
Abstract
Despite decades of research, prognosis for SCLC patients remains poor, and treatment options limited. SCLC is an immunogenic tumor with high somatic mutation rates due to tobacco exposure resulting in potential neo-antigens, the presence of suppressed immune responses, and occurrence of paraneoplastic disorders. The use of T cell immune-checkpoint inhibitors (anti-PD1: nivolumab, pembrolizumab; anti-PD-L1: atezolizumab, durvalumab; anti-CTLA-4: ipilimumab, tremelimumab) have shown promising antitumor activity with the potential to prolong survival in SCLC patients. In fact, atezolizumab when combined with chemotherapy has achieved the milestone of being the first drug to improve survival in patients with newly diagnosed extensive-stage SCLC. Other immunotherapeutic approaches evaluated in clinical trials for SCLC include the use of cytokines, cancer vaccines, antiganglioside therapies, TLR9 inhibition, anti-Notch signaling, and anti-CD47. This review discusses the rationale and clinical evidence of immunotherapy in SCLC, the conflictive clinical results of novel immunotherapeutic agents and combinatorial therapies under evaluation in SCLC patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Small cell lung cancer (SCLC) is a deadly disease that represents about 15% of all lung cancers [1]. SCLC is strongly associated with heavy tobacco exposure and is clinically characterized by a rapid growth and early metastatic widespread. SCLC molecular hallmarks consist of high mutation rates, universal TP53 and RB1 gene inactivation, and rare oncogenic drivers [2].
Around 70% of cases present with extensive-stage disease at diagnosis (ED-SCLC); the remaining 30% of patients have limited-stage disease (LS-SCLC), in which tumor involvement is confined to one hemithorax and can be treated in a tolerable radiation field [3,4,5]. The overall prognosis for patients with SCLC is poor, with a median overall survival (OS) of 15–20 months for LD-SCLC and 8–13 months for ED-SCLC [3,4,5].
First-line treatment for SCLC patients includes platinum–etoposide doublet. Despite the initial high responses (up to 75%), most of the ED-SCLC patients will progress during the first months (platinum resistant < 3 months, platinum sensitive ≥ 3 months), achieving a median progression-free survival (PFS) of only 5.5 months and a median OS of < 10 months [3,4,5]. Subsequent-line treatment options are limited. No therapy has improved on the 15–20% response rate (RR) and 30% 1-year OS provided by second-line topotecan [3,4,5]. There is no standard of care beyond second-line therapy.
Systemic therapy for SCLC patients has not changed substantially in several decades. Consequently, there is an urgent medical need to bring new treatment options to SCLC patients. This review discusses the rationale for using immunotherapy in SCLC and provides an overview of the immunotherapeutic agents under clinical investigation for SCLC.
Rationale for immunotherapy in SCLC
Adaptive immune response is the mechanism for which the immune system is capable of detecting and eradicating tumor cells [6]. SCLC has long been considered immunogenic because of the occurrence of paraneoplastic disorders, such as Lambert–Eaton myasthenic syndrome (LEMS), that result from the consequence of an immune response targeting antigens expressed by both SCLC and healthy neurons (e.g., HuD, HuC, and Hel-N1) [7]. In some instances, SCLC patients with paraneoplastic disorders have a better prognosis, perhaps because the immune response generated against the nervous system is also targeting tumor cells [8].
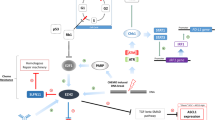
Several lines of evidence suggest that an ongoing, albeit suppressed, immune response is mounted against SCLC tumors [9, 10]. Differential tumor-infiltrating immune cell populations can affect SCLC prognosis (Fig. 1). In fact, CD4+ T immune effector T cells (Teff) are significantly higher in LD-SCLC patients than that of ED-SCLC patients including more IL-17-producing CD4+ T cells (Th17) [11]. Long-term survivors of SCLC maintained a high Teff to regulatory T (Treg) cell ratio, whereas patients with recurrent disease exhibited a low Teff to Treg cell ratio, suggesting a role of inducing Teff cells, particularly Th17 cells, while eliminating Treg cells to control systemic dissemination of SCLC [11]. In addition, expression of PD-L1 in tumor cells could be indicative of active T cell responses, and PD-L1 positive tumors have been associated with longer survival in SCLC patients [12].

Rationale for immunotherapy in SCLC. SCLC small cell lung cancer, PD-L1 programmed death-ligand 1, MHC major histocompatibility complex, TIL tumor infiltrating lymphocytes, Teff T effector, Treg T regulator, TIM-3 T cell immunoglobulin and mucin-domain containing-3, LAG3 lymphocyte-activation gene 3
SCLC is one of the tumors with the highest rate of somatic mutations [2, 13,14,15], including mutations in DNA repair mechanisms [16]. Tumors with more somatic mutations result in higher likelihood to develop tumor-specific neo-antigens that may ultimately trigger an adaptive immune response [17, 18]. Interestingly, somatic hypermutation and neoepitope formation have been associated with response to immunotherapy [18, 19]. Deficits in DNA mismatch repair genes lead to multiple gene mutations, manifested as microsatellite instability (MSI). MSI is linked to an increased benefit for PD-1 immune-checkpoint blockade [20]. In addition, tumor mutational burden (TMB) has been associated as an independent predictor of response to immunotherapy in different tumor types [21].
Despite the high somatic tumor mutational rate, SCLC has a highly immunosuppressive phenotype. SCLC cell lines and tumors are likely to have low expression of the class I major histocompatibility antigens HLA-A, B, C and beta 2-microglobulin [22]. HLA loss allows small cell lung cancer cells to evade the host immune response to the tumor and its association with intrinsic resistance to immune-checkpoint inhibitors is well established. In addition, no expression of class II major histocompatibility was found in SCLC tumors and tumor-infiltrating lymphocytes (TILs) present in SCLC tumors [23], suggesting an additional mechanism for evading the host immune response. Moreover, despite its high TMB, the level of TILs in SCLC is low and the ratio of CD8/CD3 is prominently low [24]. Moreover, immature myeloid cells have been implicated in the immunosuppressive state, making SCLC patients less likely to develop immune responses [25]. In spite of low expression of PD-L1, TIM3, and LAG3 by tumor cells, those immune checkpoints are frequently overexpressed in SCLC-associated TILs [26]. TIL expression of immune-checkpoint molecules is correlated with high FoxP3 expression and improved outcome in SCLC [26].
Immunotherapy in SCLC
There are different types of immunotherapy that have been investigated for the treatment of SCLC. We will review the use of immune-checkpoint inhibitors, cancer vaccines, and the use of cytokines. In addition, novel immunotherapeutic approaches and combinations will also be addressed (Fig. 2).

Overview of selected types of immunotherapy for SCLC. SCLC small cell lung cancer, CTLA-4 cytotoxic T-lymphocyte antigen-4, PD-1 programmed death-1, PD-L1 PD ligand-1, TLR toll-like receptor, MHC major histocompatibility complex, DLL3 Notch ligand delta-like protein 3, NK natural killer, INF interferon
Cytokines
Cytokines directly stimulate immune effector at the tumor site influencing immune cell activity. Two common cytokines are used in cancer immunotherapy and have been tested in SCLC: interferons (INFs) and interleukins (ILs) (Table 1).
Interleukin (IL)-2
High-dose IL-2 prompted to durable objective responses in a minority of patients with melanoma and renal cell carcinoma, serving as proof of principle that the immune system could eliminate cancer cells. In a phase II trial from the cancer and leukemia group B (CALGB), 4 out of 24 (17%) patients with ED-SCLC who had failed to obtain a complete remission with chemotherapy did obtain a complete remission after therapy with IL-2 [27]. However, the substantial IL-2 toxicity and the lack of efficacy in a later trial discontinued the interest of IL-2 in SCLC [28].
Interferon (IFN)
IFN inhibits tumor cell growth, stimulates the immune response, and has antiproliferative activity.
