
Abstract
Retinoblastoma, the most common intraocular malignancy of childhood arises due to mutation of the retinoblastoma gene on chromosome 13q14. In the hereditary setting this mutation is present in all germ line cells and can occur as early as during development; however it requires a mandatory second “hit” or mutation of the remaining allele for retinoblastoma to develop. The non-hereditary form arises from spontaneous mutation affecting both alleles in a somatic cell of the retina. The tumor may present with leucocoria or strabismus. The diagnosis is best made by an ophthalmologist who examines the patient under sedation. Although tissue biopsy is not routinely performed, imaging studies like ultrasound and MRI scan can serve as useful adjuncts to help in establishing the diagnosis and also aid in staging. Group A tumors are smaller than 3 mm while group B tumors are >3 mm or those located in the macula. Groups C and D tumors are associated with localized and diffuse vitreous seeds respectively. Group E tumors occupy >50% of the globe and are generally not salvagable. Despite the fact that great advances have been made in the treatment of retinoblastoma in the last two decades, a large number patients undergo procedures associated with significant morbidity such as enucleation. We recommend large multi institutional studies using newer therapeutic models and targeting novel pathways to improve the outcome in advanced stage retinoblastoma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Retinoblastoma, the most common intraocular malignancy in Pediatrics, was first described by Wardrop in 1809 as a tumor arising from the eye. In the 1800s, a variety of terms were used to describe this condition including glioma of the retina, neuroepithelioma and retinoma, until in 1926 when the term retinoblastoma was coined by Verhoeff depicting the multipotent retinal progenitor cells that give rise to the tumor [1–3].
Epidemiology
Retinoblastoma is a rare childhood cancer, occurring in approximately 1 in 15,000 to 1 in 16,600 live births in the United States with 300–350 new cases diagnosed annually [1]. Although rare cases have been reported in adults, most tumors occur in children under two, usually (75%) affecting one eye due to a spontaneous mutation [2]. Bilateral disease, just as multifocal unilateral tumors, is generally heritable in a phenotypically autosomal dominant manner with a germline mutation of the Retinoblastoma gene (RB) that has been mapped to chromosome 13q14.
Heritable retinoblastoma is often linked with a second form of cancer, most commonly osteogenic and soft tissue sarcoma, as noted in a study of 1604 patients among whom incidence of a second malignancy was recorded at 51% versus 5% for heritable and nonheritable forms of retinoblastoma respectively [3].
Genetics and Counseling
RB, the first tumor suppressor gene to be described, has been studied extensively. Briefly, it encodes a 100-kDa nuclear phosphoprotein that inhibits the E2F family of transcription factors, the downstream effect of which is cell cycle progression [4]. Mutation of RB leading to the removal of restriction of cellular proliferation is considered one factor of a “two hit” hypothesis leading to tumorigenesis. In the hereditary setting this mutation is present in all germ line cells and can occur as early as during development; however it requires a mandatory second “hit” or mutation of the remaining allele for retinoblastoma to develop [5]. The nonhereditary form arises from spontaneous mutation affecting both alleles in a somatic cell of the retina.
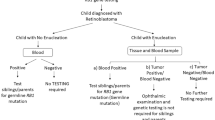
Regardless, siblings and parents of a child diagnosed with the heritable form of retinoblastoma with an identifiable mutation should be screened for the specific mutation; when present, examination under anesthesia starting at birth and every 4 mo is recommended until 5–7 y of age of every sibling and child of the patient by an experienced ophthalmologist [6].
Clinical Features
Leukocoria, an absence of the red reflex of the eye and the most common presentation of retinoblastoma in the US, may represent a large retrolental tumor, commonly detached from the retina. In contrast to the developed world where most cases are detected early, in developing countries the tumor is often detected in an enlarged eye with locally invasive disease. Strabismus is the second most presenting feature, and either presentation mandates an urgent referral to an ophthalmologist. Vitreous hemorrhage, hyphema, glaucoma and heterochromia of the iris remain other less common presenting features.
