
Abstract
The potential role of the mitochondrial genome has recently attracted interest because of its high mutation frequency in tumors. Different aspects of mtDNA make it relevant for cancer‘s biology, such as it encodes a limited but essential number of genes for OXPHOS biogenesis, it is particularly susceptible to mutations, and its copy number can vary. Moreover, most ROS in mitochondria are produced by the electron transport chain. These characteristics place the mtDNA in the center of multiple signaling pathways, known as mitochondrial retrograde signaling, which modifies numerous key processes in cancer. Cybrid studies support that mtDNA mutations are relevant and exert their effect through a modification of OXPHOS function and ROS production. However, there is still much controversy regarding the clinical relevance of mtDNA mutations. New studies should focus more on OXPHOS dysfunction associated with a specific mutational signature rather than the presence of mutations in the mtDNA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Spotlight on mtDNA, why now? Characteristics that makes it relevant for cancer
Although a relationship between mitochondria, metabolism, and cancer was originally proposed by Warburg nearly a century ago [1], interest in the field has grown rapidly in recent years [2]. Mitochondria are semiautonomous organelles containing their own DNA, and are present in the vast majority of eukaryotic cells. Mitochondria play vital roles in a variety of cellular functions, including metabolism, energy production through the oxidative phosphorylation (OXPHOS) system, reactive oxygen species (ROS) generation and signaling, apoptosis, and calcium homeostasis [3]. Massive sequencing efforts in tumor and healthy tissue control pairs have identified numerous mutations in the mitochondrial DNA (mtDNA) of tumor cells, suggesting the involvement of mtDNA in malignant transformation; however, the pathological relevance of these findings remains controversial [4–6].
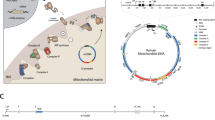
Human mtDNA is a circular double-stranded DNA molecule of approximately 16 kb in length that encodes a limited but essential number of genes for OXPHOS biogenesis [7] (Fig. 1a). The OXPHOS system transfers electrons from reduced cofactors to molecular oxygen and pumps protons from the mitochondrial matrix into the intermembrane space, generating a proton gradient that is used by complex V to generate ATP (Fig. 1b). Thus, the type and number of mtDNA mutations occurring can impact multiple facets of cellular bioenergetics, which is considered a cancer hallmark [2]. mtDNA is particularly susceptible to mutations compared to nuclear DNA. This phenomenon has been attributed to its proximity to sources of ROS, and consequent ROS-mediated oxidative damage, as well as the absence of nucleosome protection [8]. Over the past few years, however, earlier theories of oxidative-induced mtDNA mutations have been questioned by studies showing that defective replication plus less efficient repair machinery are largely responsible for somatic mtDNA mutations [9, 10]. Regardless of the source of these mutations, the increased mutation rate in mtDNA favors the appearance of variants that may result in a clonal advantage for cancer progression. Moreover, because mtDNA does not contain introns or intergenic spaces, the vast majority of mtDNA mutations affect coding regions necessary for OXPHOS function. Indeed, even mutations in the non-coding D-LOOP region can affect OXPHOS function by altering mitochondrial DNA copy number [11].

Mitochondrial DNA, oxidative phosphorylation system, and ROS metabolism. a Human mtDNA molecule encodes 37 genes, including: 7 subunits of complex I (ND1, 2, 3, 4, 4L, 5, and 6), 1 subunit of complex III (Cyt b), 3 subunits of complex IV (COX I, II, and III), 2 subunits of complex V (ATP6 and ATP8), 2 rRNAs (12S and 16S), and 22 tRNAs. In the last years, two small peptides have been described in the ORF of the 12S and 16S rRNAs, revealing new mitochondrial-derived peptides (MDPs) with signaling functions. b Oxidative phosphorylation (OXPHOS) system is composed of complex I (NADH dehydrogenase–CoQ reductase, CI), complex II (succinate dehydrogenase, CII), complex III (ubiquinone-cytochrome c oxidoreductase, CIII), complex IV (cytochrome oxidase, CIV), and complex V (ATP synthase, CV), plus two electron carriers: coenzyme Q (CoQ) and cytochrome C (CytC). Complexes I–IV transfer electrons from reduced cofactors to molecular oxygen to produce water through a chain of redox reactions, simultaneously pumping protons from the matrix to the intermembrane space to generate a proton gradient. Some of these reactions produce ROS as a byproduct, mainly CI and CIII. The proton gradient is dissipated across the mitochondrial inner membrane (MIM) back to the matrix, passing through complex V generating ATP. Note that CoQ also receives electrons from dihydroorotate dehydrogenase (DHOD) and electron transfer flavoprotein-ubiquinone oxidoreductase or electron transfer flavoprotein-dehydrogenase (ETFDH). Subunits encoded by mtDNA are in colors corresponding to panel A. c ROS homeostasis. Superoxide is mostly produced by the OXPHOS system and NOX proteins. Superoxide is transformed to hydrogen peroxide (H2O2) by superoxide dismutases (SODs). The H2O2 produced plays a central role in ROS dynamics, since it can generate the highly reactive hydroxyl radical (·OH), which produces oxidative damage and produces modifications in proteins by thiol oxidation, thus initiating complex signaling cascades, or be safely converted to water (H2O) by detoxification enzymes, such as catalase, peroxiredoxins (PRX), or glutathione peroxidases (GPX)
A key feature of the mitochondrial genome is its high copy number in cells (10–10,000 copies), a condition known as polyplasmia, and two scenarios are possible during cellular division, since mitochondria are randomly distributed between daughter cells: homoplasmy (identical molecules) or heteroplasmy (coexistence of different mtDNA variants). This property of mitochondria is key to understand the cellular consequences of mtDNA mutations. Thus, the final phenotype of the cell will depend not only on the severity of the mutation and the gene affected, but also on the percentage of heteroplasmy, a phenomenon known as the “threshold effect”. mtDNA copy number is strictly regulated and can vary among different tissues and also in response to environmental conditions to ensure that OXPHOS function is appropriate to the needs of the cell [12]. Interestingly, this adaptation mechanism is also used by tumor cells [13, 14], and correlations exist between changes in mtDNA copy number and the onset of different types of cancer, with higher levels associated with an increased risk for lymphoma, but a lower risk factor for bone cancer [15]. Moreover, it is also possible that the mtDNA copy number can vary with tumor progression or in response to treatments as a decline in the mtDNA content has been associated with lung cancer progression after neoadjuvant chemotherapy [16].
