
Abstract
Many members of the heat shock protein family act in unison to refold or degrade misfolded proteins. Some heat shock proteins also directly interfere with apoptosis. These homeostatic functions are especially important in proteinopathic neurodegenerative diseases, in which specific proteins misfold, aggregate, and kill cells through proteotoxic stress. Heat shock protein levels may be increased or decreased in these disorders, with the direction of the response depending on the individual heat shock protein, the disease, cell type, and brain region. Aging is also associated with an accrual of proteotoxic stress and modulates expression of several heat shock proteins. We speculate that the increase in some heat shock proteins in neurodegenerative conditions may be partly responsible for the slow progression of these disorders, whereas the increase in some heat shock proteins with aging may help delay senescence. The protective nature of many heat shock proteins in experimental models of neurodegeneration supports these hypotheses. Furthermore, some heat shock proteins appear to be expressed at higher levels in longer-lived species. However, increases in heat shock proteins may be insufficient to override overwhelming proteotoxic stress or reverse the course of these conditions, because the expression of several other heat shock proteins and endogenous defense systems is lowered. In this review we describe a number of stress-induced changes in heat shock proteins as a function of age and neurodegenerative pathology, with an emphasis on the heat shock protein 70 (Hsp70) family and the two most common proteinopathic disorders of the brain, Alzheimer’s and Parkinson’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Proteotoxic stress responses in neurodegeneration and aging
Neurodegenerative diseases are primarily disorders of protein misfolding. For this reason, many authors refer to them as proteinopathies (Angot et al. 2010; Dickson 2009; Jellinger 2008, 2009; Uversky 2009; Xilouri and Stefanis 2010). Proteinopathies are characterized by the formation of fibrillar, amyloid-like structures that are insoluble in detergents, have a high content of proteinaceous β-sheets, and bind lipophilic dyes (Dobson 2003; Muchowski and Wacker 2005). The intermediate structures formed during the process of protein aggregation, oligomers and protofibrils, are known to be potent neurotoxins (Muchowski and Wacker 2005; Ross and Poirier 2004). In addition, non-enzymatic protein modifications by oxidants and glycating agents negatively affect protein structure and function, thereby eliciting severe proteotoxic stress (Nowotny et al. 2014). Recent evidence further suggests that the pathology in neurodegenerative disorders is transmissible from cell to cell and that neurons in specific brain regions die because of this overwhelming proteotoxic stress (Bae et al. 2012; Danzer et al. 2009; Desplats et al. 2009; Hansen et al. 2011; Luk et al. 2012a, b; Volpicelli-Daley et al. 2011). However, cells have evolved multiple systems to blunt the catastrophic impact of proteotoxic stress (Morimoto 2008, 2011). For example, the initial response to misfolded proteins in an otherwise healthy cell is to refold them back into their native shapes. This is accomplished by a large, interdependent network of chaperone and co-chaperone proteins that help prevent amyloid assembly. The best-studied examples of chaperones are the highly conserved heat shock family of proteins. Members of the heat shock protein family are induced by thermal stress, hence the name “heat shock” protein. However, many non-thermal stimuli also denature proteins, such as oxidative stress, mitochondrial collapse, hypoxia, irradiation, endoplasmic reticulum stress, and physical trauma, and these stimuli often also induce heat shock protein synthesis (Aridon et al. 2011; Lanneau et al. 2010). Heat shock proteins are so essential for life and well conserved across species that the genes encoding some of these proteins have been termed “vitagenes” by Vittorio Calabrese and his colleagues (Calabrese et al. 2011, 2012; Cornelius et al. 2013).