Diagnosis
Ophthalmoscopic examination under anesthesia that permits visualization of the tumor and the retina is usually followed up with imaging such as ultrasound and/or CT scan of the orbit. MRI is helpful if optic nerve involvement is suspected while calcification, suggestive of retinoblastoma, is best delineated on a CT scan. Imaging of the brain also helps detect the so-called “trilateral retinoblastoma” in which bilateral retinoblastoma is accompanied by a tumor of the pineal gland [7].
In the event that metastasis is suspected due to complaints such as vomiting, loss of weight, proptosis, focal neurological deficit etc. a full work up should be pursued including bone marrow examination, spinal fluid analysis and bone scan.
A complete blood count, serum electrolytes and baseline assessment of renal and liver function are useful tests prior to initiating chemotherapy. An aqueous humor to serum LDH ratio of greater than one has been suggested to be consistent with retinoblastoma, although this is no longer used [8].
Differential Diagnosis
Pseudoretinoblastoma, a term that encompasses other diseases in the differential diagnosis includes toxocara endophthalmitis, persistent hyperplastic primary vitreous (PHPV) and Coats’ disease. A careful attention to history e.g., exposure to the larvae of the nematode T. canis can provide valuable clues to the correct diagnosis. Other conditions in the differential include retinopathy of prematurity, congenital cataract and coloboma.
Cytology
Retinoblastoma is a rare tumor in that a tissue sample is routinely not obtained to establish diagnosis, principally because the radiographic and physical findings are typical but also from an urge to avoid extraocular seeding, a situation that clearly worsens prognosis. However under select circusmtances some authors have shown that fine needle aspiration biopsy (FNAB), can provide tissue diagnosis in the ambiguous setting without extraocular spread [9].
Pathology
Gross Features
Retinoblastoma has the gross appearance of a chalky white friable mass that can either grow into the vitreal cavity in an endophytic manner or into the subretinal space in an exophytic fashion. While most tumors show features of both types, a third pattern, diffuse infiltrating retinoblastoma, poses a diagnostic dilemma and is seen mostly in older children. Interestingly, some cases of retinoblastoma regress spontaneously, a finding that is also associated with central retinal artery occlusion [10, 11].
Histology
Under the microscope, retinoblastoma may demonstrate areas of extensive necrosis and scattered calcification. The viable tumor cells are small, hyperchromatic and mitotically active with high nuclear cytoplasmic ratio; these cells surround blood vessels forming a characteristic pseudorossette. Flexner-Wintersteiner rosettes, frequently found in retinoblastomas, contain cells with projecting photoreceptors, an observation that has led to the hypothesis that the “cell of origin” in these tumors is an immature one capable of differentiating into photoreceptors.
High Risk Features
Localized Disease
Unilateral localized disease has an excellent visual prognosis in sharp contrast to bilateral disease where extent of tumor involvement and available treatment modalities are important deciding factors for residual vision [12].
Metastatic Disease
Remnant of tumor in the optic nerve stump , so called cut end disease is well recognized to confer a high risk of metastasis [13]. Many experts also believe that involvement of the optic nerve posterior to the lamina cribrosa and “massive” choroidal disease impart an increased risk of recurrence [14, 15].
Trilateral Retinoblastoma
As mentioned above, this represents most commonly a pineoblastoma arising in the first 5 y in patients diagnosed with bilateral retinoblastoma. Fortunately, this is found in only 3% of all cases of retinoblastoma, detected at a mean interval of 22 mo from the diagnosis of retinoblastoma [7]. Systemic chemotherapy for retinoblastoma has been suggested to reduce the incidence of pineoblastoma ; in a study of 142 patients treated with systemic chemotherapy none of whom developed pineoblastoma [16], however this finding is not universally accepted by all experts.
Extraocular Retinoblastoma
Extraocular retinoblastoma may be confined to the soft tissue surrounding the eye, or may extend to the optic nerve away from the margin of resection. Further spread may occur with leptomeningeal disease as well as distant metastasis to the lungs, bones and bone marrow. The Children’s Cancer Group considers microscopic involvement of the scleral emissaries as Class I disease, while microscopic involvement of the cut end of the optic nerve is considered Class II disease. Orbital disease in biopsy is considered Class III and leptomeningeal dissemination is regarded as Class IV, with Class V being reserved for distant metastasis. For Class I–III disease, a combination of chemotherapy (with multiple agents such as Vincristine, Cyclophophamide, Doxorubicin and platinum), followed by enucleation and further chemotherapy and local control with radiation is considered standard of care [17]. Although anecdotal reports of successful treatment of retinoblastoma that has spread in the CNS or further to the bone, bone marrow and/or the liver exist, generally these tumors are considered to have a dismal prognosis [17]; more recently, reports on small number of patients have documented successful treatment of patients using high dose myeloablative chemotherapy followed by autologous stem cell rescue [18]. Regardless a multicenter trial needs to be performed to best outline the treatment of such patients.