Recently, Reznik et al. analyzing data from the Cancer Genome Atlas (TCGA) consortium found alterations in mtDNA copy number in many tumor types when compared with adjacent normal tissue, with decreased tumor mtDNA levels observed for kidney (clear cell and papillary subtypes), breast, bladder, liver, head and neck squamous cell cancer, esophageal cancers, and increased levels observed only for lung adenocarcinoma [17]. In addition, the authors also found a correlation between mtDNA copy number and the incidence of key driver mutations. While interestingly, a clear limitation of these studies is the lack mtDNA mutation co-analysis, which may contribute to a more comprehensive evaluation.
mtDNA variants in cancer
mtDNA is strictly maternally transmitted [18], and throughout evolution, mtDNA has accumulated a high number of “neutral” nucleotide substitutions with no apparent consequences for OXPHOS function. In silico approaches have allowed the classification of the evolution of human mtDNA into distinct lineages or so-called mitochondrial haplogroups, comprising specific combinations of these polymorphisms. Although assumed to be neutral, mtDNA haplogroups have been linked to evolutionary adaptations to different climatic conditions. For example, haplogroups from polar regions have a slightly uncoupled electron transport chain (ETC) that increases heat production [19]. In addition, mtDNA haplogroups have been epidemiologically associated with different diseases, such as Alzheimer´s or Parkinson´s diseases [20].
The implication of mtDNA haplogroups to cancer is not entirely understood, but recent data have shown their associations with different types of cancer, including gastric [21], cervical [22], lung [23], breast [24–26], pancreatic [27], thyroid [28], and prostate and renal cancer [29]. The importance of mtDNA haplogroups has further been demonstrated in cellular models, suggesting an important role in OXPHOS performance [30], in modifying expression of the NAD-dependent deacetylase SIRT3 [31], and in the assembly of OXPHOS complexes [32]. Therefore, mtDNA haplogroups can influence OXPHOS function to favor or protect against the development of certain pathologies [33].
In addition to polymorphisms, about 300 mutations have thus far been identified in the mitochondrial genome, which are associated with classical mitochondrial diseases [8], and many somatic substitutions are known in human cancers with yet unclear consequences [4–6]. In both cases, changes have been shown to affect the function of ribosomal RNAs, transfer RNAs (tRNAs), and protein coding genes. Mutations in the latter principally affect a single OXPHOS complex, whereas mutations in tRNAs alter the translational capacity of all 13 mtDNA-encoding proteins, impacting four out of five OXPHOS complexes and resulting in a diversity of functional deficiencies [34]. Frame-shift mutations caused by insertions or deletions in mtDNA can be variable in length and can affect any region. These mutations typically have a serious impact on mitochondrial function. Interestingly, deletions in the D-LOOP region, which is involved in mtDNA replication and transcription, are highly represented in cancers with respect to other pathologies [35].
Moreover, a relationship has been described between cancer and proteins involved in mtDNA maintenance, such as POLG [36, 37], POLG2 [38], helicases [39], and DGUOK [40]. For example, due to an OXPHOS defect caused by hampered mtDNA replication, mutant POLG DNA polymerase expressed in breast cancer cells increases in vitro tumorigenicity [41], and mice heterozygous for the DNA helicase SUV3 develop tumors at multiple sites [42].
Although functional studies are required to fully understand the role of these mtDNA changes in cancer, clinical studies suggest that mtDNA mutations may influence disease prognosis [43–45], and indicate that severe mtDNA mutations are less frequent in cancer patients [5, 6].
Clearly, more comprehensive studies are necessary, taking into account different variables altogether, such as the type of mutation, its functional impact, heteroplasmy levels, tumor grade, relationship with nuclear mutations, as well as the role of low percentage germline mutations in the origin of the disease.
mtDNA and mitochondrial ROS
ROS are chemically reactive molecules containing oxygen which, in high amounts, can oxidize other molecules. Although several sources of ROS exist in cells, mitochondria are one of the main contributors to the ROS levels, thus determining the cellular redox status.
