
Abstract:
Pseudomonas aeruginosa is an opportunistic pathogen causing severe respiratory infections. Acylated homoserine lactones (AHLs) are self-generated diffusible signal molecules that mediate population density dependent gene expression (quorum sensing, QS) in a variety of Gram-negative bacteria, and several virulence genes of bacterial pathogens are known to be controlled by QS. Hence, fitness mutant of virulent factors is beneficial for natural selection. In this study, strains of P. aeruginosa isolated from chronic lung infection of cystic fibrosis patients, were screened for AHLs production by using indicator strains of Chromobacterium violaceum CV026 and Agrobacterium tumefaciens strain At136. Four AHLs defective strains were selected from fifty-three clinical isolates. PCR analysis revealed that only one isolate was negative for lasR gene. These four AHLs defective isolates produced less virulence factors and forming less biofilm than PAO1. Only isolate PA41 produce little more pyocyanin than PAO1. The results indicate that, despite the pivotal role of QS in the pathogenesis of P. aeruginosa infections, AHLs-deficient strains are still capable of causing infections in human.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pseudomonas aeruginosa is an opportunistic pathogen that causes extensive morbidity and mortality in individuals who are immunocompromised or have underlying medical conditions such as urinary tract, respiratory tract and skin infections and primarily causes of nosocomial infections. This pathogen can secret many virulence factors (elastase, phospholipase C, lecitinase, rhamnolipids, toxins, hemolysins) as well as secondary metabolites (pyocyanin cyanide), and another factor contributing to pathogenesis of P. aeruginosa is its tendancy to form organized communities, known as biofilms when it attaches to biotic or abiotic surfaces. It is considered that P. aeruginosa virulence factors and biofilm formation is coordinated by a cell density monitoring mechanism termed Quorum Sensing (QS). The two well-defined QS systems in P. aeruginosa, namely las and rhl, rely on N-acyl homoserine lactone (AHL) signal molecules, also termed autoinducers. The las system is comprised of the transcriptional regulatory protein LasR, its cognate autoinducer molecule N-(3-oxododecanoyl) homoserine lactone (3–O–C12–HSL) and the AHL synthase LasI. Similarly, the rhl system consists of RhlR, its cognate autoinducer molecule N-butyryl homoserine lactone (C4–HSL) and the AHL synthase RhlI. The two QS systems of P. aeruginosa are hierarchically linked. The las system positively regulates the expression of both rhlR and rhlI [1].
The importance of QS in the virulence of P. aeruginosa has been shown in various animal studies in models of burn wound infection and pneumonia. These studies indicate that the QS systems of P. aeruginosa play an important role during respiratory tract infections and burn wound infections in the mouse mode. However, QS–deficient mutants, mainly strains deficient in lasI, rhlI, double lasI/rhlI, or lasR, generated in standard laboratory strains have been reported to be attenuated in virulence in mouse models of pneumonia and burn wound infection, as well as in a rat model of chronic lung infection [2]. Since the QS systems control the production of different virulence factors, biofilm formation and drug resistant, it is possible that the loss of production of AHLs severely compromises the ability of P. aeruginosa to cause infections in humans, and quorum quenching may be a new way to combat these bacterial infection without influence their growth [3, 4]. In this paper, we isolated clinical AHLs defective P. aeruginosa strains from chronic lung infections of cystic fibrosis patients and further characterized the isolates with respect to virulence factors production and biofilm formation.
Materials and Methods
Isolation and Identification of P. aeruginosa
In this study, samples were collected from 85 patients including both sexes with different ages, who suffered from chronic lung infection in cystic fibrosis (CF) patients at Lishui People’s Hospital, Zhejiang province. P. aeruginosa was identified biochemically from routinely obtained specimens by means of the Vitek ATB Expression System, version 2.7.8 (BioMérieux Deutschland GmbH, Nürtingen, Germany), which uses 32 biochemical reactions. Bacterial isolates were stored as suspensions in a 10 % (wt/vol) sterilized milk solution containing 12.5 % (vol/vol) glycerol at −70 °C until tests were performed.