Staging
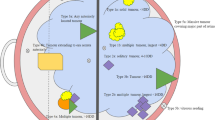
Intraocular retinoblastoma, confined within the eye has excellent prognosis with reported survival rates of above 90% [12]. Unfortunately in developing countries this rapidly growing tumor often spills from the sclera into the orbit or beyond the optic nerve leading to seeding in the vitreous cavity or extension into the sub retinal space, thereby decreasing survival rates to around 50% [19]. The Reese–Ellsworth staging method, first developed in the 1950s was used extensively until better understanding of the disease and newer treatment modalities led to subsequent staging systems e.g., the Essen system in the 1980’s. The International Classification for Intraocular Retinoblastoma (ICIRB), currently in use by the Children’s Oncology Group (COG), utilizes the size, the location of the tumor and focal and diffuse seeding to help guide treatment. Briefly, group A tumors are retinal tumors smaller than 3 mm and outside the macula, while group B tumors are >3 mm or those located in the macula. Groups C and D tumors are associated with localized and diffuse vitreous seeds respectively. Group E tumors occupy >50% of the globe and are generally described as unsalvagable.
Treatment
The treatment of retinoblastoma is complex as it involves multiple factors including prognosis of vision, age of the patient, size and location of the tumor and the presence or absence of vitreous and sub retinal seeding. A wide variety of modalities are available (as outlined below) but often a combination of therapies is necessary to optimize individualized care.
External Beam Radiation Therapy (EBRT)
The role of radiation therapy in treating retinoblastoma has constantly evolved since the first use of EBRT in treating a case of retinoblastoma in 1903 [20]. Retinoblastoma, by virtue of its radiosensitivity, has historically been treated with external beam radiation therapy at a dosage of 45–50 Gy given in 1.5–2 Gy/fraction, with a success rate above 85% at eye preservation. However over the last decade and a half, several adverse effects associated with radiation have come forth such as recurrence of tumor. Cosmetic defects like midface hypoplasia are seen especially in children radiated before the age of 1 y [21]. In addition the association between radiation and the risk for a second cancer, usually seen in the field of radiation is now estimated by some studies to be as high as 6 fold [22]. In view of these observations, radiation therapy is now reserved as a salvage therapy, or for those who progress while receiving chemotherapy. Several new modalities have now emerged such as Intensity-Modulated Radiation Therapy (IMRT) and three-dimensional (3D) conformal radiation therapy, which allow for careful planning using 3-D CT images with computerized dose calculations to identify the dose intensity pattern that best conforms to the shape of the tumor. This approach allows for maximum delivery of target dose to the tumor while minimizing radiation to surrounding normal tissues, thereby helping physicians in avoiding some of the well recognized adverse effects of radiation [23]. A review performed at the authors’ Institution (MD Anderson Cancer Center) comparing photon three dimensional conformal radiation therapy (3D-CRT), electron therapy, IMRT and standard (nonintensity modulated) proton therapy, found that protons were most optimal in treating retinoblastomas, among several other cancers [24]. Protons enable delivering of superior target dose with sparing of normal structures which may well translate into fewer acute and late toxicities.
Enucleation
Despite significant advances in treatment options, enucleation or removal of the eye leaving the muscles intact remains the treatment of choice in situations where the tumor has diffusely seeded or it is massive or if there is evidence of tumor invasion into the optic nerve, choroid, or anterior chamber with little hope of residual vision. Extreme caution is employed by the surgeon to avoid tumor spillage as well as to avoid excess pressure on the thin sclera while hooking the muscles [16]. However , the trend to enucleate has been declining steadily over the last few decades as we have transitioned from enucleating every unilaterally diseased eye towards approaching a more conservative vision-sparing approach. Historically, for bilateral disease the more advanced eye was enucleated and radiation was used to treat the lesser involved eye. In developing countries however, exenteration or removal of the eye with all its contents is sometimes still pursued in cases of significant tumor burden in the surrounding areas [25]. Overall, 75% of unilateral retinoblastomas in developing countries are enucleated since typically the disease is detected at an advanced stage. Following enucleation an orbital implant (polymethylmethacrylate or hydroxyapatite) is placed to preserve the natural appearance of the orbit and to enable the prosthesis to be mobile [16].