Traditionally, the origin of mitochondrial ROS was attributed entirely to the ETC; however, ROS production from several other mitochondrial enzymes has been described [46–48]. Most ROS in mitochondria are produced by reduction of oxygen (O2) to superoxide anion (O2 −) by complexes I and III [49]. Complex I releases the O2 − into the mitochondrial matrix, whereas complex III releases O2 − on both sides of the membrane. This evidently generates a different signaling potential depending on the origin of ROS [49]. Two O2 − molecules can then be converted to one molecule of hydrogen peroxide (H2O2) by different isoforms, mitochondrial, and cytoplasmic of the enzyme superoxide dismutase (SOD). H2O2, in turn, can accept an additional electron by the Fenton reaction to yield the highly reactive hydroxyl radical (OH−), or it can be reduced to H2O by various enzymes, such as glutathione peroxidases, peroxiredoxins, or catalases (Fig. 1b, c). H2O2 has the ability to cross biological membranes and is significantly more stable than other ROS allowing H2O2 to act as second messenger through oxidation of cysteine residues in proteins. Cysteine residues exist in equilibrium between the reduced thiol (Cys-SH) and the oxidized thiolate (Cys-S) forms; the latter may react with H2O2 to give a residue Cys-SOH. The oxidation of cysteine residues can directly affect the catalytic center of the protein or indirectly modify its activity by affecting regulatory residues, or its ability to interact with other molecules. These properties make ROS important signaling molecules, acting at multiple levels and regulating numerous physiological and pathological key processes involving proteins, such as GAPDH [50], NOX1 [51], ERK [52], NF-kB [53], AKT [54], HIF1α [55] and SRC [56], among others.
Conditions that alter the electron transport flow through the ETC, such as ETC complex inhibition [51], mutations in ETC subunits [57], the presence of regulatory elements [58], as well as defects in the assembly of individual complexes or supercomplexes [59], are associated with an increased production of mitochondrial ROS. In addition, some physiological processes, such as the induction of the complex I NDUFA4L2 subunit by hypoxia [60], the expression of uncoupling proteins [61], or ROS themselves modulating the transition between active and inactive forms of complex I, can also regulate the production of mitochondrial ROS [62].
The increased production of mitochondrial ROS has been proposed as a pathological mechanism in different mitochondrial and degenerative diseases [63], as well as a key element in the development of cancer [52, 64, 65].
The cybrid model to study mtDNA variants in cancer
Cytoplasmic hybrids, also known as transmitochondrial cybrids or cybrids, represent a model widely used to study the effects of mtDNA variants on cell physiology and human pathology. Cybrids are generated by fusing mtDNA-depleted cells (ρ0 cells) with donor cytoplasts, typically platelets, or enucleated fibroblasts [66] (Fig. 2). Cybrid analysis has been employed to determine the metabolic consequences of pathological OXPHOS defects, which are of key importance for elucidating pathogenic mechanisms of different mtDNA mutations [67].

Overall strategy for studies using transmitochondrial cybrid cell lines in cancer. Tumoral cell lines previously depleted of mtDNA (known as rho0 or ρ0 cells) are fused with mitochondria containing different mtDNAs (usually using platelets or cytoplasts of enucleated cells as source). After fusion, cybrid cells harboring the same nucleus from the parental ρ0 and mtDNA from the exogenous source are clone-selected and expanded. Although there are disputes about the role of mtDNAs variants in cancer biology among different studies, it seems clear that mtDNA is able to modify tumoral properties
The first studies involving mtDNA variants and cancer were carried out before the development of ρ0 cells and cybrid technology. In these seminal studies, the tumorigenic properties of cells could be modified by cytoplasm from normal cells [68, 69]. Later, both somatic mtDNA mutations found in cancer cells and pathogenic mtDNA mutations were studied using transmitochondrial cybrids. In a pioneering study, Hayashi et al. demonstrated that the tumorigenicity of HeLa cells depended on the presence of mtDNA, but the modulation of the phenotype was unaffected by mtDNA mutations [70]. Subsequently, the same authors showed that the metastatic potential of different mouse tumor cells was strictly dependent on mutated mtDNA. In these experiments, some mutations acted through an ROS-dependent mechanism, while others acted in a ROS-independent manner [64, 71].
The MT-ATP6 m.8993T>G mutation introduced into PC3 prostate cancer ρ0 cells gives rise to cells that generate tumors more efficiently and produce higher levels of ROS than those receiving wild-type mtDNA [72]. Two additional mutations in the MT-ATP6 gene, m.8993T>G and m.9176T>C, identified in patients with encephalomyopathy, have also been demonstrated to be critical for the capacity of transmitochondrial cybrids to generate tumors. Mutant mtDNAs conferred to cybrids from ρ0 HeLa cells an advantage in the early stage of tumor growth compared to wild-type mtDNA. These results also suggested that mutated mtDNA contributes to the promotion of tumors by preventing apoptosis [73].