Despite the integrated network of chaperones and co-chaperones, proteins cannot always be folded back into their native shapes, especially when they are severely damaged. If the protein-saving systems fail to refold proteins, the irreparably damaged proteins can still be ubiquitinated and targeted to the 26S proteasome for clearance, again with the help of a retinue of chaperones and co-chaperones. The ubiquitin-proteasome system then degrades misfolded proteins into peptides via three types of catalytic sites: chymotrypsin-like, trypsin-like, and caspase-like (Goldberg 2003, 2007; Kisselev et al. 2003; Kisselev and Goldberg 2005; Lecker et al. 2006). The resulting short peptides are subsequently recycled into amino acids by peptidases in the cytosol. A second alternative to failed protein refolding is the process of lysosomal autophagy. In this process, proteins, carbohydrates, and lipids are engulfed by acidified lysosomes for macro- or microautophagic degradation (Cuervo 2004; Cuervo et al. 2005; Kaushik and Cuervo 2006; Wong and Cuervo 2010). Misfolded proteins with a KFERQ motif can also be guided by heat shock proteins into the lysosome via translocation through the lysosomal-associated membrane transporter (LAMP2a) (Arias and Cuervo 2011; Kaushik and Cuervo 2009; Massey et al. 2004, 2006; Orenstein and Cuervo 2010; Xilouri and Stefanis 2010). The latter process is known as chaperone-mediated autophagy because of its critical dependence on heat shock proteins. Under physiological conditions, these three systems, chaperone-guided protein refolding, ubiquitin-proteasomal degradation, and lysosomal autophagy are sufficient to handle mild fluctuations in the environmental milieu. However, under pathological conditions, these systems can become overwhelmed and then the affected neurons begin to die. The vast majority of neurons in the brain can never be replaced once they have disappeared. Therefore, improving chaperone defenses to help reduce proteotoxicity and subsequent neurodegeneration is an urgent priority for the aging population of the United States.
Although the neurodegeneration in Parkinson’s and Alzheimer’s disease is inexorable in its progression, it is also delayed in onset and surprisingly slow, unfolding only over the course of several decades towards the end of life. This is even true for familial forms of these disorders, despite the presence of genetic mutations from birth onwards. How can the stressed brain stem the tide of cell death for so long? One possibility is that the proteotoxic stress in these diseases is relatively mild and that neurons and glia can battle it for long periods of time, perhaps by raising pro-survival defenses such as the heat shock proteins. Given that neuronal systems in the brain are highly redundant and that it requires considerable cell loss before neurobehavioral syndromes finally emerge, a protective response to mild stress might well explain the length of time before clinical symptoms become manifest. However, defense systems may eventually become overwhelmed, lose heat shock protein expression, succumb to proteotoxicity, and ultimately self-destruct. Some brain regions are also likely to have higher heat shock protein expression than others, leading to heterogenous patterns of neurodegeneration and the signature topographical maps of protein inclusions that characterize neurodegenerative disorders. Studies that demonstrate higher or lower levels of heat shock proteins in the surviving tissue in neurodegenerative conditions are consistent with these possibilities and are therefore discussed in this review. Most commonly, there appear to be straightforward increases or decreases of heat shock proteins in neurodegenerative conditions and in aging, with the responses varying by heat shock protein, disease, cell type, brain region, etc. However, on occasion we have also observed a somewhat biphasic expression pattern in the heat shock protein response to age-related stress (Gleixner et al. 2014). If one assumes that age-related stress accrues over time, our findings bear some resemblance to the biphasic phenomenon of ‘hormesis,’ defined in the toxicological literature as low dose stimulation and high dose inhibition (Calabrese 2010, 2013; Calabrese and Blain 2011; Calabrese et al. 2010; Mattson 2008). A biphasic stress response to aging in the expression of some heat shock proteins would also be consistent with Hans Selye’s observations long ago that organisms can adapt to mild, short duration stress (eustress) but are weakened by severe, chronic stress (distress) (Selye 1975).