Radioactive Plaques (Brachytherapy)
Newer techniques such as radioactive plaques using Iodine or Ruthenium to deliver approximately 4000–4500 cGy to the apex of the tumor have enabled the avoidance of large doses of radiation that have now come to be recognized to cause significant impairments including several fold increase in the risk of a second malignancy and disruption of bone growth with secondary cosmetic defects. Several advantages of this modality such as focal delivery, short duration of treatment and reduced damage to surrounding orbital tissue make this the treatment of choice for patients with groups B and C disease. Out of 60 tumors treated with plaque radiotherapy alone, there was no incidence of a second cancer over a 5 y follow up, as observed by Shields et al, offering a clear advantage over external beam radiation [26]. In the same group recurrence at 1 y was 12% compared to recurrence rates of 8, 25 and 34% for those pretreated with chemoreduction, external beam radiotherapy and a combination of the two, respectively [26]. Nonproliferative and proliferative retinopathies and maculopathy are the most commonly observed adverse effects of plaque therapy which is limited to tumors not greater than 16 mm at base and 8 mm in thickness.
Laser Photoablation and Cryotherapy
Both laser and cryotherapy are delivered over two or three monthly sessions targeting small tumors less than 3.5 mm in thickness. The former uses an argon laser or xenon arc to induce coagulation of blood vessels supplying the tumor while the latter employs triple freeze thaw techniques delivered over one or two sessions [27, 28].
Thermo and Chemothermotherapy
Since heat can sensitize tumors to chemotherapy or radiation, delivery of heat to the eye using the diode or YAG laser to a temperature between 42°C to 60°C has been used to complement chemotherapy (chemothermotherapy) or radiotherapy (thermoradiotherapy). One study of 394 tumors showed complete tumor control using chemoreduction plus thermotherapy or cryotherapy [29].
Chemotherapy
Systemic Chemotherapy
Retinoblastomas belonging to groups B, C and D have been treated with multiple chemotherapy agents such as Carboplatin, Vincristine and Etoposide. So-called chemoreduction has helped to preserve vision and achieve shrinkage of a tumor that is not amenable to local control measures alone. The chemosensitivity of retinoblastomas is also utilized to prevent tumor recurrence where risk factors for recurrence/metastatic disease are present (e.g., tumor cells positive in optic nerve stump) in addition to situations like frank metastatic disease. As mentioned above, some theorize that systemic chemotherapy reduces the incidence of trilateral retinoblastomas [16]. In any case, chemotherapy generally requires adjuvant therapies such as laser thermotherapy, photocoagulation, cryotherapy or brachytherapy. Report from a Canadian study of 43 patients (66 eyes) over a mean follow up of 7.5 y showed success (defined as tumor control with chemotherapy/focal therapy without EBRT and without enucleation) rates between 77 and 100% for groups A-C disease [30]. Interestingly this group also utilized cyclosporine A to overcome emergence of chemoresistance without any significant toxicity. In a British study of a select group of 14 patients with Reese-Ellsworth grade V bilateral disease, 70% of the eyes could be preserved using a combination of chemo and radiotherapy. Six to 8 cycles of chemotherapy with Vincristine, Etoposide, and Carboplatin resulted in complete tumor response in 72% of 78 patients treated with chemotherapy alone with macular tumors showing a high response rate of 84% [31].
Intra-arterial Chemotherapy
Several reports have been published documenting the injection of chemotherapy (Melphalan, Carboplatin) directly into the ophthalmic artery thereby potentially avoiding some of the systemic toxicities encountered with intravenous administration of chemotherapeutic agents [32, 33]. In a study of 95 eyes, ocular event free survival rate of over 80% was noted for eyes treated with intra-arterial chemotherapy as primary management with a mean of 3.1 infusions per eye [32].