Similar conclusions were reached using the nuclear background of the 143B osteosarcoma cell line. Cybrids carrying a frameshift mutation of MT-ND5 with different mutation load levels, 72 % heteroplasmy, and nearly mutant homoplasmy, exhibited striking differences in their tumorigenic properties. While cybrids harboring the heteroplasmic MT-ND5 mtDNA mutation produced tumors with significantly enhanced growth, tumor formation was inhibited in homoplasmic cybrids. These differences could also be attributed to an alteration of ROS production and apoptosis [74], and indicate that a severe impairment of mitochondrial function disrupts the development of tumors. Further studies with the 143B cellular background demonstrated that the transplantation of mitochondria from the benign breast epithelial cell line MCF10A, and from the moderately metastatic breast cancer cell line MDAMB-468, both reversed the tumorigenic properties of parental cells, indicating that benign mitochondria can revert the oncogenic potential of 143B cells. In addition, microarray studies have suggested that several oncogenic pathways observed in cybrids with cancerous mitochondria are inhibited in cybrids with non-cancerous mitochondria [75].
Contrastingly, recently published findings seem to indicate that mild mtDNA mutations do not increase tumorigenic potential. Cybrids harboring the m.3460G>A mtDNA mutation in complex I, with a mild functional impairment, had an equivalent tumorigenic potential to control cells, whereas cybrids with a severe complex I functional deficiency displayed a reduced tumorigenic potential [76]. These results are consistent with previous studies from the same group, in which they showed that mitochondrial function was necessary for the metabolic switch mediated by HIF1α and consequent tumorigenic behavior. In these studies, the tumorigenic capacity was lost through a high mutation load that profoundly affected mitochondrial function. Tumorigenicity was then restored when the mutation was complemented by the expression of the wild-type protein, recovering the mitochondrial function and the activation of HIF1α [77].
We have found that 143B ρ0 cells devoid of mtDNA, cybrids harboring wild-type mtDNA and cybrids causing severe mitochondrial dysfunction do not produce tumors. In contrast, cybrids containing mild mutant mtDNAs exhibit different tumorigenic capacities that are dependent on OXPHOS dysfunction [57]. These observed differences in tumorigenicity correlate with an enhanced resistance to apoptosis and high levels of ROS production. Nevertheless, the overall capacity of the different cybrid cell lines to generate tumors is most likely a consequence of a complex array of pro-oncogenic and anti-oncogenic factors associated with mitochondrial dysfunction [57]. In a similar manner, Yuan and coworkers found that missense and nonsense mutations in MT-ND6 promote tumorigenicity of the lung adenocarcinoma cell line A549 and are associated with reduced survival rate in patients [44].
The importance of maintaining some OXPHOS function for tumor progression has been elegantly demonstrated recently using metastatic murine tumor models depleted of mtDNA [78]. The authors showed that tumor ρ0 cell lines exhibited a long lag to tumor formation that was associated with the acquisition of mtDNA from host cells. Strikingly, they also noted a stepwise recovery of OXPHOS function throughout the process of tumor progression, from low levels in primary tumor cells derived from ρ0 cells, to full restoration in metastatic lung cells. This new mechanism of horizontal mtDNA transfer would expand the metabolic reprogramming capacity of tumor cells, a crucial process in situations, where profound changes in the tumor microenvironment take place, such as those occurring during therapeutic treatment or metastatic colonization [79].
Mitochondrial retrograde signaling and cancer
From cybrids studies, we have learned that the relevance of mitochondrial function and mtDNA in tumorigenicity involves complex signaling processes mediated not only by OXPHOS function, but also by ions, proteins, metabolites, and ROS [80–82]. One of the most illustrative examples of the influence of mtDNA on OXPHOS performance and nuclear reprogramming was shown by Pickard et al. in a study demonstrating how in a set of cybrids with increased heteroplasmy of the mutation m.3243G>A from a MELAS patient (0, 20, 30, 50, 60, 90 and 100 % of m.3243G>A mutated copies), the active status of several pathways (including glycolysis, antioxidant, and signaling pathways) changes according to the OXPHOS defect [83]. Interestingly, the pattern of many of these changes follows a U-shape, with similar alterations for the 0 and 100 % mutations containing cybrids but varying in between, which would reflect the different effect of mild mutations versus severe mutations or wild-type molecules in tumor promotion.
An increasing number of factors are now recognized as being responsive to mitochondrial function, including calcium [84], IkBβ [85], NOX [86], SRC [56], iron-sulfur cluster-containing proteins [87], AKT [88] and HIF1α [76], among others (Fig. 3). Moreover, for many of these, ROS are reported to play a key role, being one of the mitochondrial products described frequently in diverse publications in the area. Despite the increasing number of pathways implicated in mitochondrial retrograde signaling, a great deal of information is still to be discovered. For example, in the last years, a novel series of small open reading frames (ORFs) in the mtDNA sequence encoding polypeptides with signaling functions have been described [89, 90]. For example, humanin has anti-apoptotic activity [91] and has been implicated in cancer chemoresistance [92]. Another mechanism recently discovered is the so-called “moonlighting” of mitochondrial proteins in the nucleus. These proteins promote a rapid response to changes in OXPHOS function or ROS production and may directly link metabolic activity to genome integrity and gene expression [93].

