AVK and VVK are the pediatrician and pediatric endocrinologists involved in the management of these children. VR and VE are involved in the collection of data. VE, SAC, HKP and AVK are involved in statistical analysis of the data. All authors contributed to the manuscript and AVK shall act as a guarantor for the study.
Conflict of Interest
None.
Role of Funding Source
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khadilkar, V.V., Prasad, H.K., Ekbote, V.H. et al. Response of Indian Growth Hormone Deficient Children to Growth Hormone Therapy: Association with Pituitary Size. Indian J Pediatr 82, 404–409 (2015). https://doi.org/10.1007/s12098-014-1412-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-014-1412-9