Stem Cell Transplant
High-dose chemotherapy followed by autologous stem cell rescue has been reported in the literature to treat systemic retinoblastoma [18, 34]. In a Chilean study, 11 children with metastatic disease involving the bone marrow, bone, orbit and CNS were conditioned using carboplatin and etoposide with thiotepa, melphalan or cyclophosphamide; seven have remained disease free after a median follow up of 39 mo, suggesting that stem cell transplant may be a considerable option for such patients [18].
Future Directions
Several exciting new approaches are currently under trial in the treatment of retinoblastomas. One such model using recombinant adenovirus coding for the retinoblastoma gene was able to suppress proliferation in retinoblastoma cell lines [35]. Scleral reservoirs of chemotherapy have also shown promise in animal models. Such experimental models hold great promise in the future of treating retinoblastoma.
References
Young JL, Smith MA, Roffers SD, et al. Retinoblastoma. In: Ries LAG, Smith MA, Gurney JG, et al., eds. Cancer incidence and survival among children and adolescents: united states SEER program 1975–1995. Bethesda: National Cancer Institute; 1999.
Berkeley JS, Kalita BC. Retinoblastoma in an adult. Lancet. 1977;2:508–9.
Wong FL, Boice JD, Abramson DH, et al. Cancer incidence after retinoblastoma—Radiation dose and sarcoma risk. JAMA. 1997;278:1262–7.
Dyer MA, Bremner R. The search for the retinoblastoma cell of origin. Nat Rev Cancer. 2005;5:91–101.
Knudson AG Jr, Hethcote HW, Brown BW. Mutation and childhood cancer: a probabilistic model for the incidence of retinoblastoma. Proc Natl Acad Sci USA. 1975;72:5116–20.
Hurwitz RL, Shields CL, Shields JA, Chevez-Barrios P, Hurwitz MY, Chintagumpala MM. Retinoblastoma. In: Pizzo PA, Poplack DG, eds. Principles and practice of pediatric oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 865–86.
Bader JL, Meadows AT, Zimmerman LE, et al. Bilateral retinoblastoma with ectopic intracranical retinoblastoma: trilateral retinoblastoma. Cancer Genet Cytogenet. 1982;5:203–13.
Piro PA Jr, Abramson DH, Ellsworth RM, Kitchin D. Aqueous humor lactate dehydrogenase in retinoblastoma patients. Clinicopathologic correlations. Arch Ophthalmol. 1978;96:1823–5.
Karcioglu ZA. Fine needle aspiration biopsy (FNAB) for retinoblastoma. Retina. 2002;22:707–10.
Lindley J, Smith S. Histology and spontaneous regression of retinoblastoma. Trans Ophthalmol Soc UK. 1974;94:953–67.
Kao LY, Yang ML. Spontaneous regression of retinoblastoma in a Taiwan series. J Pediatr Ophthalmol Strabismus. 2005;42:228–32.
Stiller CA. Population-based survival rates for childhood-cancer in Britain, 1980–91. BMJ. 1994;309:1612–6.
Messmer EP, Heinrich T, Hopping W, Desutter E, Havers W, Sauerwein W. Risk-factors for metastases in patients with retinoblastoma. Ophthalmology. 1991;98:136–41.
Shields CL, Shields JA, Baez K, Cater JR, De Potter P. Optic-nerve invasion of retinoblastoma—metastatic potential and clinical risk-factors. Cancer. 1994;73:692–8.
Shields CL, Shields JA, Baez KA, Cater J, De Potter PV. Choroidal invasion of retinoblastoma—metastatic potential and clinical risk-factors. Br J Ophthalmol. 1993;77:544–8.
Shields CL, Meadows AT, Shields JA, Carvalho C, Smith AF. Chemoreduction for retinoblastoma may prevent intracranial neuroblastic malignancy (trilateral retinoblastoma). Arch Ophthalmol. 2001;119:1269–72.
Antoneli CBG, Ribeiro KB, Rodrigue-Galindo C, et al. The addition of ifosfamide/etoposide to cisplatin/teniposide improves the survival of children with retinoblastoma and orbital involvement. J Pediatr Hematol Oncol. 2007;29:700–4.