IFN-α2a combined with first-line chemotherapy was evaluated in a randomized trial involving 90 SCLC patients [29]. Compared to chemotherapy, IFN-α added to chemotherapy did not only increase the RR but also prolonged OS. This benefit seemed to be limited to patients with limited disease.
In a non-blinded, randomized, phase II study, 164 patients with SCLC were randomized to receive chemotherapy or immunotherapy plus chemotherapy [30]. Immunotherapy was divided into three arms: IFN-α, IFN-γ, and IFN-α plus IFN-γ. No differences in response and survival were observed, with the exception of the IFN-α arm but only in patients with limited stage, with few patients in this group to draw formal conclusions (n = 41). Tolerance was also worse in the combination arms (characterized by fever, anorexia and fatigue), and also with more neutropenia with IFN-γ [30].
Maintenance with IFNs after radical treatment was initially suggested to have a role in survival for SCLC [31]. However, in several randomized trials, both IFN-γ and IFN-α2a failed to prolong survival in SCLC patients that achieved complete remission after induction chemotherapy ± consolidation radiotherapy [32,33,34].
In patients with recurrent SCLC, the addition to paclitaxel of INF-α plus modulation of BCL-2 by 13-cis-retinoic acid did not improve clinical outcomes in a single-arm phase II trial [35].
Vaccines
Vaccines harness the adaptive immune recognition of a specific tumor antigen to effect antitumor responses. There are different types of cancer vaccines that have been evaluated in SCLC patients.
Antigen vaccines
One approach is to use immunotherapy directed towards cell surface antigens that are selectively and highly expressed on SCLC tumors.
Ganglioside fucosyl GM1 is a monosialoganglioside with limited expression in normal tissues, but with high expression on the surface of tumor cells in SCLC [36]. Vaccination with a synthetic version of fucosyl GM1 conjugated to keyhole limpet hemocyanin (KLH) was evaluated in patients with SCLC after a major response to initial therapy. Synthetic fucosyl GM1–KLH conjugate at a dose of 30 μg induced an IgM antibody response against fucosyl GM1 and tumor cells expressing fucosyl GM1 [37]. The most common toxicity was injection site reaction observed in 87% of patients.
Polysialic acid (polySA) is a polymer side chain bound to the neural cell adhesion molecule that is extensively expressed on the surface of SCLC cells. KLH-conjugated N-propionylated-(NP) polySA vaccine induced a robust antibody response. However, peripheral neuropathy and ataxia were limiting toxicities at a dose of 30 μg [38]. A de-escalation dosing study established NP-polySA-KLH 10 μg as the lowest optimally immunogenic dose [39]. Self-limited grade 3 ataxia of unclear etiology was seen in 1 of 18 patients. The future plan is to incorporate NP-polySA into a polyvalent vaccine against SCLC with four glycolipid antigens also widely expressed in SCLC-GD2, GD3, fucosylated GM1, and globo H.
Dendritic cell vaccines
Active immunotherapy using dendritic cells (DCs) to deliver tumor antigens has also been evaluated in SCLC. The tumor suppressor gene, p53, has many features of an ideal tumor-associated antigen [40, 41], with encouraging results both preclinically and in humans with anti-wildtype-p53 cancer therapy [41,42,43]. DCs transduced with adenovirus expressing the wild-type p53 gene has been used in patients with ED-SCLC that received at least a prior platinum therapy [25]. Despite vaccination resulted in the development of p53-specific T cell responses in just over half of the treated patients (57.1%), only one radiological response was observed out of the 29 patients treated with the vaccine. Two important observations were made: first, patients with an increased presence of immature myeloid cells were less likely to develop responses to vaccination; second, an unusually high response rate to chemotherapy post-progression to the vaccine. Based on these results, the optimal use of p53 cancer vaccine might be in combination with chemotherapy rather than as single agent. Moreover, a recent phase II trial (NCT03406715) is evaluating whether adding a dendritic cell-based p53 vaccine to nivolumab and ipilimumab combination checkpoint inhibition will improve outcomes among patients with recurrent SCLC.
Immune-checkpoint inhibitors
The cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death-1 (PD-1) pathways are the most extensively studied immune-checkpoint pathways (Tables 2, 3) [9, 44,45,46,47]. Preclinical data demonstrate that treatment with antibodies specific for CTLA-4 can restore an immune response through increased accumulation and survival of memory T cells and depletion of Tregs [45]. The use of monoclonal antibodies (mAbs) to block either PD-1 or PD-L1 prevents the downregulation of T cell effector function, allowing T cells to mediate tumor cell death [48].
Ipilimumab
Ipilimumab is a fully human IgG1 anti-CTLA-4 monoclonal antibody.
Ipilimumab has been evaluated in combination with chemotherapy in newly diagnosed ED-SCLC in two phase II trials and in a phase III randomized clinical trial.
CA184-041 is a phase II randomized, double-blind trial, where 130 patients were randomized 1:1:1 to receive carboplatin plus paclitaxel in addition to placebo (control arm), or iplilimumab 10 mg/kg concurrent, or after 2 doses of carboplatin/paclitaxel (phased) [49]. In the phased ipilimumab arm, median immune-related (ir) PFS (primary endpoint) was 6.4 months vs. 5.3 months (P = 0.03), and median OS was 12.9 months vs. 9.9 months (P = 0.13) compared to paclitaxel and carboplatin alone. However, no improvement in efficacy endpoints (including irPFS, mWHO-PFS, OS, and tumor response) was noted with concurrent ipilimumab. Ipilimumab was associated with known immune-mediated adverse events. Grade 3/4 treatment-related adverse events (TRAEs) were higher in phased regimen (50%) vs. control (30%), although the rate of treatment discontinuation was similar in phased (5%) vs. control (9%).
An open-label, single arm, phase II trial evaluated the safety and efficacy of first-line ipilimumab (10 mg/kg iv) combined with carboplatin plus etoposide (ICE) in 42 patients with ED-SCLC [50]. The 1-year PFS by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. was 15.8% (primary endpoint). Median PFS was 6.9 months (95% CI 5.5–7.9), median irPFS was 7.3 months (95% CI 5.5–8.8) and median OS was 17.0 months (95% CI 7.9–24.3). Objective response by RECIST was 72.4%, and 84.8% by the immune-related response criteria (irRC). Grade 3 or higher adverse events developed in 89.7%, 69.2% related to ipilimumab. Ad hoc analysis related the presence of autoantibodies at baseline with improved outcomes and severe neurological toxicity.
CA184-156 is a randomized, double-blind phase III study that evaluated the efficacy and safety of ipilimumab or placebo plus etoposide and platinum in 1132 patients with newly diagnosed ED-SCLC [51]. There was no difference in median OS (primary endpoint) between patients receiving chemotherapy plus ipilimumab (n = 478) versus chemotherapy plus placebo (n = 476): 11.0 months vs. 10.9 months, respectively (hazard ratio 0.94, 95% CI 0.81–1.09, P = 0.3775). No differences in efficacy in terms of PFS or tumor responses were observed with the addition of ipilimumab. Diarrhea, rash, and colitis were more frequent with ipilimumab and the rate of treatment-related discontinuation was higher with chemotherapy plus ipilimumab (18% vs. 2% with chemotherapy plus placebo).
Based on the negative results of ipilimumab combined to chemotherapy, additional studies are currently evaluating ipilimumab in combination with anti-PD1 inhibitors in SCLC.
Nivolumab
Nivolumab is a fully human IgG4 monoclonal antibody directed against PD-1.
CheckMate 032 is a phase I/II multicentre, multi-arm, open-label trial that included a cohort of patients with SCLC [52]. Patients with SCLC progressing after at least one platinum-containing therapy were allocated to three treatment arms: nivolumab plus ipilimumab [1 mg/kg + 1 mg/kg iv (n = 3), 1 mg/kg + 3 mg/kg iv (n = 61), and 3 mg/kg + 1 mg/kg iv (n = 54)] versus nivolumab monotherapy (3 mg/kg iv) (n = 98). Nivolumab plus ipilimumab was administered every 3 weeks for four cycles followed by nivolumab 3 mg/kg iv every other week. The primary end point was objective response per RECIST v1.1.