Mitochondrial retrograde signaling caused by mtDNA variants. Schematic representation of the different pathways involved in the mitochondrial retrograde signal that may affect tumorigenic behavior described in the literature. We propose that small changes in OXPHOS function and ROS production trigger a complex mitochondrial retrograde response that ultimately enhances the tumorigenic phenotype: (↑) and (↓) indicate increase/activation or decrease/inhibition, respectively; (+) and (−) indicate increase/activation or decrease/inhibition mediated by ROS, respectively (modified from [57])
There is also the possibility that mtDNA changes are involved in the phenomenon of reprogramming of cancer stem cells (CSCs), since some metabolic phenotypes (metabotypes) are more prone to maintain a higher proportion of CSCs [94–96]. This could in part explain the observed differences in tumorigenicity depending on the mtDNA, as CSCs have been associated with a greater tumorigenic potential and with increased resistance to treatment [97, 98].
Concluding remarks
The rediscovery of the Warburg effect together with the finding of a higher frequency of mtDNA mutations in cancer and the recognition of mitochondrial signaling to the nucleus to control cellular reprogramming at different levels have placed mtDNA under the spotlight in cancer studies (Table 1).
Tumor cells use or modify cellular tools for their own benefit. The Warburg effect itself is the shift from OXPHOS to glycolytic metabolism under aerobic conditions to promote cell growth and proliferation, a mechanism shared with healthy proliferative cells in embryonic tissues [99]. It is, therefore, not surprising that mtDNA mutations affecting OXPHOS function, a common process in aging [9, 100], would represent an advantage for tumor cells.
Although controversy remains regarding the clinical relevance of mtDNA mutations, the majority of cybrid studies support the notion that moderate mutations promote tumorigenic phenotypes through different mechanisms, with increased ROS production representing a common factor in most cases. In addition, cybrid studies demonstrate that a complete lack of OXPHOS function is detrimental for tumorigenesis; an assertion that seems to be confirmed by studies in patients [5, 6].
It makes sense that cancer cells require some mitochondrial function, since it is essential for the basic cellular processes (particularly for proliferative cells with higher metabolic intermediates and energy requirements [101]). In addition, slightly impaired mitochondrial function that results in advantages in the processes of metabolic adaptation and ROS-mediated cellular signaling, likely also benefit cancer cells.
Regardless of the biological significance of these mutations, their presence could be used as tumor markers [102] in plasma [103–106], urine [107, 108], CSF [109], or NAF [110]. These mutations are easily detected in body fluids, because the mtDNA has a higher copy number than nuclear DNA; however, their clinical application is still to be validated. Monitoring mtDNA mutations in patients would be very interesting for cases without known driver mutations in those cancer types with a high number of somatic mtDNA mutations. There is no mutational hotspot associated with different cancer types; hence, sequencing the entire mitochondrial genome would be required to detect potential mutations. The lack of hotspots is most likely due to the nature of the OXPHOS system, in which different mutations produce the same defect.
For all the aforementioned reasons, we believe that further studies should focus more on finding a mutational signature based on OXPHOS alterations rather than a hotspot in a particular region, since the former is associated with a particular defect which is ultimately responsible for the retrograde signal and, therefore, the modulation of tumor behavior. These studies will shed light on the relationships between the different mtDNA mutations and other tumor variables, and this knowledge may allow for the development of new therapies and improved diagnosis or prognosis of cancer patients.
Abbreviations
- OXPHOS:
-
Oxidative phosphorylation
- ROS:
-
Reactive oxygen species
- mtDNA:
-
Mitochondrial DNA
References
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. Elsevier Inc.; 2011;144:646–74.
Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012/03/20 ed. Elsevier Inc.; 2012;148:1145–59.
Larman TC, DePalma SR, Hadjipanayis AG, Protopopov A, Zhang J, Gabriel SB, et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci USA. 2012;109:14087–91.
Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik-Zainal S, Ramakrishna M, et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife. 2014;3:1–28.
Stewart JB, Alaei-Mahabadi B, Sabarinathan R, Samuelsson T, Gorodkin J, Gustafsson CM, et al. Simultaneous DNA and RNA mapping of somatic mitochondrial mutations across diverse human cancers. PLoS Genet. 2015;11:e1005333.
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65.
Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012;226:274–86.
Itsara LS, Kennedy SR, Fox EJ, Yu S, Hewitt JJ, Sanchez-Contreras M, et al. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014;10:e1003974.
Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radic Biol Med Elsevier. 2015;85:250–8.
Liou C-W, Lin T-K, Chen J-B, Tiao M-M, Weng S-W, Chen S-D, et al. Association between a common mitochondrial DNA D-loop polycytosine variant and alteration of mitochondrial copy number in human peripheral blood cells. J Med Genet. 2010;47:723–8.
Campbell CT, Kolesar JE, Kaufman B a. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta. Elsevier B.V.; 2012;1819:921–9.
Guo J, Zheng L, Liu W, Wang X, Wang Z, Wang Z, et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011;71:2978–87.
van Osch FHM, Voets AM, Schouten LJ, Gottschalk RWH, Simons CCJM, van Engeland M, et al. Mitochondrial DNA copy number in colorectal cancer: between tissue comparisons, clinicopathological characteristics and survival. Carcinogenesis. 2015;36:1502–10.
Mi J, Tian G, Liu S, Li X, Ni T, Zhang L, et al. The relationship between altered mitochondrial DNA copy number and cancer risk: a meta-analysis. Sci. Rep. Nature Publishing Group; 2015;5:10039.