Palma J,Sasso DF, Dufort G, et al. Successful treatment of metastatic retinoblastoma with high-dose chemotherapy and autologous stem cell rescue in South America. Bone Marrow Transplant. 2011. May 23 [Epub ahead of print]
Shields CL, Shields JA. Basic understanding of current classification and management of retinoblastoma. Curr Opin Ophthalmol. 2006;17:228–34.
Hilgartner HL. Report of case of double glioma treated with x-ray 1903. Tex Med. 2005;101:10.
Peylan-Ramu N, Bin-Nun A, Skleir-Levy M, et al. Orbital growth retardation in retinoblastoma survivors: work in progress. Med Pediatr Oncol. 2001;37:465–70.
Eng C, Li FP, Abramson DH, et al. Mortality from 2nd tumors among long-term survivors of retinoblastoma. J Nat Cancer Inst. 1993;85:1121–8.
Reisner ML, PaisViégas CM, Grazziotin RZ, et al. Retinoblastoma—Comparative analysis of external radiotherapy techniques, including an IMRT technique. Int J Radiat Oncol Biol Phys. 2007;67:933–41.
Lee CT, Bilton SD, Famiglietti RM, et al. Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma: how do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys. 2005;63:362–72.
Mohan ER, Verma M, Kumar K, et al. Clinico-pathological profile of orbital exenteration in a tertiary eye care institute: 7 year experience. Bangalore; AIOC Proceedings. 2008.
Shields CL, Shields JA, Cater J, Othmane I, Singh AD, Micaily B. Plaque radiotherapy for retinoblastoma—Long-term tumor control and treatment complications in 208 tumors. Ophthalmology. 2001;108:2116–21.
Shields JA, Shields CL, Parsons H, Giblin ME. The role of photocoagulation in the management of retinoblastoma. Arch Ophthalmol. 1990;108:205–8.
Shields JA, Parsons H, Shields CL, Giblin ME. The role of cryotherapy in the management of retinoblastoma. Am J Ophthalmol. 1989;108:260–4.
Shields CL, Mashayekhi A, Cater J, Shelil A, Meadows AT, Shields JA. Chemoreduction for retinoblastoma. Analysis of tumor control and risks for recurrence in 457 tumors. Am J Ophthalmol. 2004;138:329–37.
Chan HSL, Gallie BL, Munier FL, Popovic MB. Chemotherapy for retinoblastoma. Ophthalmol Clin North Am. 2005;18:55–63.
Gombos DS, Kelly A, Coen PG, Kingston JE, Hungerford JL. Retinoblastoma treated with primary chemotherapy alone: the significance of tumour size, location, and age. Br J Ophthalmol. 2002;86:80–3.
Gobin YP, Dunkel IJ, Marr BP, Brodie SE, Abramson DH. Intra-arterial chemotherapy for the management of retinoblastoma: four-year experience. Arch Ophthalmol. 2011;129:731–7.
Vajzovic LM, Murray TG, Aziz-Sultan MA, et al. Supraselective intra-arterial chemotherapy: evaluation of treatment-related complications in advanced retinoblastoma. Clin Ophthalmol. 2011;5:171–6.
Tsuruta T, Aihara Y, Kanno H, et al. High-dose chemotherapy followed by autologous and allogeneic peripheral blood stem cell transplantation for recurrent disseminated trilateral retinoblastoma. Childs Nerv Syst. 2011;27:1019–24.
Demers GW, Harris MP, Wen SF, Engler H, Nielsen LL, Maneval DC. A recombinant adenoviral vector expressing full-length human retinoblastoma susceptibility gene inhibits human tumor cell growth. Cancer Gene Ther. 1998;5:207–14.
Acknowledgements
The authors are grateful to Ayesha Ray for editing and helping with preparation of the manuscript.
Contributions
Tribhawan Vats conceptualized the article; the manuscript was written by AK and DG, critically analyzed and finalized by DG.
Conflict of Interest
None.
Role of Funding Source
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ray, A., Gombos, D.S. Retinoblastoma: An Overview. Indian J Pediatr 79, 916–921 (2012). https://doi.org/10.1007/s12098-012-0726-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-012-0726-8