Objective response was achieved in 14/61 (23%) receiving nivolumab 1 mg/kg plus ipilimumab 3 mg/kg; 10/54 (19%) receiving nivolumab 3 mg/kg plus ipilimumab 1 mg/kg, 1/3 (33%) receiving nivolumab 1 mg/kg plus ipilimumab 1 mg/kg, and 10/98 (10%) patients in the nivolumab monotherapy arm.
The median duration of response (DoR) was 17.9 months for nivolumab monotherapy, and 14.2 months with nivolumab 1 mg/kg plus ipilimumab 3 mg/kg. Patients with ongoing responses at 2 years were 45% for nivolumab monotherapy and 36% in the combination. PD-L1 expression was assessable in 148 of 216 patient samples (69%), of which 25 (17%) had tumoral PD-L1 expression ≥ 1%. Responses were observed regardless of platinum sensitivity, line of therapy, or PD-L1 status. Two-year OS rates were 14% for nivolumab monotherapy and 26% in the combination arm, with a median OS of 4.1 (95% CI 3.0–6.8) and 7.8 (95% CI 3.6–14.2), respectively. In a randomized, phase II cohort from CheckMate 032 to further evaluate nivolumab ± ipilimumab, the initial efficacy of 242 patients was consistent with that in the non-randomized cohort [53].
Grade ≥ 3 toxicities occurred in 18/61 (30%) in the nivolumab 1 mg/kg plus ipilimumab 3 mg/kg group, 10/54 (19%) in the nivolumab 3 mg/kg plus ipilimumab 1 mg/kg, and 13/98 (13%) in the nivolumab monotherapy. Six (6%) patients in the nivolumab 3 mg/kg group, seven (11%) in the nivolumab 1 mg/kg plus ipilimumab 3 mg/kg group, and four (7%) in the nivolumab 3 mg/kg plus ipilimumab 1 mg/kg group discontinued treatment due to TRAEs. Four patients who received nivolumab plus ipilimumab died from TRAEs (myasthenia gravis, pneumonitis, encephalitis and hepatitis), and one patient who received nivolumab monotherapy died from treatment-related pneumonitis.
Based on the antitumour activity with durable responses and safety profile reported in CheckMate 032 trial, nivolumab ± ipilimumab is already included in the NCCN guidelines for the treatment of relapsed SCLC [54].
In an attempt to better predict clinical outcomes in SCLC patients treated with nivolumab ± ipilimumab, whole-exome sequencing was performed to evaluate the impact of tumor mutational burden (TMB) [15]. TMB was evaluable in 53% of the 401 ITT population. Patients with high TMB (cutoff 248 mut/Mb) had improved ORR, PFS, and OS compared with low/medium TMB for both nivolumab monotherapy and nivolumab + ipilimumab. Furthermore, nivolumab + ipilimumab appeared to provide a greater clinical benefit compared with nivolumab monotherapy in the high TMB tertile. Therefore, TMB is a potentially relevant biomarker that warrants cutoff optimization and prospective validation.
Several trials are currently evaluating nivolumab ± ipilimumab, including two randomized, phase III clinical trials: nivolumab versus chemotherapy (topotecan or amrubicin) in patients with relapsed SCLC (CheckMate 331, NCT02481830) [55]; nivolumab alone, nivolumab 1 mg/kg in combination with ipilimumab 3 mg/kg, or placebo as consolidation/maintenance therapy after completion of platinum-based first-line chemotherapy in patients with ED-SCLC (Checkmate 451, NCT02538666) [56]. However, in two recent press releases (October 12th, and November 26th, 2018, respectively), Bristol-Myers Squibb announced both CheckMate 331 and 451 studies did not meet their primary endpoint of overall survival, while disclose of results is still pending. Furthermore, a phase II trial of consolidation with nivolumab and ipilimumab in LS-SCLC after chemo-radiotherapy is ongoing (STIMULI, NCT02046733) [57].
Pembrolizumab
Pembrolizumab is an anti-PD1 humanized IgG4 antibody.
Pembrolizumab is being extensively studied in SCLC (Table 2): (1) in monotherapy in relapsed SCLC; (2) in combination with other chemotherapies in relapsed SCLC (e.g., paclitaxel, irinotecan, or amrubicin); (3) in combination with other immune-modulating agents; (4) concurrent or sequencial to radiation therapy and chemotherapy in patients with LS- or ED-SCLC; (5) face to face randomized clinical trials against standard of care topotecan in second-line, and to platinum plus etoposide in first-line ED-SCLC.
Pembrolizumab’s antitumor activity was initially evaluated in refractory, PD-L1-positive SCLC in a phase Ib trial (KEYNOTE 028) [58]. PD-L1 (22C3 antibody clone) was considered positive if membranous PD-L1 expression ≥ 1% of tumor and associated inflammatory cells or positive staining in stroma. The primary endpoint was ORR as per RECIST v1.1. Forty-six patients from 145 evaluable patients screened for PD-L1 expression were positive (31.7%). In total, 24 patients with PD-L1-positive ED-SCLC received pembrolizumab 10 mg/kg every 2 weeks up to 24 weeks or until progression. Up to 87.5% of patients enrolled in the trial received ≥ 2 previous lines, representing a heavily pretreated population. The ORR was 33.3% (95% CI 16%–55%) with a median time to response of 2 months (range 1.7–3.7 months), and a median DoR of 19.4 months (range 3.6–20+ months), that highlight both the rapid onset and the DoR to immunotherapy compared to short-lasting responses to chemotherapy in this setting. The median PFS and OS were 1.9 months (95% CI 1.7–5.9 months) and 9.7 months (95% CI 4.1 months—not reached), respectively. The 6- and 12-month OS rates were 66.0% and 37.7%, respectively. The most common adverse events were asthenia (n = 7), fatigue (n = 7), and cough (n = 6). Two patients experienced grade ≥ 3 TRAEs: one patient had elevated bilirubin, and one patient had asthenia, grade 5 colitis, and intestinal ischemia. The safety profile is consistent with previously known in other tumor types. There was no relationship between higher PD-L1 expression and frequency of response (P = 0.235).
A phase II basket trial is evaluating predictive biomarkers for pembrolizumab in 11 different tumor types (KEYNOTE 158, NCT02628067). The initial results from the SCLC cohort have been recently communicated [59]. Pembrolizumab was administered at a fixed dose of 200 mg iv every 3 weeks for a maximum of 2 years. In the overall SCLC patient cohort (n = 107), included few patients with carcinoid tumor (n = 1), and large cell neuroendocrine carcinoma (n = 7), the ORR (primary endpoint) was 18.7% (95% CI 11.8–27.4). Median DoR was not reached (range 2.1+ to 18.7+ months) and 73% of patients had a DoR of 12 months or longer. Median PFS was 2.0 months (95% CI 1.9–2.1), and median OS was 8.7 months (95% CI 5.6–12.0). PD-L1 (22C3) was found positive in 47% using a combined positive score (CPS) of ≥ 1 (defined as the ratio of PD-L1 positive cells including tumor cells, lymphocytes and macrophages to the total number of tumor cells x 100). None of the patients was MSI-H. Antitumor activity was particularly promising among patients PD-L1 positive (n = 42) compared to those PD-L1 negative (n = 50): ORR of 35.7% (95% CI 21.6–52.0) vs. 6% (95% CI 1.3–16.5), and median OS of 14.9 months (95% CI 5.6-NR) vs. 5.9 months (95% CI 3.3–10.1), with 12-month OS rates of 66.0% vs. 30.7%, respectively. TRAEs occurring in 10 percent or more of patients were fatigue (14%), pruritus (12%), hypothyroidism (12%), decreased appetite (10%) and nausea (10%). Thirteen patients had grade 3–4 TRAEs; two deaths occurred due to TRAEs (pneumonia and encephalopathy).