Our recent study of the impact of age on heat shock protein expression in the dorsal striatum and ventral mesencephalon also supports the notion that the effects of age-related stress vary depending upon the individual heat shock protein and the specific brain region in question (Gleixner et al. 2014). In addition to the identity of the heat shock protein and the brain region of interest, the direction of the stress response is also likely to vary with cell type. That is, some cells may be able to adapt better to severe stress than others. In this context, it is worth noting that the tissue assayed after death in neurodegenerative conditions includes those cells that have managed to resist severe proteotoxic stress through endogenous defenses, perhaps explaining why heat shock protein expression is often higher in diseased tissue (see below). Consistent with this notion, we have collected evidence in primary cortical astrocytes that the remaining glia that manage to survive severe proteotoxic stress successfully engage natural antioxidant and chaperone defense systems (Titler et al. 2013). Our findings are consistent with previous studies showing that astrocytes upregulate several heat shock proteins in neurodegenerative conditions (Dabir et al. 2004; Durrenberger et al. 2009; Kawamoto et al. 2007; Lowe et al. 1992; Renkawek et al. 1993, 1994a, b, 1999; Seidel et al. 2012; Shinohara et al. 1993; Wilhelmus et al. 2006). In our study, severe stress was defined as stress that was lethal to some fraction of the astrocyte population. The remaining astrocyte survivors required greater levels of proteotoxic injury before they died in response to a second proteotoxic hit, supporting the hypotheses that exposure to severe stress rendered them more resistant than before and that some types of stressed cells can become progressively harder to kill. Astrocytes are well known for providing metabolic, trophic, and antioxidant support to neurons (Allen and Barres 2009; Barres 2008; Zhang and Barres 2010) and are thought to engulf aggregated proteins such as α-synuclein and β-amyloid from the extracellular space (Lee et al. 2010a; Wyss-Coray et al. 2003). Healthier astrocytes that are less susceptible to proteotoxic stress should be able to support neighboring neurons more effectively. If this form of glial plasticity can be generalized to humans, it might help delay the onset and slow the progression of neuron loss in neurodegenerative disorders. However, this phenomenon of ‘survival of the fittest astrocytes’ cannot be generalized to all cell types. In some systems, the cells that survive severe stress are not resistant, but are actually sensitized to subsequent stress, so that the two hits of severe stress are synergistic in their toxic effects in these populations (Boger et al. 2010; Carvey et al. 2006; Gao and Hong 2011; Manning-Bog and Langston 2007; Sulzer 2007; Unnithan et al. 2012, 2013; Weidong et al. 2009; Zhu et al. 2001, 2007). In short, one might expect the direction of the stress response, i.e., whether it will elicit pro-survival or pro-death responses, to depend on cell type, heat shock protein expression, antioxidant defenses, dose and duration of the injury, previous exposures to other stressors, brain region, animal age, nutrient intake, physical activity levels, disease stage, and numerous other endogenous and environmental variables (Leak 2014).
In the following discussion of heat shock proteins, we will focus mostly on the heat shock protein 70 family as it is so well studied. The present review is not meant to be an exhaustive description of all heat shock proteins as a function of age/disease or of their specific roles in all experimental models of neurodegenerative disorders. We have further narrowed our discussion of neurodegenerative disorders to Alzheimer’s and Parkinson’s disease, as they are the two most common of these conditions. Finally, we have included several examples of age-related changes in heat shock proteins because aging is the major risk factor for these disorders and is known to elicit mild proteotoxic stress, as shown by an overall decline in proteasomal clearance capacity and autophagic activity (Conconi et al. 1996; Conconi and Friguet 1997; Cuervo 2008; Friguet et al. 2000; Keller et al. 2000a, b, 2002, 2004; Koga et al. 2011; Nowotny et al. 2014). Morimoto, Cuervo, and others have recently argued that the efficiency of the entire proteostasis network declines with age (Labbadia and Morimoto 2014; Lopez-Otin et al. 2013; Morimoto and Cuervo 2014), even though several heat shock proteins are upregulated with age (see below). This suggests that the ability of chaperones to be further induced by additional stress may be impaired in aged organisms (Arumugam et al. 2010; Blake et al. 1991; Labbadia and Morimoto 2014; Pardue et al. 1992; Soti and Csermely 2000), although whether this is a general rule has been questioned by some (Walters et al. 2001). One might also speculate that basal levels of some heat shock proteins are increased with aging as a compensatory adaptation to loss of efficiency of the proteostasis network.