Micro-AHL Cross-feeding Bioassay
To analyse AHLs production, cross-feeding bioassay method was used [5]. The assay relies on the induction of purple pigment production by the Chromobacterium violaceum strain CV026 and Agrobacterium tumefaciens strain At136 subsequent to diffusion of the AHLs into the medium produced by the test organisms. For this purpose, strain CV026 or At136 was streaked at the center of the nutrient agar plate, the target bacteria were streaked on the same plate against CV026 or At136 line, if the target bacteria have AHLs-producing ability, diffusible AHLs produced by the target bacteria induces strain CV026 to produce pigment or At 136 produce blue pigment when X-gal exist. Assays were performed in triplicate in three different occasions.
PCR Analysis of the QS Genes
Chromosomal DNA was extracted from PAO1 and the clinical isolates and utilized as templates in PCR experiments. A single bacterial colony was obtained from a fresh culture on LB agar and suspended in 50 ml of lysis solution (%1 TritonX-100, 10 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA). The suspension was boiled at 100°°C for 15 min. After centrifugation at 14,000 rpm for 5 min, 3 ml of the supernatant containing the bacterial DNA was used as a template for PCR amplification. Oligonucleotide primers lasR1 (50-atggccttggttgacggtt-30) and lasR2 (50-gcaagatcagagagtaataagaccca-30), lasI1 (50-atgatcgtacaaattggtcggc-30) and lasI2 (50-gtcatgaaaccgccagtcg-30), rhlR1 (50-caatgaggaatgacggaggc-30) and rhlR2 (50-gcttcagatgaggcccagc-30), rhlI1 (50-cttggtcatgatcgaattgctc-30) and rhlI2 (50-acggctgacgacctcacac-30) were used to amplify lasR, lasI, rhlR and rhlI genes, respectively [6]. The amplification mixture for each gene fragment contained bacterial DNA, two primers at 400 nM each, 250 mM dNTPs (each) (Shenggong, Shanghai, China) and 1× reaction buffer supplied by the manufacturerwith 1.5 mM MgCl2 and 1U of Taq DNApolymerase (Shenggong, Shanghai, China) in a final volume of 50 ml. PCR conditions for the amplification step were: 30 cycles of 95 1C for 45 s, 55 1C for 45 s and 72 1C for 1 min. DNA fragments were detected on 1 % agarose gels with ethidium bromide staining.
Detection of QS Controlled Phenotypes
Elastase Activity and Proteases Production
Elastase activity was measured using the elastin Congo red (ECR, Sigma) assay. Cells were grown in LB broth at 37 °C for 14 h. A 100-μl aliquot of bacterial supernatant was added to 900 μl of ECR buffer (100 mM Tris, 1 mM CaCl2, pH 7.5) containing 20 mg of ECR and incubated with shaking at 37 °C for 3 h. Insoluble ECR was removed by centrifugation, and the absorption of the supernatant was measured at 495 nm. LB medium was used as a negative control.
Protease production was detected on 1.2 % agar plates supplemented with 10 % (v/v) sterile skimmed milk (105 °C for 30 min). The bacterial supernatant was doped on the hole in the skim milk agar plates and incubated at 37 °C for 24–36 h. Proteolytic activity was measured by the clearing zone around the hole.
Production of Rhamnolipids
LB agar plates containing 0.2 g cetyltrimethylammoniumbromide and 5 mg methylene blue l−1 were inoculated with 2 μl of an overnight LB culture of P. aeruginosa strains. After an overnight incubation at 37 °C, the diameter of the clearing zone around the bacterial spots was measured as evidence of rhamnolipid production [7].
Pyocyanin Assay
Pyocyanin was extracted from culture supernatants and measured by the method of Essar et al. [8]. A 3-ml volume of chloroform was added to 5 ml of culture supernatant and mixed. The chloroform layer was transferred to a fresh tube and mixed with 1 ml of 0.2 M HCl. After centrifugation, the top layer (0.2 M HCl) was removed. The amount of pyocyanin within the extracted layer was determined by measuring A520.
Biofilm Assay
A quantitative biofilm formation experiment was per formed in a microtiter plate as described previously [9], with minor modification. Briefly, bacteria were grown on LB agar, and several colonies were gently re-suspended in LB (with or without the appropriate antibiotic); 100-μl aliquots were placed in a microtiter plate (polystyrene) and incubated 48 h at 28 °C without shaking. After the bacterial cultures were poured out, the plate was washed extensively with water, fixed with 2.5 % glutaraldehyde, washed once with water, and stained with a 0.4 % crystal violet solution. After solubilization of the crystal violet with ethanol-acetone (80:20, vol/vol) the absorbance at 590 nm was determined using a microplate reader (Bio-Rad, Hercules, Calif).