Pembrolizumab has also been tested as maintenance therapy in ED-SCLC patients upon completion of first-line treatment in a phase II study (NCT02359019) [60]. With a mPFS of 1.4 months, maintenance pembrolizumab did not achieve its primary endpoint. Only 3/30 tumors evaluable (10%) had PD-L1 expression (≥ 1%) in the tumor cells. Patients with tumors with PD-L1 expression at the stromal interface had better outcomes: PFS 5.5 vs. 1.3 months and OS 10.1 vs. 7.2 months.
Finally, pembrolizumab is being evaluated in patients with newly diagnosed ED-SCLC in combination with chemotherapy (cisplatin/carboplatin plus etoposide) in an ongoing phase III, randomized, double-blind, placebo-controlled trial (KEYNOTE 604, NCT03066778) [61]. Pembrolizumab 200 mg iv every 3 weeks or placebo is administered in combination to chemotherapy and thereafter until 2 years. Coprimary endpoints are PFS per RECIST 1.1 as assessed by blinded independent central review (BICR) and OS.
Atezolizumab
Atezolizumab is humanized, IgG1 monoclonal antibody that binds to PD-L1 and blocks interactions with the PD-1 and B7-1 receptors.
Atezolizumab showed encouraging single-agent activity in pretreated ED-SCLC patients in a phase Ia study of atezolizumab for locally advanced/metastatic solid tumors [62]. A total of 17 patients with ED-SCLC were enrolled in the study to receive atezolizumab 15 mg/kg or 1200 mg every 3 weeks; 65% of the patients were heavily pretreated (≥ 3 prior therapies). Confirmed ORR by RECIST was 6% and 24% by irRC. mPFS by RECIST was 1.5 months (95% CI 1.2–2.7), and mOS was 5.9 months (95% CI 4.3–20.1). Grade > 2 TRAEs occurred in two patients (12%): grade 3 pneumonitis resulted in treatment discontinuation for one patient; and one patient experienced a grade 5 hepatic failure. PD-L1 expression (IHC) was overall low, consistent with published data. A trend towards greater clinical benefit was seen for higher expression (≥ median) of PD-L1 mRNA and T effector (Teff) gene signature (CD8A, GZMA, GZMB, EOMES, CXCL9, CXCL10, TBX21).
IFCT-1603 study is a randomized non-comparative phase II trial in second-line SCLC, where atezolizumab as single agent did not show any efficacy signal in unselected population [63]. A total of 73 patients were assigned 2:1 to atezolizumab (n = 49) or chemotherapy (either topotecan or retreatment with carboplatin plus etoposide if indicated, n = 24). Only one patient in the atezolizumab arm [2.3%, CI (0.0; 6.8)] had a confirmed response at 6 weeks and did not meet the primary endpoint of the study. Moreover, progression-free survival was significantly shorter with atezolizumab compared to chemotherapy: 1.4 months, (CI 1.2–1.5) and 4.2 months (CI 1.5–5.9), respectively [HR 2.26 (1.30–3.93); p = 0.004]. Retrospective analysis on PD-L1 expression did not predict for patients with sustained disease control. No new safety signals were reported.
IMpower133 (NCT02763579) is a global, phase I/III, randomized, multicenter, double-blinded, placebo-controlled trial that evaluated the efficacy and safety of first-line atezolizumab in combination with carboplatin plus etoposide in treatment-naive patients with ED-SCLC [65]. A total of 403 eligible patients regardless of PD-L1 expression status were randomized 1:1 to receive four 21-day cycles of atezolizumab (1200 mg IV) or placebo in combination with carboplatin (AUC 5 mg/mL/min IV, d1) and etoposide (100 mg/m2, d1–3 IV), followed by maintenance therapy with atezolizumab or placebo until progression per RECIST v1.1. This study met its coprimary endpoints of OS and investigator-assessed PFS at its first interim analysis [65]. After a median follow-up of 13.9 months, median OS was 12.3 months (95% CI 10.8–15.9) in the atezolizumab arm compared with 10.3 months (95% CI 9.3–11.3) in the placebo arm (HR 0.70, 95% CI 0.54–0.91, P = 0.0069). Median PFS was 5.2 months (95% CI 4.4–5.6) in the atezolizumab group compared with 4.3 months (95% CI 4.2–4.5) in the placebo group (HR 0.77, 95% CI 0.62–0.96, P = 0.017). Atezolizumab was associated with a higher 6-month PFS rate (30.9% vs. 22.4%), and a more than doubling 12-month PFS rate (12.6% vs. 5.4%) compared with placebo. Exploratory analysis of blood-based TMB lacked for prediction on OS and PFS for prespecified cutoffs. Safety was consistent with the known chemotherapy and atezolizumab. The most common grade 3 or 4 TRAEs were neutropenia (23%), anemia (14%), decreased neutrophil count (14%), and thrombocytopenia (10%). Immune-related adverse events occurred in 40% in the atezolizumab group and in 24.5% in the placebo group, with rash (19%), hypothyroidism (13%), and hepatitis (7%) being the most common. Hence, atezolizumab in combination with chemotherapy is the first positive phase III trial to increase survival for the initial treatment of ED-SCLC in decades and represents a new standard of care for this disease.
Durvalumab–tremelimumab
Durvalumab is an anti-PD-L1 human IgG1 mAb. Tremelimumab (formerly ticilimumab, CP-675,206) is a fully human IgG2 monoclonal antibody targeting CTLA-4 (CD152). Durvalumab or the combination of tremelimumab plus durvalumab is currently under evaluation in SCLC.
BALTIC (NCT02937818) is a phase II, open-label, multi-arm study to determine preliminary efficacy of novel combinations of immunotherapies or DNA damage repair inhibitors in platinum-refractory ED-SCLC (progressed during, or within 90 days of completing first-line platinum-based chemotherapy) [66]. Arm A of this study will evaluate durvalumab 1500 mg + tremelimumab 75 mg iv q4w for 4 doses, followed by durvalumab monotherapy 1500 mg iv q4w. The primary end point is ORR. The secondary end points are DoR, PFS, OS, safety and tolerability.
CASPIAN (NCT03043872) is a phase III, randomized, multicenter, open-label study to determine the efficacy of durvalumab or durvalumab and tremelimumab in combination with platinum-based chemotherapy for first-line treatment in patients with ED-SCLC [67]. Patients will be randomized 1:1:1 to receive durvalumab (1500 mg) + tremelimumab (75 mg) iv every 3 weeks + chemotherapy; durvalumab (1500 mg) iv q3w + chemotherapy; or chemotherapy alone. Co-primary endpoints are investigator-assessed PFS per RECIST v1.1 and OS. Both BALTIC and CASPIAN studies are ongoing, and the first results are expected for 2019–2020.
Durvalumab–olaparib
High mutation load and genomic instability are two key features of SCLC. Preclinical data show that the inhibition of the enzyme poly-ADP ribose polymerase (PARP) upregulates PD-L1 expression and may further enhance the cancer-associated immunosuppression [68]. Olaparib is FDA-approved oral PARP inhibitor for germline BRCA-mutated (gBRCAm) advanced ovarian cancer, and gBRCAm breast cancer. MEDIOLA (NCT02734004) is a phase I/II, open-label, basket trial of durvalumab in combination with olaparib in advanced solid tumors [69]. Patients with relapsed SCLC received olaparib monotherapy (300 mg bid) for 4 weeks, then olaparib (300 mg bid) plus durvalumab (1500 mg iv q4w) until disease progression. Primary objectives were disease control rate (DCR) at 12 weeks, safety and tolerability. DCR at 12 weeks was 29% (7/38 patients) and did not meet the prespecified futility threshold. ORR was 11%, with all responses occurred prior to the addition of durvalumab (median DoR 4.4 months). Median PFS was 3.0 months (95% CI 2.4–4.6), and median OS was 8.8 months (95% CI 5.6-NC). The most frequent grade ≥3 AEs in this study were anemia (39.5%) and lymphopenia (13.2%) and TRAEs were in line with those previously reported in olaparib and durvalumab studies. PD-L1 expression in both tumor cells and immune cells was relatively low. CD3/CD8/PD-L1 IHC did not significantly correlate with clinical outcomes.