Heat shock protein classification and function
Heat shock proteins have been classified into six major families according to their molecular mass in kilodaltons: the small heat shock proteins (<34 kDa; eg. αB-crystallin, Hsp27, Hsp32), Hsp40 (35–54 kDa), Hsp60 (55–64 kDa), Hsp70 (65–80 kDa), Hsp90 (81–99 kDa), and Hsp100 (>100 kDa) (Aridon et al. 2011). As mentioned above, the best studied of these heat shock proteins belong to the Hsp70 family, which has both stress inducible and constitutive members. Hsp70 proteins interact with hydrophobic peptide segments of unstructured proteins in an ATP-dependent manner. They are comprised of a C-terminal substrate-binding domain that recognizes the unstructured polypeptide segments, and an N-terminal ATPase domain that helps the protein alternate between an ATP-bound, open state with low substrate affinity and an ADP-bound closed state with high substrate affinity (Mayer and Bukau 2005). In support of the notion that heat shock protein upregulation in aged animals is compensatory in nature, higher basal levels of Hsp70 appear to be characteristic of longer-lived mammalian and avian species (Salway et al. 2011). Furthermore, humans exhibit 43-fold higher levels of Hsp70 than non-stressed rats and 14-fold higher levels than heat-shocked rats (Pardue et al. 2007). In addition, the constitutive member of this family, heat shock cognate 70 (Hsc70) is 1.5 fold more highly expressed in humans than rats (Pardue et al. 2007). Although authors have often used the terms Hsp70 and Hsc70 interchangeably, ‘Hsp70’ is typically used to refer to the major family members Hsp70A1 and Hsp70A2, which are identical or close to identical in amino acid sequence and are coded by the HSPA1A and HSPA1B genes, respectively. Hsp70 and Hsc70 share almost 90 % amino acid homology and exhibit quite similar biochemical properties (Brocchieri et al. 2008; Daugaard et al. 2007; Kabani and Martineau 2008). Some of the studies discussed below in reference to Hsp70 may therefore actually be reporting findings on Hsc70. Other than Hsp70A1, Hsp70A2, and Hsc70, which has both major (coded by HSPA8) and minor (coded by HSPA2) forms, the remaining Hsp70 proteins include the endoplasmic reticulum-associated glucose regulated protein 78 (GRP78, also known as BiP; coded by the HSPA5 gene), and mitochondrial Hsp70 (mtHsp70, also known as mortalin, glucose-regulated protein 75, and PBP74; coded by the HSPA9B gene). Several of the other Hsp70 family members are false entries, pseudogenes, or testes specific.
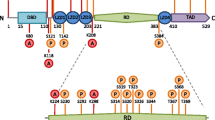
Once transcribed in response to stress, Hsp70 proteins form one of several complexes with other chaperones and co-chaperones. For example, the ATPase cycle of Hsp70 is modulated by Hsp40, which elicits an increase in ATPase activity (Bukau and Horwich 1998; Minami et al. 1996). Depending on the constituents of the complex, the complex either 1) refolds and rescues misfolded proteins or 2) guides proteins to the proteasome or lysosome for degradation if they are damaged beyond repair (Fig. 1). Hsp70 and Hsc70 overlap in the formation of these complexes and we will therefore simply refer to Hsp70 here. In the first scenario, Hsp70 binds Hip, Hop, and Hsp90, and helps refold the denatured protein in an ATP-dependent manner (Esser et al. 2004). Hip (or ST13) stands for Hsp70-interacting protein and prolongs the half-life of ADP-Hsp70-substrate complexes (Hohfeld et al. 1995; Hohfeld and Jentsch 1997). Because this interaction stabilizes the ADP-bound state of Hsp70, it increases its affinity for substrate proteins (Hohfeld et al. 1995). For example, Hip stabilizes the complex formed by Hsp70 and α-synuclein, thereby sustaining Hsp70-mediated anti-amyloid activity (Roodveldt et al. 2009). Notably, Hip is reportedly consistently reduced in levels in the blood of Parkinson’s patients (Scherzer et al. 2007).