Results and Discussion
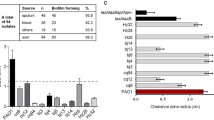
Eighty-five samples were collected from chronic lung infection of cystic fibrosis patients, at Lishui People’s Hospital, Zhejiang province, and Fifty-three isolates of P. aeruginosa were selected to screening the QS deficient. Among these isolates, strong positive reactions were observed in most of the strains, but four of them (PA-10, PA-28, PA-39 and PA-41) can not induce C. violaceum CV026 to produce purple pigment, this indicated that they can not produce short chain AHLs. (Fig. 1). The failure of four isolates did not activate violacein production in the C. violaceum CV026 may be due to the loss of any one of the QS system genes. Therefore QS-associated genes, lasR, lasI, rhlI and rhlR genes were checked by PCR. Oligonucleotide primers used for the PCR experiments are given in the methodology section. PAO1 was used as the positive control. PCR analysis revealed that three isolates (Pa28, 39 and 41) contained lasR, lasI, rhlR and rhlI genes while Pa10 was negative for lasR gene (Table 1).

Cross-feeding assays for detection of AHLs (A) violacein production by C. violaceum as an indication of short-chain AHLs
These four AHLs-dificient isolates were decreased in all virulence factors production and biofilm formation (Table 1). The production of elastase by the AHLs defective isolates was examined by the elastin Congo red assay. In vitro levels of elastase produced by these four clinical isolates were compared with wild-type strain PAO1, all these isolates produced less elastase, protease and rhamnolipid than PAO1 (Fig. 2; Table 1). Three of these strains produced less pyocyanin than PAO1, only PA41 produced little more pyocyanin than PAO1 (Fig. 3). All four AHLs deficient strains formed almost no biofilm compared to strain PAO1.

Elastase assay of the culture supernatants of PAO1 and AHLs deficient clinical isolates

Pyocyanin production of the culture supernatants of PAO1 and AHLs deficient clinical isolates
The essential role of QS in the pathogenesis of P. aeruginosa in respiratory tract infections has been demonstrated in animal models although its importance in human lung infections remains unclear because QS-deficient isolates have been isolated from such infections [10]. Clinical studies suggested that QS systems are fully functional within infected tissues, especially the lungs of CF patients, who were chronically infected with P. aeruginosa. In several animal models, QS mutants are significantly attenuated in their virulence compared with the wild type [11, 12]. In these studies, inadequacy of QS deficient strains to establish successful infection was proposed to be associated with reduced production of virulence factors. Lazenby et al. [13] have shown that a quadruple knockout of lasIR and rhlIR of P. aeruginosa PAO1 retains wild-type twitching motility and has equivalent infectivity and persistence to PAO1 in a mouse model of lung infection.
In the present study, we measured the production of autoinducers C12–HSL and C4–HSL using CV026 and At136 and QS-dependent virulence factors in clinical isolates. Most of these isolates were QS proficient, and can produce virulence factors and form biofilm [14]. Among the 53 isolates from chronic lung infection in cystic fibrosis (CF) patients, we identified four isolates that were defective in producing AHLs and some of the virulence factors, but these isolates still cause respiratory tract infections in humans. PCR analysis of these isolates for the presence of QS-genes revealed that only one isolate was negative for lasR gene. Several other studies also showed that the clinical isolates loss of any single virulence factor appeared to be compensated by other virulence factors during infection. However the impact of the loss of both C4–HSL and C12–HSL signaling molecules and several virulence factors on the pathogenesis of P. aeruginosa clinical isolates has been reported in very few studies. Schaber et al. [6] identified one QS deficient clinical isolate which lost all virulence factors tested, yet still caused a wound infection. Similar to our findings, Dénervaud et al. [15] identified three P. aeruginosa strains, obtained from intubated patients, which were defective in the production of both signaling molecules and extracellular virulence factors. Results of these two studies suggested that besides known virulence factors, there may be additional factors yet uncharacterized involved in the pathogenesis of P. aeruginosa.
Another possibility that may lead a QS deficient strain to cause infection is the presence of multiple P. aeruginosa strains in the infection site. A single patient may be infected by both QS proficient and deficient strains of P. aeruginosa. QS deficient strains could profit from the extracellular enzymes produced by QS proficient partners. Production of signaling molecules and/or QS-regulated factors by QS proficient strains may enable a QS deficient strain to take part in an infection.