Other immunotherapy agents and combinations with novel therapies
TLR9 agonist
Lefitolimod (MGN1703) is a DNA-based agonist of the Toll-like receptor 9 (TLR9) expressed in dendritic cells, which initiates immune surveillance activating IFN-α secretion and thereby stimulating monocytes, NK cells, T cells and NKT cells [70]. Its efficacy and safety profile were evaluated in the phase II trial-IMPULSE, where 102 patients with ED-SCLC were randomized to receive lefitolimod maintenance therapy (twice weekly, 60 mg, subcutaneously) or local standard of care, after objective response to four cycles of platinum-based first-line induction chemotherapy. Analysis of results showed no OS advantage in the ITT population (primary endpoint), but with an OS benefit signal in the subgroup of patients with a low number of activated CD86+ B cells (284 days vs. 231.5, HR 0.59, 95% CI 0.29–1.21). The hypothesis postulated is that a lower count of activated/regulatory B cells produces less inhibition of lefitolimod-induced antitumor effect. Lefitolimod showed a favorable safety profile, with cough (25%), asthenia (13.3%) and headache (21.7%) as the most common symptoms reported [71].
Notch signaling
The inhibitory Notch ligand Delta-like protein 3 (DLL3) has generated great interest in SCLC [2, 17]. There are several clinical studies evaluating antibodies targeting DLL3 (an atypical Notch receptor family ligand), highly expressed on the cell surface of SCLC and other neuroendocrine tumors. Rovalpituzumab Tesirine (Rova-T) is an antibody–drug conjugate composed of a humanized DLL3-specific IgG1 monoclonal antibody linked to a toxic DNA cross-linking agent that induces cell death upon internalized [72]. A phase I study of Rova-T in relapsed metastatic SCLC patients, showed promising results in both sensitive and refractory disease, in second and third line of treatment. PFS was 3.1 months (95% CI 2.7–4.1 months), and median OS of 4.6 months (95% CI 3.9–7.1 months). Exploratory analysis showed the greatest benefit in the subgroup of patients with a high level of DLL3 expression (IHC ≥ 50%), with a PFS and OS of 4.5 (95% CI 3.0–5.4) and 5.8 (95% CI 4.4–11.6) months, respectively, and a 38% RR compared to 18% in the overall population [73]. The recommended phase II dose and schedule is 0.3 mg/kg every 6 weeks, and the most frequent grade 3 or worse TRAEs were thrombocytopenia (12%), pleural effusion (8%), skin reaction (8%), and increased lipase (7%).
Multiple clinical studies with Rova-T are ongoing in SCLC, including a phase I study of the efficacy of Rova-T in the first-line setting in series or in combination with frontline chemotherapy in DLL3+ subjects (NCT02819999); a phase III with Rova-T as maintenance therapy after first-line platinum-based chemotherapy (MERU trial, NCT03033511); a second-line phase III trial compared to topotecan in DLL3-high tumors (TAHOE trial, NCT03061812); and a phase I/II study investigating the safety and efficacy of Rova-T in combination with nivolumab or nivolumab + ipilimumab in progressive disease after at least one platinum-based chemotherapy (NCT03026166). The results of a phase II single-arm pivotal study in third line DLL3-expressing SCLC has been recently presented (TRINITY trial, NCT02674568). Rova-T showed single-agent activity with a 16% ORR and 5.6-month median OS. Toxicity profile was consistent with the previously known from Rova-T, as serosal effusions (28% pleural, 12% pericardial), photosensitivity reactions (35%), peripheral edema (26%), fatigue (28%), and thrombocytopenia (22%). There were ten cases (3%) of drug-related deaths, including generalized edema, pneumonitis, ascites, liver injury, pleural effusion, pneumothorax, respiratory failure, and sepsis. Overall, the study failed to meet the primary endpoint threshold of 25% for best ORR [74].
Tarextumab (TRXT, OMP-59R5) is a fully human IgG2 monoclonal antibody targeting the Notch 2 and 3 receptors. Tarextumab in combination with platinum-based therapy failed to improve PFS, OS, and ORR in previously untreated ED-SCLC in a phase II study (PINNACLE), while patients treated with tarextumab experienced more toxicity (diarrhea, thrombocytopenia, fatigue, anemia, and nausea) [75]. Biomarker analysis of the Notch pathway gene activation did not find any predictive marker for tarextumab efficacy.
Passive immunotherapy with antiganglioside therapy
Fucosyl GM1
BMS-986012 is a first-in-class fully human immunoglobulin G1 mAb with enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) that binds with high affinity and specificity to fucosyl GM1 and exhibited preliminary activity in SCLC [76]. A phase I/II dose-escalation and expansion study of BMS-986012 in combination with nivolumab in patients with relapsed/refractory SCLC showed responses in 5 of 27 patients treated (ORR of 19%) but this antitumor activity was limited to patients with platinum-sensitive SCLC [77]. Most patients treated with BMS-986012 + nivolumab experienced low-grade (grade 1–2) TRAEs (pruritus was the most common adverse event). There was one case of grade 3 hepatic failure that led to discontinuation that was attributable to nivolumab. Overall, there was no evidence of clinically meaningful additive efficacy or toxicity over BMS-986012 or nivolumab monotherapy [77].
GD2 ganglioside
Dinutuximab is a disialoganglioside (GD2)-binding human/mouse chimeric mAb, that binds to the glycolipid GD2, a tumor-associated antigen expressed on cells surfaces, and induces cell lysis through ADCC and complement-dependent cytotoxicity [78, 79]. Dinutuximab was first approved for treatment of pediatric patients with neuroblastoma [80, 81]. A recent multicenter, open-label, randomized, phase II/III study is evaluating the efficacy and safety of dinutuximab in combination with irinotecan for second-line treatment in subjects with relapsed or refractory SCLC (NCT03098030).
Anti-CD47
Therapies targeting the CD47 axis have demonstrated success in preclinical models and are currently under investigation in clinical trials for both solid and haematologic malignancies [82, 83]. CD47 is a cell-surface molecule that promotes immune evasion by engaging signal-regulatory protein alpha (SIRPα), which serves as an inhibitory receptor on macrophages. CD47 is part of the innate immune system and mediates a “don’t eat me” signal that contributes to the resistance of tumor cells to be eradicated by phagocytosis [84]. Weiskopf et al. demonstrated that CD47 is highly expressed on the surface of human SCLC cells; and the disruption of the interaction of CD47 with SIRPα using anti-CD47 antibodies induces macrophage-mediated phagocytosis of human SCLC cells and cytotoxic T cell activation, and this would be the rationale for its potential use as an immunotherapy agent in SCLC [85].
Conclusions
SCLC is a challenging disease in the need of new therapeutic opportunities. The use of immunotherapy can benefit SCLC patients by generating effective antitumor responses in the host. Inspite of considering SCLC an immunogenic tumor with high somatic mutation rates due to tobacco exposure, SCLC also displays a very immunosuppressive phenotype that hinder therapeutic advances in immunotherapy. This may explain, in part, the recent conflicting results of the efficacy of immune-checkpoint inhibitors in SCLC.