Lin C-S, Wang L-S, Tsai C-M, Wei Y-H. Low copy number and low oxidative damage of mitochondrial DNA are associated with tumor progression in lung cancer tissues after neoadjuvant chemotherapy. Interact CardioVasc Thorac Surg. 2008;7:954–8.
Reznik E, Miller ML, Şenbabaoğlu Y, Riaz N, Sarungbam J, Tickoo SK, et al. Mitochondrial DNA copy number variation across human cancers. Elife. 2016;5:1–20.
Pyle A, Hudson G, Wilson IJ, Coxhead J, Smertenko T, Herbert M, et al. Extreme-depth re-sequencing of mitochondrial DNA Finds no evidence of paternal transmission in humans. PLoS Genet. 2015;11:e1005040.
Ruiz-Pesini E, Mishmar D, Brandon M. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science (80-.). 2004;303:223–7.
Coskun P, Wyrembak J, Schriner SE, Chen H-W, Marciniack C, Laferla F, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta. Elsevier B.V.; 2012;1820:553–64.
Wang C, Wang Y, Wang H, Zhang R, Guo Z. Mitochondrial DNA haplogroup N is associated good outcome of gastric cancer. Tumour Biol. 2014;35:12555–9.
Kabekkodu SP, Bhat S, Mascarenhas R, Mallya S, Bhat M, Pandey D, et al. Mitochondrial DNA variation analysis in cervical cancer. Mitochondrion. © Elsevier B.V. and Mitochondria Research Society. All rights reserved.; 2014;16:73–82.
Wang Z, Choi S, Lee J, Huang Y-T, Chen F, Zhao Y, et al. Mitochondrial variations in non-small cell lung cancer (NSCLC) survival. Cancer Inf. 2015;14:1–9.
Weigl S, Paradiso A, Tommasi S. Mitochondria and familial predisposition to breast cancer. 2013;195–203.
Blein S, Bardel C, Danjean V, McGuffog L, Healey S, Barrowdale D, et al. An original phylogenetic approach identified mitochondrial haplogroup T1a1 as inversely associated with breast cancer risk in BRCA2 mutation carriers. Breast Cancer Res. 2015;17:61.
Bai R-K, Leal SM, Covarrubias D, Liu A, Wong L-JC. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007;67:4687–94.
Lam ET, Bracci PM, Holly E a, Chu C, Poon A, Wan E, et al. Mitochondrial DNA sequence variation and risk of pancreatic cancer. Cancer Res. 2012;72:686–95.
Fang H, Shen L, Chen T, He J, Ding Z, Wei J, et al. Cancer type-specific modulation of mitochondrial haplogroups in breast, colorectal and thyroid cancer. BMC Cancer. 2010;10:421.
Booker LM, Habermacher GM, Jessie BC, Sun QC, Baumann AK, Amin M, et al. North American White Mitochondrial Haplogroups in Prostate and Renal Cancer. J Urol. 2006;175:468–73.
Gómez-Durán A, Pacheu-Grau D, López-Gallardo E, Díez-Sánchez C, Montoya J, López-Pérez MJ, et al. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum Mol Genet. 2010;19:3343–53.
D’Aquila P, Rose G, Panno ML, Passarino G, Bellizzi D. SIRT3 gene expression: a link between inherited mitochondrial DNA variants and oxidative stress. Gene Elsevier B.V.; 2012;497:323–9.
Pello, Martin MA, Carelli V, Nijtmans LG, Achilli A, Pala M, et al. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum Mol Genet. 2008;17:4001–11.
Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, et al. Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: implications for population susceptibility to diseases. Biochim. Biophys. Acta. Elsevier B.V.; 2014;1842:208–19.
Abbott J a, Francklyn CS, Robey-Bond SM. Transfer RNA and human disease. Front Genet. 2014;5:158.
Damas J, Samuels DC, Carneiro J, Amorim A, Pereira F. Mitochondrial DNA rearrangements in health and disease—a comprehensive study. Hum Mutat. 2014;35:1–14.
Datta S, Ray A, Roy R, Roy B. Association of DNA sequence variation in mitochondrial DNA polymerase with mitochondrial DNA synthesis and risk of oral cancer. Gene. Elsevier B.V.; 2016;575:650–4.
Popanda O, Seibold P, Nikolov I, Oakes CC, Burwinkel B, Hausmann S, et al. Germline variants of base excision repair genes and breast cancer: a polymorphism in DNA polymerase gamma modifies gene expression and breast cancer risk. Int J Cancer. 2013;132:55–62.
Ratanajaraya C, Nishiyama H, Takahashi M, Kawaguchi T, Saito R, Mikami Y, et al. A polymorphism of the POLG2 gene is genetically associated with the invasiveness of urinary bladder cancer in Japanese males. J Hum Genet. 2011;56:572–6.
Ding L, Liu Y. Borrowing Nuclear DNA Helicases to Protect Mitochondrial DNA. Int J Mol Sci. 2015;16:10870–87.
Gandhi VV, Samuels DC. Correlated tissue expression of genes of cytoplasmic and mitochondrial nucleotide metabolisms in normal tissues is disrupted in transformed tissues. Nucleosides, Nucleotides Nucleic Acids. 2012;31:112–29.
Singh KK, Ayyasamy V, Owens KM, Koul MS, Vujcic M. Mutations in mitochondrial DNA polymerase-γ promote breast tumorigenesis. J. Hum. Genet. Nature Publishing Group. 2009;54:516–24.