Schematic of Hsp70 (red) and a few of its cofactors. Under physiological conditions, misfolded proteins can induce Hsp70 gene expression and they may be degraded by the ubiquitin proteasome system (UPS) if they are irreparably damaged. During substrate degradation, CHIP binds the C-terminal of Hsp70 and acts as an E3 ubiquitin ligase to mark the protein for removal. During this process, Bag1 binds the ATPase domain at the N-terminal and serves to link the Hsp70/cofactor complex with the UPS. On the other hand, when proteins can be refolded to their native structure and salvaged, Bag-1 binding is blocked by the cochaperone Hip and CHIP binding is blocked by Hop. Hsp40 and Hsp90 may also bind the protein refolding complex and promote ATP-dependent folding activity. Not shown in this figure is the dimerization of some of these proteins. Under conditions of severe proteotoxic stress or loss of homeostasis, proteins may not undergo degradation or refolding and are sequestered in fibrillar deposits, such as Lewy bodies in Parkinson’s disease
Hip and Hop co-chaperones are both known as folding cofactors. Hop (Hsp70/90-organizing protein) is an adaptor that transfers client proteins from Hsp70 to Hsp90 (Scheufler et al. 2000). The tetratrico-peptide repeat-1 (TPR1) domain of Hop recognizes Hsp70 and the TPR2 domain recognizes Hsp90. Hsp90 is found at high levels even in unstressed cells; it has numerous client proteins that it stabilizes, folds, activates, and assembles and it affects the rate of folding of denatured proteins (Aridon et al. 2011; Johnson and Brown 2009; Pearl and Prodromou 2006). When proteins are damaged beyond repair and cannot be refolded, Hsp70 and Hsp90 both bind to the co-chaperone CHIP (carboxy terminus of Hsp70-interacting protein) so that the protein can be targeted for degradation (Ballinger et al. 1999; Connell et al. 2001). CHIP effectively blocks Hsp40-stimulated ATPase and refolding activities of Hsp70 (Ballinger et al. 1999). CHIP binds to the carboxyl terminus of Hsp70 (Esser et al. 2004) and can also bind to Hsp90 via its TPR-binding domain (Salminen et al. 2011). CHIP serves as an E3 ubiquitin ligase by attaching a polyubiquitin chain to the protein, thereby targeting the protein for proteasomal degradation (Jiang et al. 2001). The proteasomal degradation process facilitated by CHIP also requires binding of the nucleotide exchange factor Bcl-2-associated athanogene (BAG-1) to the ATPase, N-terminal domain of Hsp70 (Fig. 1) (Esser et al. 2004). In addition to serving as an E3 ubiquitin ligase, CHIP also specifically targets proteins for chaperone-mediated autophagy by binding to Hsc70, which then guides damaged proteins containing a KFERQ motif toward the lysosome (Arias and Cuervo 2011; Shin et al. 2005). Thus, CHIP can triage irreparably damaged proteins between the lysosome and proteasome (Kalia et al. 2011; Shin et al. 2005; Tetzlaff et al. 2008). Not surprisingly, CHIP deficiency induces a decline in proteasomal activity and accelerates cellular senescence (Min et al. 2008).
Hsp70 proteins also play an integral role in the inhibition of caspase-dependent and caspase-independent apoptosis (Kennedy et al. 2014; Lanneau et al. 2008). For example, Hsp70 has the capacity to neutralize apoptosis-inducing factor (AIF) (Kroemer 2001; Ravagnan et al. 2001), a mitochondrial intermembrane protein which translocates to the nucleus to induce chromatin condensation and DNA fragmentation (Cande et al. 2002). The ATPase domain of Hsp70 can also bind directly to the caspase recruitment domain of apoptotic protease activation factor 1 (Apaf1), thereby preventing Apaf1 oligomerization and inhibiting procaspase-9 recruitment to the apoptosome (Beere et al. 2000; Saleh et al. 2000). Furthermore, Hsp70 inhibits many pro-death signaling molecules, such as apoptosis signal-regulating kinase (Ask1) (Park et al. 2002), p38, and c-Jun N-terminal kinase (JNK) (Gabai et al. 2000; Park et al. 2001).