Treatment with immune-checkpoint inhibitors against CTLA-4 and PD-1/PD-L1 are likely to change the paradigm of treatment of SCLC in a similar manner that they are already doing in NSCLC. Nivolumab ± ipilimumab, and pembrolizumab have already shown compelling activity in relapsed SCLC with the potential to prolong survival in this patient population. However, without patient selection, immune-checkpoint inhibitors given as monotherapy seem to be inferior to standard chemotherapy in relapsed SCLC so far. The same holds true when administered in the maintenance setting after first-line chemotherapy induction. In contrast, atezolizumab when combined with chemotherapy has achieved the milestone of being the first drug to improve survival in patients with newly diagnosed ED-SCLC. Overall, the side effects observed from immune-checkpoint inhibitors in SCLC are similar to the observed in other indications such as in NSCLC.
The search for biomarkers of clinical benefit to immune-checkpoint inhibitors in SCLC remains elusive. The role of selection of SCLC patients for anti-PD1/PD-L1 therapies according to PD-L1 expression is currently unknown and must await further analysis [86]. The use of TMB has the potential to identify those patients more likely to benefit from immunotherapy in SCLC and warrants prospective validation.
The results of ongoing large randomized trials along with future research and novel immunotherapeutics and new combinations are expected to define the final role of immunotherapy in the treatment algorithm for SCLC [87].
References
Alvarado-Luna G, Morales-Espinosa D. Treatment for small cell lung cancer, where are we now?—a review. Transl Lung Cancer Res. 2016;5(1):26–38.
George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. https://doi.org/10.1038/nature14664.
Rudin CM, Ismaila N, Hann CL, Malhotra N, Movsas B, Norris K, et al. Treatment of small-cell lung cancer: American society of clinical oncology endorsement of the American college of chest physicians guideline. J Clin Oncol. 2015;33(34):4106–11. https://doi.org/10.1200/jco.2015.63.7918.
Früh M, De Ruysscher D, Popat S, Crinò L, Peters S, Felip E, et al. Small-cell lung cancer (SCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl_6):99–105. https://doi.org/10.1093/annonc/mdt178.
Artal Cortés Á, Dómine Gómez M, Font Pous A, García Campelo R, Cobo Dolls M, Isla Casado D. SEOM clinical guidelines for the treatment of small-cell lung cancer. Clin Transl Oncol. 2010;12(1):27–31. https://doi.org/10.1007/s12094-010-0463-2.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. https://doi.org/10.1016/j.immuni.2013.07.012.
Darnell RB. Onconeural antigens and the paraneoplastic neurologic disorders: at the intersection of cancer, immunity, and the brain. Proc Natl Acad Sci USA. 1996;93(10):4529–36.
Maddison P, Newsom-Davis J, Mills KR, Souhami RL. Favourable prognosis in Lambert-Eaton myasthenic syndrome and small-cell lung carcinoma. Lancet. 1999;353(9147):117–8. https://doi.org/10.1016/S0140-6736(05)76153-5.
Spigel DR, Socinski MA. Rationale for chemotherapy, immunotherapy, and checkpoint blockade in SCLC: beyond traditional treatment approaches. J Thorac Oncol. 2013;8(5):587–98. https://doi.org/10.1097/JTO.0b013e318286cf88.
Wang W, Hodkinson P, McLaren F, Mackean MJ, Williams L, Howie SEM, et al. Histologic assessment of tumor-associated CD45(+) cell numbers is an independent predictor of prognosis in small cell lung cancer. Chest. 2013;143(1):146–51. https://doi.org/10.1378/chest.12-0681.
Koyama K, Kagamu H, Miura S, Hiura T, Miyabayashi T, Itoh R, et al. Reciprocal CD41 T-cell balance of effector CD62Llow CD41 and CD62LhighCD251 CD41 regulatory T cells in small cell lung cancer reflects disease stage. Clin Cancer Res. 2008;14(21):6770–9. https://doi.org/10.1158/1078-0432.Ccr-08-1156.
Ishii H, Azuma K, Kawahara A, Yamada K, Imamura Y, Tokito T, et al. Significance of programmed cell death-ligand 1 expression and its association with survival in patients with small cell lung cancer. J Thorac Oncol. 2015;10(3):426–30. https://doi.org/10.1097/JTO.0000000000000414.
Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104. https://doi.org/10.1038/ng.2396. https://www.nature.com/articles/ng.2396#supplementary-information.
Weiss GJ, Byron SA, Aldrich J, Sangal A, Barilla H, Kiefer JA, et al. A prospective pilot study of genome-wide exome and transcriptome profiling in patients with small cell lung cancer progressing after first-line therapy. PLoS One. 2017;12(6):e0179170. https://doi.org/10.1371/journal.pone.0179170.
Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell. 2018;33(5):853. https://doi.org/10.1016/j.ccell.2018.04.001.
Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. 2017;17:725. https://doi.org/10.1038/nrc.2017.87. https://www.nature.com/articles/nrc.2017.87#supplementary-information.
Sabari JK, Lok BH, Laird JH, Poirier JT, Rudin CM. Unravelling the biology of SCLC: implications for therapy. Nat Rev Clin Oncol. 2017;14:549. https://doi.org/10.1038/nrclinonc.2017.71.
Yarchoan M, Johnson III BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17:209. https://doi.org/10.1038/nrc.2016.154.
Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol. 2017. https://doi.org/10.3389/fimmu.2017.01679.
Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin Cancer Res. 2016;22(4):813–20. https://doi.org/10.1158/1078-0432.Ccr-15-1678.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–608. https://doi.org/10.1158/1535-7163.Mct-17-0386.
Doyle A, Martin WJ, Funa K, Gazdar A, Carney D, Martin SE, et al. Markedly decreased expression of class I histocompatibility antigens, protein, and mRNA in human small-cell lung cancer. J Exp Med. 1985;161(5):1135–51.
He Y, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, Yu H, et al. MHC class II expression in lung cancer. Lung Cancer. 2017;112:75–80. https://doi.org/10.1016/j.lungcan.2017.07.030.
Schalper KA, Carvajal-Hausdorf DE, McLaughlin JF, Altan M, Chiang AC, Velcheti V et al. Objective measurement and significance of PD-L1, B7-H3, B7-H4 and TILs in small cell lung cancer (SCLC). J Clin Oncol. 2016;34(15_suppl):8566. https://doi.org/10.1200/jco.2016.34.15_suppl.8566.
Antonia SJ, Mirza N, Fricke I, Chiappori A, Thompson P, Williams N, et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res. 2006;12(3 Pt 1):878–87. https://doi.org/10.1158/1078-0432.CCR-05-2013.
Rivalland G, Walkiewicz M, Wright GM, John T. Small cell lung cancer: the immune microenvironment and prognostic impact of checkpoint expression. J Clin Oncol. 2017;35(15_suppl):8569. https://doi.org/10.1200/jco.2017.35.15_suppl.8569.
Clamon G, Herndon J, Perry MC, Ozer H, Kreisman H, Maher T, et al. Interleukin-2 activity in patients with extensive small-cell lung cancer: a phase II trial of Cancer and Leukemia Group B. J Natl Cancer Inst. 1993;85(4):316–20.
Clamon G, Herndon J, Akerley W, Green M. Subcutaneous interleukin-2 as initial therapy for patients with extensive small cell lung cancer. Lung Cancer. 1998;19(1):25–9. https://doi.org/10.1016/S0169-5002(97)00070-6.
Zarogoulidis K, Ziogas E, Papagiannis A, Charitopoulos K, Dimitriadis K, Economides D, et al. Interferon alpha-2a and combined chemotherapy as first line treatment in SCLC patients: a randomized trial. Lung Cancer. 1996;15(2):197–205.
Zarogoulidis K, Ziogas E, Boutsikou E, Zarogoulidis P, Darwiche K, Kontakiotis T, et al. Immunomodifiers in combination with conventional chemotherapy in small cell lung cancer: a phase II, randomized study. Drug Des, Dev Ther. 2013;7:611–7. https://doi.org/10.2147/dddt.S43184.
Mattson K, Niiranen A, Pyrhonen S, Holsti LR, Holsti P, Kumpulainen E, et al. Natural interferon alfa as maintenance therapy for small cell lung cancer. Eur J Cancer. 1992;28a(8–9):1387–91.