In conclusion, the results of this study indicate that the QS systems deficient P. aeruginosa strains still can cause infections of the respiratory tract. These findings do not contradict the theory that QS plays a major role in P. aeruginosa pathogenicity, to reconcile these findings, there may be other virulence factors which may not be stringently controlled by QS in addition to known virulence factors, an alternative explanation is that QS-deficient variants may be social cheaters.
References
Schuster M, Greenberg EP (2006) A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol 296:73–81
Pearson JP, Pesci EC, Iglewski BH (1997) Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J Bacteriol 179:5756–5767
Kalia VC (2013) Quorum sensing inhibitors: an overview. Biotechnol Adv 31:224–245
Kalia VC, Purohit HJ (2011) Quenching the quorum sensing system: potential antibacterial drug targets. Crit Rev Microbiol 37:121–140. doi:10.3109/1040841X.2010.532479
Chu WH, Vattem DA, Maitin V, Barnes MB, McLean RJ (2011) Bioassays of quorum sensing compounds using Agrobacterium tumefaciens, and Chromobacterium violaceum. Methods Mol Biol 692:3–19
Schaber JA, Carty NL, McDonald NA, Graham ED, Cheluvappa R, Griswold JA, Hamood AN (2004) Analysis of quorum sensing-deficient clinical isolates of Pseudomonas aeruginosa. J Med Microbiol 53:841–853
Pinzon NM, Ju LK (2009) Improved detection of rhamnolipid production using agar plates containing methylene blue and cetyl trimethylammonium bromide. Biotechnol Lett 31:1583–1588
Essar DW, Eberly L, Hadero A, Crawford I (1990) Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. J Bacteriol 172:884–900
Jabalameli F, Mirsalehian A, Khoramian B, Aligholi M, Khoramrooz SS, Asadollahi P, Taherikalani M, Emaneini M (2012) Evaluation of biofilm production and characterization of genes encoding type III secretion system among Pseudomonas aeruginosa isolated from burn patients. Burns 38:1192–1197
Gupta P, Gupta RK, Harjai K (2013) Multiple virulence factors regulated by quorum sensing may help in establishment and colonisation of urinary tract by Pseudomonas aeruginosa during experimental urinary tract infection. Indian J Med Microbiol 31:29–33
Kondo A, Hirakata Y, Gotoh N, Fukushima K, Yanagihara K, Ohno H, Higashiyama Y, Miyazaki Y, Nishide K, Node M, Yamada Y, Kohno S, Kamihira S (2006) Quorum sensing system lactones do not increase invasiveness of a MexAB-OprM efflux mutant but do play a partial role in Pseudomonas aeruginosa invasion. Microbiol Immunol 50:395–401
Mittal R, Sharma S, Chhibber S, Harjai K (2006) Contribution of quorum sensing systems to virulence of Pseudomonas aeruginosa in an experimental pyelonephritis model. J Microbiol Immunol Infect 39:302–309
Lazenby JJ, Griffin PE, Kyd J, Whitchurch CB, Cooley MA (2013) A quadruple knockout of lasIR and rhlIR of Pseudomonas aeruginosa PAO1 that retains wild-type twitching motility has equivalent infectivity and persistence to PAO1 in a mouse model of lung infection. PLoS ONE 8:e60973. doi:10.1371/journal.pone.0060973
Wang HF, Tu FP, Gui ZH, Lu XH, Chu WH (2013) Antibiotic resistance profiles and quorum sensing-dependent virulence factors in clinical isolates of Pseudomonas aeruginosa. Indian J Microbiol 53:163–167
De′nervaud V, TuQuoc P, Blanc D, Favre-Bonte′ S, Krishnapillai V, Reimmann C, Haas D, Van Delden C (2004) Characterization of cell-to-cell signaling-deficient Pseudomonas aeruginosa strains colonizing intubated patients. J Clin Microbiol 42:554–562
Conflict of interest
We declare that we have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhenqiu Shang and Huafu Wang have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Shang, Z., Wang, H., Zhou, S. et al. Characterization of N-Acyl-homoserine Lactones (AHLs)-Deficient Clinical Isolates of Pseudomonas aeruginosa . Indian J Microbiol 54, 158–162 (2014). https://doi.org/10.1007/s12088-014-0449-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12088-014-0449-9