Chen P-L, Chen C-F, Chen Y, Guo XE, Huang C-K, Shew J-Y, et al. Mitochondrial genome instability resulting from SUV3 haploinsufficiency leads to tumorigenesis and shortened lifespan. Oncogene. 2013;32:1193–201.
Yadav N, Chandra D. Mitochondrial DNA mutations and breast tumorigenesis. Biochim. Biophys. Acta - Rev. Cancer. Elsevier B.V.; 2013;1836:336–44.
Yuan Y, Wang W, Li H, Yu Y, Tao J, Huang S, et al. Nonsense and Missense Mutation of Mitochondrial ND6 gene promotes cell migration and invasion in human lung adenocarcinoma. BMC Cancer. 2015;15:346.
Xu H, He W, Jiang H-G, Zhao H, Peng X-H, Wei Y-H, et al. Prognostic value of mitochondrial DNA content and G10398A polymorphism in non-small cell lung cancer. Oncol Rep. 2013;30:3006–12.
Hey-Mogensen M, Goncalves RLS, Orr AL, Brand MD. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic Biol Med. 2014;72:149–55.
Fisher-Wellman KH, Gilliam LAA, Lin C-T, Cathey BL, Lark DS, Neufer PD. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radic Biol Med. 2013;65:1201–8.
Mailloux RJ. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol Elsevier. 2015;4C:381–98.
Bleier L, Wittig I, Heide H, Steger M, Brandt U, Dröse S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic Biol Med Elsevier. 2015;78:1–10.
Peralta D, Bronowska AK, Morgan B, Dóka É, Van Laer K, Nagy P, et al. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat Chem Biol 2015;11.
Desouki MM, Kulawiec M, Bansal S, Das GC, Singh KK. Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol. Ther. Landes Bioscience Inc.; 2005;4:1367–73.
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010/04/28 ed. 2010;107:8788–93.
Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2010/12/29 ed. 2011;21:103–15.
Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet. 2011/09/06 ed. 2011;20:4605–16.
Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. Elsevier Inc.; 2010;40:294–309.
Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014;8:754–66.
Cruz-Bermúdez A, Vallejo C, Vicente-blanco RJ, Gallardo ME, Fernandez-Moreno MA, Quintanilla M, et al. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget. 2015;6:13628–43.
Formentini L, Sanchez-Arago M, Sanchez-Cenizo L, Cuezva JM. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol Cell. 2012;45:731–42.
Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid Redox Signal. 2013/04/16 ed. 2013;19:1469–80.
Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordóñez Á, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14:768–79.
Mailloux RJ, Harper M-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. Elsevier Inc.; 2011;51:1106–15.
Dröse S, Brandt U, Wittig I. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim. Biophys. Acta. Elsevier B.V.; 2014;1844:1344–54.
Tuppen H a L, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta. Elsevier B.V.; 2010;1797:113–28.
Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–4.
Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14:709–21.
King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science (80-.). 1989/10/27 ed. 1989;246:500–3.
Yarham JW, Al-Dosary M, Blakely EL, Alston CL, Taylor RW, Elson JL, et al. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat. 2011;32:1319–25.
Howell a N, Sager R. Tumorigenicity and its suppression in cybrids of mouse and Chinese hamster cell lines. Proc Natl Acad Sci USA. 1978;75:2358–62.
Hayashi J, Werbin H, Shay JW. Effects of normal human fibroblast mitochondrial DNA on segregation of HeLaTG Mitochondrial DNA and on tumorigenicity of HeLaTG cells. Cancer Res. 1986;46:4001–6.
Hayashi J, Takemitsu M, Nonaka I. Recovery of the missing tumorigenicity in mitochondrial DNA-less HeLa cells by introduction of mitochondrial DNA from normal human cells. Somat Cell Mol Genet. 1992;18:123–9.
Imanishi H, Hattori K, Wada R, Ishikawa K, Fukuda S, Takenaga K, et al. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS One. 2011;6:e23401.
Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci USA. 2005;102:719–24.
Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, et al. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–63.
Park JS, Sharma LK, Li H, Xiang R, Holstein D, Wu J, et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum Mol Genet. 2009;18:1578–89.
Kaipparettu BA, Ma Y, Wong LJ. Functional effects of cancer mitochondria on energy metabolism and tumorigenesis: utility of transmitochondrial cybrids. Ann N Y Acad Sci. 2010;1201:137–46.
Iommarini L, Kurelac I, Capristo M, Calvaruso MA, Giorgio V, Bergamini C, et al. Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum Mol Genet. 2014;23:1453–66.
Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013;1:11.
Tan AS, Baty JW, Dong L-F, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. Elsevier Inc.; 2015;21:81–94.
Berridge MV, Dong L, Neuzil J. Mitochondrial DNA in tumor initiation, progression, and metastasis: role of horizontal mtDNA transfer. Cancer Res. 2015;75:3203–8.
Wallace DC. Mitochondria and cancer. Nat. Rev. Cancer. Nature Publishing Group; 2012;12:685–98.
Horan MP, Gemmell NJ, Wolff JN. From evolutionary bystander to master manipulator: the emerging roles for the mitochondrial genome as a modulator of nuclear gene expression. Eur J Hum Genet. 2013;21:1335–7.