Jett JR, Maksymiuk AW, Su JQ, Mailliard JA, Krook JE, Tschetter LK, et al. Phase III trial of recombinant interferon gamma in complete responders with small-cell lung cancer. J Clin Oncol. 1994;12(11):2321–6. https://doi.org/10.1200/jco.1994.12.11.2321.
Kelly K, Crowley JJ, Bunn PA Jr, Hazuka MB, Beasley K, Upchurch C, et al. Role of recombinant interferon alfa-2a maintenance in patients with limited-stage small-cell lung cancer responding to concurrent chemoradiation: a Southwest Oncology Group study. J Clin Oncol. 1995;13(12):2924–30. https://doi.org/10.1200/jco.1995.13.12.2924.
van Zandwijk N, Groen HJ, Postmus PE, Burghouts JT, ten Velde GP, Ardizzoni A, et al. Role of recombinant interferon-gamma maintenance in responding patients with small cell lung cancer. A randomised phase III study of the EORTC Lung Cancer Cooperative Group. Eur J Cancer. 1997;33(11):1759–66.
Pillai RN, Aisner J, Dahlberg SE, Rogers JS, DiPaola RS, Aisner S, et al. Interferon alpha plus 13-cis-retinoic acid modulation of BCL-2 plus paclitaxel for recurrent small-cell lung cancer (SCLC): an Eastern Cooperative Oncology Group study (E6501). Cancer Chemother Pharmacol. 2014;74(1):177–83. https://doi.org/10.1007/s00280-014-2427-7.
Shengle Z, Carlos C-C, Zhang S, Reuter VE, Sucharita A, Bradley HW, et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int J Cancer. 1997;73(1):42–9. https://doi.org/10.1002/(sici)1097-0215(19970926)73:1%3c42:aid-ijc8%3e3.0.co;2-1.
Krug LM, Ragupathi G, Hood C, Kris MG, Miller VA, Allen JR, et al. Vaccination of patients with small-cell lung cancer with synthetic fucosyl GM-1 conjugated to keyhole limpet hemocyanin. Clin Cancer Res. 2004;10(18 Pt 1):6094–100. https://doi.org/10.1158/1078-0432.CCR-04-0482.
Krug LM, Ragupathi G, Ng KK, Hood C, Jennings HJ, Guo Z, et al. Vaccination of small cell lung cancer patients with polysialic acid or N-propionylated polysialic acid conjugated to keyhole limpet hemocyanin. Clin Cancer Res. 2004;10(3):916–23.
Krug LM, Ragupathi G, Hood C, George C, Hong F, Shen R, et al. Immunization with N-propionyl polysialic acid-KLH conjugate in patients with small cell lung cancer is safe and induces IgM antibodies reactive with SCLC cells and bactericidal against group B meningococci. Cancer Immunol Immunother. 2012;61(1):9–18. https://doi.org/10.1007/s00262-011-1083-6.
Chada S, Mhashilkar A, Roth JA, Gabrilovich D. Development of vaccines against self-antigens: the p53 paradigm. Curr Opin Drug Discov Devel. 2003;6(2):169–73.
Vierboom MP, Nijman HW, Offringa R, van der Voort EI, van Hall T, van den Broek L, et al. Tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. J Exp Med. 1997;186(5):695–704.
Zwaveling S, Vierboom MP, Ferreira Mota SC, Hendriks JA, Ooms ME, Sutmuller RP, et al. Antitumor efficacy of wild-type p53-specific CD4(+) T-helper cells. Cancer Res. 2002;62(21):6187–93.
Chiappori AA, Soliman H, Janssen WE, Antonia SJ, Gabrilovich DI. INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opin Biol Ther. 2010;10(6):983–91. https://doi.org/10.1517/14712598.2010.484801.
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–9. https://doi.org/10.1038/nature10673.
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–42. https://doi.org/10.1038/nri3405.
Apetoh L, Ladoire S, Coukos G, Ghiringhelli F. Combining immunotherapy and anticancer agents: the right path to achieve cancer cure? Ann Oncol. 2015;26(9):1813–23. https://doi.org/10.1093/annonc/mdv209.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252. https://doi.org/10.1038/nrc3239.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. https://doi.org/10.1146/annurev.immunol.26.021607.090331.
Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24(1):75–83. https://doi.org/10.1093/annonc/mds213.
Arriola E, Wheater M, Galea I, Cross N, Maishman T, Hamid D, et al. Outcome and biomarker analysis from a multicenter phase 2 study of ipilimumab in combination with carboplatin and etoposide as first-line therapy for extensive-stage SCLC. J Thorac Oncol. 2016;11(9):1511–21. https://doi.org/10.1016/j.jtho.2016.05.028.
Reck M, Luft A, Szczesna A, Havel L, Kim SW, Akerley W, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol. 2016;34(31):3740–8. https://doi.org/10.1200/JCO.2016.67.6601.
Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(7):883–95. https://doi.org/10.1016/s1470-2045(16)30098-5.
Hellmann MD, Ott PA, Zugazagoitia J, Ready NE, Hann CL, De Braud FG, et al. Nivolumab (nivo) ± ipilimumab (ipi) in advanced small-cell lung cancer (SCLC): first report of a randomized expansion cohort from CheckMate 032. J Clin Oncol. 2017;35(15_suppl):8503. https://doi.org/10.1200/jco.2017.35.15_suppl.8503.
National Comprehensive Cancer Network. Small Cell Lung Cancer (Version 2.2018). https://www.nccn.org/professionals/physician_gls/pdf/sclc.pdf. Accessed 15 Jun 2018.
Horn L, Reck M, Gettinger SN, Spigel DR, Antonia SJ, Rupnow BA, et al. CheckMate 331: An open-label, randomized phase III trial of nivolumab versus chemotherapy in patients (pts) with relapsed small cell lung cancer (SCLC) after first-line platinum-based chemotherapy (PT-DC). J Clin Oncol. 2016;34(15_suppl):TPS8578. https://doi.org/10.1200/jco.2016.34.15_suppl.tps8578.
Ready N, Owonikoko TK, Postmus PE, Reck M, Peters S, Pieters A, et al. CheckMate 451: A randomized, double-blind, phase III trial of nivolumab (nivo), nivo plus ipilimumab (ipi), or placebo as maintenance therapy in patients (pts) with extensive-stage disease small cell lung cancer (ED-SCLC) after first-line platinum-based doublet chemotherapy (PT-DC). J Clin Oncol. 2016;34(15_suppl):TPS8579. https://doi.org/10.1200/jco.2016.34.15_suppl.tps8579.
De Ruysscher D, Pujol JL, Popat S, Reck M, Le Pechoux C, Liston A, et al. STIMULI: a randomised open-label phase II trial of consolidation with nivolumab and ipilimumab in limited-stage SCLC after standard of care chemo-radiotherapy conducted by ETOP and IFCT. Ann Oncol. 2016;27(suppl_6):1430TiP-TiP. https://doi.org/10.1093/annonc/mdw389.08.
Ott PA, Elez E, Hiret S, Kim DW, Morosky A, Saraf S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the phase Ib KEYNOTE-028 Study. J Clin oncol. 2017;35(34):3823–9. https://doi.org/10.1200/JCO.2017.72.5069.
Chung HC, Lopez-Martin JA, Kao SC-H, Miller WH, Ros W, Gao B et al. Phase 2 study of pembrolizumab in advanced small-cell lung cancer (SCLC): KEYNOTE-158. J Clin Oncol. 2018;36(15_suppl):8506-. https://doi.org/10.1200/jco.2018.36.15_suppl.8506.
Gadgeel SM, Pennell NA, Fidler MJ, Halmos B, Bonomi P, Stevenson J, et al. Phase II study of maintenance pembrolizumab in patients with extensive-stage small cell lung cancer (SCLC). J Thorac Oncol. 2018. https://doi.org/10.1016/j.jtho.2018.05.002.
Rudin CM, Shen L, Pietanza MC. KEYNOTE-604: Phase 3 trial of pembrolizumab plus etoposide/platinum (EP) for first-line treatment of extensive stage small-cell lung cancer (ES-SCLC). Ann Oncol. 2017;28(suppl_5):mdx386.008. https://doi.org/10.1093/annonc/mdx386.008.
Sequist LV, Chiang A, Gilbert J, Gordon M, Conkling PR, Thompson D, et al. Clinical activity, safety and predictive biomarkers results from a phase Ia atezolizumab (atezo) trial in extensive-stage small cell lung cancer (ES-SCLC). Ann Oncol. 2016;27(suppl_6):1425PD-PD. https://doi.org/10.1093/annonc/mdw389.03.
Pujol JL, Greillier L, Audigier Valette C, Moro-Sibilot D, Uwer L, Hureaux J, et al. A randomized non-comparative phase II study of anti–PD-L1 atezolizumab or chemotherapy as second-line therapy in patients with small cell lung cancer: results from the IFCT-1603 trial. Ann Oncol. 2018;29(Suppl 8):1664O. https://doi.org/10.1093/annonc/mdy298.
Horn L, Reck M, Mok TSK, Johnson M, Waterkamp D, Lam S, et al. A Phase III study of atezolizumab with carboplatin plus etoposide in patients with extensive-stage small cell lung cancer (IMpower133). Ann Oncol. 2016;27(suppl_6):1431TiP-TiP. https://doi.org/10.1093/annonc/mdw389.09.
Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220–9. https://doi.org/10.1056/nejmoa1809064.
Pawel JV, Vynnychenko I, Jiang H, Huang Y, Dennis PA. A phase II, open-label, multi-arm study of novel combinations of immunotherapies or DDR inhibitors in platinum-refractory, extensive disease small-cell lung cancer (ED-SCLC): BALTIC. J Clin Oncol. 2017;35(15_suppl):TPS8585-TPS. https://doi.org/10.1200/jco.2017.35.15_suppl.tps8585.
Paz-Ares L, Jiang H, Huang Y, Dennis P. CASPIAN: phase 3 study of first-line durvalumab ± tremelimumab + platinum-based chemotherapy vs chemotherapy alone in ED-SCLC. J Thorac Oncol. 2017;12(11):S2398. https://doi.org/10.1016/j.jtho.2017.11.015.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–20. https://doi.org/10.1158/1078-0432.CCR-16-3215.
Krebs M, Ross K, Kim S, De Jonge M, Barlesi F, Postel-Vinay S, et al. An open-label, multitumor phase II basket study of olaparib and durvalumab (MEDIOLA): results in patients with relapsed SCLC. J Thorac Oncol. 2017;12(11):S2044–S5. https://doi.org/10.1016/j.jtho.2017.09.1040.
Schmidt M, Hagner N, Marco A, Konig-Merediz SA, Schroff M, Wittig B. Design and structural requirements of the potent and safe TLR-9 agonistic immunomodulator MGN1703. Nucleic Acid Ther. 2015;25(3):130–40. https://doi.org/10.1089/nat.2015.0533.
Thomas M, Ponce-Aix S, Navarro Mendivil A, Riera Knorrenschild J, Schmidt M, Krikow M, et al. Top-line data from the randomized phase 2 IMPULSE study in small-cell lung cancer (SCLC): Immunotherapeutic maintenance treatment with lefitolimod. Ann Oncol. 2017;28(suppl_5):mdx386. https://doi.org/10.1093/annonc/mdx386.
Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med. 2015;7(302):302ra136. https://doi.org/10.1126/scitranslmed.aac9459.
Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017;18(1):42–51. https://doi.org/10.1016/s1470-2045(16)30565-4.
Carbone DP, Morgensztern D, Moulec SL, Santana-Davila R, Ready N, Hann CL, et al. Efficacy and safety of rovalpituzumab tesirine in patients with DLL3-expressing, ≥ 3rd line small cell lung cancer: results from the phase 2 TRINITY study. J Clin Oncol. 2018;36(15_suppl):8507. https://doi.org/10.1200/jco.2018.36.15_suppl.8507.
Daniel DB, Rudin CM, Hart L, Spigel DR, Edelman MJ, Goldschmidt J, et al. Results of a randomized, placebo-controlled, phase 2 study of tarextumab (TRXT, anti-Notch2/3) in combination with etoposide and platinum (EP) in patients (pts) with untreated extensive-stage small-cell lung cancer (ED-SCLC). Ann Oncol. 2017;28(suppl_5):mdx386.004. https://doi.org/10.1093/annonc/mdx386.004.
Chu QSC, Markman B, Leighl N, Krug L, Rudin C, Lathers D, et al. A phase 1/2 trial of a monoclonal antibody targeting fucosyl GM1 in relapsed/refractory small cell lung cancer (SCLC): safety and preliminary efficacy. Ann Oncol. 2016;27(suppl_6):1427PD. https://doi.org/10.1093/annonc/mdw389.05.
Chu QSC, van Herpen C, Leighl NB, Markman B, Clarke S, Juergens RA, et al. Initial results of BMS-986012, a first-in-class fucosyl-GM1 mAb, in combination with nivolumab, in pts with relapsed/refractory (rel/ref) small-cell lung cancer (SCLC). Ann Oncol. 2017;28(suppl_5):mdx386.002. https://doi.org/10.1093/annonc/mdx386.002.
Mueller BM, Romerdahl CA, Gillies SD, Reisfeld RA. Enhancement of antibody-dependent cytotoxicity with a chimeric anti-GD2 antibody. J Immunol. 1990;144(4):1382–6.
Navid F, Santana VM, Barfield RC. Anti-GD2 antibody therapy for GD2-expressing tumors. Curr Cancer Drug Targets. 2010;10(2):200–9.
Dhillon S. Dinutuximab: first global approval. Drugs. 2015;75(8):923–7. https://doi.org/10.1007/s40265-015-0399-5.
Castel V, Segura V, Canete A. Treatment of high-risk neuroblastoma with anti-GD2 antibodies. Clin Transl Oncol. 2010;12(12):788–93. https://doi.org/10.1007/s12094-010-0600-y.
Weiskopf K. Cancer immunotherapy targeting the CD47/SIRPalpha axis. Eur J Cancer. 2017;76:100–9. https://doi.org/10.1016/j.ejca.2017.02.013.
Xiang Y-R, Liu L. Eating cancer cells by blocking CD47 signaling: cancer therapy by targeting the innate immune checkpoint. Cancer Transl Med. 2017;3(6):200–8. https://doi.org/10.4103/ctm.ctm_26_17.
Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. 2012;109(17):6662–7. https://doi.org/10.1073/pnas.1121623109.
Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest. 2016;126(7):2610–20. https://doi.org/10.1172/JCI81603.
Navarro A, Felip E. Pembrolizumab in advanced pretreated small cell lung cancer patients with PD-L1 expression: data from the KEYNOTE-028 trial: a reason for hope? Transl Lung Cancer Res. 2017;6(Suppl 1):S78–83. https://doi.org/10.21037/tlcr.2017.10.04.
Giaccone G, Debruyne C, Felip E, Chapman PB, Grant SC, Millward M, et al. Phase III study of adjuvant vaccination with Bec2/bacille Calmette-Guerin in responding patients with limited-disease small-cell lung cancer (European Organisation for Research and Treatment of Cancer 08971-08971B; Silva Study). J Clin Oncol. 2005;23(28):6854–64. https://doi.org/10.1200/jco.2005.17.186.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AC has received honorary/consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Eli Lilly and Company, Novartis, Merck Sharp & Dohme, and Bristol-Myers Squibb. The rest of the authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent is not applicable to this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Calles, A., Aguado, G., Sandoval, C. et al. The role of immunotherapy in small cell lung cancer. Clin Transl Oncol 21, 961–976 (2019). https://doi.org/10.1007/s12094-018-02011-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-018-02011-9