Ward PS, Thompson CB. metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. Elsevier. 2012;21:297–308.
Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci USA. 2014;111:E4033–42.
Amuthan G, Biswas G, Zhang SY, Klein-Szanto a, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 2001;20:1910–20.
Tang W, Chowdhury AR, Guha M, Huang L, Van Winkle T, Rustgi AK, et al. Silencing of IkBβ mRNA causes disruption of mitochondrial retrograde signaling and suppression of tumor growth in vivo. Carcinogenesis. 2012;33:1762–8.
Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004;64:3580–5.
Veatch JR, McMurray M a, Nelson ZW, Gottschling DE. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell. Elsevier Ltd; 2009;137:1247–58.
Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol. 2006;175:913–23.
Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21:443–54.
Yen K, Lee C, Mehta H, Cohen P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J Mol Endocrinol. 2013;50:R11–9.
Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003/05/07 ed. 2003;423:456–61.
Mottaghi-Dastjerdi N, Soltany-Rezaee-Rad M, Sepehrizadeh Z, Roshandel G, Ebrahimifard F, Setayesh N. Genome expression analysis by suppression subtractive hybridization identified overexpression of Humanin, a target gene in gastric cancer chemoresistance. Daru. 2014/01/10 ed. 2014;22:14.
Monaghan RM, Whitmarsh AJ. mitochondrial proteins moonlighting in the nucleus. Trends Biochem. Sci. Elsevier Ltd; 2015;xx:1–8.
Ye X-Q, Li Q, Wang G-H, Sun F-F, Huang G-J, Bian X-W, et al. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int J Cancer. 2011;129:820–31.
Menendez JA, Alarcón T. Metabostemness: a new cancer hallmark. Front. Oncol. 2014;4:262.
Guha M, Srinivasan S, Ruthel G, Kashina AK, Carstens RP, Mendoza A, et al. Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene. Macmillan Publishers Limited; 2014;33:5238–50.
Yang M, Yan M, Zhang R, Li J, Luo Z. Side population cells isolated from human osteosarcoma are enriched with tumor-initiating cells. Cancer Sci. 2011;102:1774–81.
Martins-Neves SR, Lopes ÁO, do Carmo A, Paiva A a, Simões PC, Abrunhosa AJ, et al. Therapeutic implications of an enriched cancer stem-like cell population in a human osteosarcoma cell line. BMC Cancer. BioMed Central Ltd; 2012;12:139.
Heiden M Vander, Cantley L, Thompson C. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (80-.). 2009;324:1029–34.
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23.
Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3:1.
Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, et al. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science (80-.). 2000;287:1997–9.
Takeuchi H, Fujimoto A, Hoon DSB. Detection of mitochondrial DNA alterations in plasma of malignant melanoma patients. Ann N Y Acad Sci. 2004;1022:50–4.
Okochi O, Hibi K, Uemura T, Inoue S, Takeda S, Kanek T, et al. Detection of mitochondrial DNA alterations in the serum of hepatocellular carcinoma patients. Clin Cancer Res. 2002;8:2875–8.
Hibi K, Nakayama H, Yamazaki T, Takase T, Taguchi M, Kasai Y, et al. Detection of mitochondrial DNA alterations in primary tumors and corresponding serum of colorectal cancer patients. Int J Cancer. 2001;94:429–31.
Yu M, Wan YF, Zou QH. Cell-free circulating mitochondrial DNA in the serum: a potential non-invasive biomarker for Ewing’s Sarcoma. Arch Med Res Elsevier Inc; 2012;43:389–94.
Duberow DP, Brait M, Hoque MO, Theodorescu D, Sidransky D, Dasgupta S, et al. High-performance detection of somatic D-loop mutation in urothelial cell carcinoma patients by polymorphism ratio sequencing. 2016;94:1015–1024.
Jerónimo C, Nomoto S, Caballero OL, Usadel H, Henrique R, Varzim G, et al. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene. 2001;20:5195–8.
Wong LJC, Lueth M, Li XN, Lau CC, Vogel H. Detection of mitochondrial DNA mutations in the tumor and cerebrospinal fluid of medulloblastoma patients. Cancer Res. 2003;63:3866–71.
Zhu W, Qin W, Bradley P, Wessel A, Puckett CL, Sauter ER. Mitochondrial DNA mutations in breast cancer tissue and in matched nipple aspirate fluid. Carcinogenesis. 2005;26:145–52.
Acknowledgements
Work in the authors’ laboratories is supported by “Instituto de Salud Carlos III” [PI13/01806 and PIE14/0064 to M.P., PI10/0703 and PI13/00556 to R.G. and PI04/1001 to M.A.F.M.]; “Comunidad Autónoma de Madrid” [S2010/BMD-2402 to R.G.]; “Fundación Mutua Madrileña” [10.04.02.0064 to M.A.F.M.]. We thank Dr. Bruno Sainz Jr. for helpful suggestions to the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Cruz-Bermúdez, A., Vicente-Blanco, R.J., Gonzalez-Vioque, E. et al. Spotlight on the relevance of mtDNA in cancer. Clin Transl Oncol 19, 409–418 (2017). https://doi.org/10.1007/s12094-016-1561-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-016-1561-6