The endoplasmic reticulum plays an integral role in protein folding and transport and is rich in molecular chaperones, such as GRP78 and GRP94 (Brown and Naidoo 2012; Schroder and Kaufman 2005; Viana et al. 2012). Proteotoxic stress in the lumen of the endoplasmic reticulum can lead to the unfolded protein response (UPR) and endoplasmic reticulum-associated protein degradation (ERAD). The UPR is initially protective and clears damaged proteins through a series of signaling pathways, but it can also trigger apoptosis when the damage is excessive. Under physiological conditions, GRP78 holds on to the luminal N-termini of transmembrane, stress-sensitive proteins such as PRKR-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and serine/threonine-protein kinase/endoribonuclease (IRE1), serving to inactivate them (Brown and Naidoo 2012; Schroder and Kaufman 2005; Viana et al. 2012). When proteins within the lumen of the endoplasmic reticulum become damaged and misfolded, GRP78 must be freed from these positions to bind the hydrophobic regions of unstructured proteins. As a result, GRP78 is no longer able to hold and disengage the stress transducers, and the gears of the UPR are set in motion. In ERAD, aberrant proteins that cannot be refolded are exported to the cytoplasm and targeted to the 26S proteasome or lysosome for proteasomal or autophagic clearance. Under conditions of severe endoplasmic reticulum stress, pro-death signaling molecules such as p38 and JNK and transcription factors such as CCAAT-enhancer-binding protein homologous protein (CHOP) are also upregulated, so that dysfunctional cells can be cleared from the organism by apoptosis (Brown and Naidoo 2012; Schroder and Kaufman 2005; Viana et al. 2012). Proteotoxic stress in the endoplasmic reticulum may therefore trigger a biphasic response, with pro-survival UPR and ERAD responses predominating with mild injury, but UPR-initiated apoptosis predominating with severe injury.
Hsp32 is also known as heme oxygenase 1 (HO1) and differs from the other heat shock proteins in that it plays a more direct role in antioxidant defense. Hsp32 is induced by stress via the transcription factor nuclear factor erythroid 2 related factor 2 (Nrf2) (Zhang et al. 2013). Nrf2 is normally bound to kelch-like ECH-associated protein 1 (Keap1) homodimers in the cytoplasm under physiological conditions. Keap1 binding ensures that Nrf2 is tagged with ubiquitin and degraded by the proteasome. However, Keap1 has highly reactive cysteine residues that are exquisitely sensitive to electrophilic and oxidative stress (Yamamoto et al. 2008). Oxidation of these cysteine residues quickly initiates Nrf2/Keap1 dissociation so that Nrf2 can escape proteasomal degradation and translocate into the nucleus (McMahon et al. 2006; Zhang et al. 2013). Activated, nuclear Nrf2 then binds the antioxidant response element (ARE) in the promoter region of the Hsp32 gene as well as many other pro-survival genes, such as those involved in glutathione metabolism. Upon induction, Hsp32 plays a major role in the breakdown of toxic heme into the antioxidant biliverdin and the prosurvival molecule carbon monoxide (Aztatzi-Santillan et al. 2010; Grochot-Przeczek et al. 2012; Schipper 2000; Schipper et al. 2009; Wu et al. 2011). Biliverdin is further broken down into bilirubin, which also has potent antioxidant properties. Hsp32 is also induced by proteotoxic stress (Posimo et al. 2013; Titler et al. 2013), probably because oxidative and proteotoxic stress are inextricably intertwined and propel each other forward (Cecarini et al. 2007; Dasuri et al. 2013; Ding et al. 2006; Keller et al. 2000a, 2004).
Heat shock proteins in neurodegenerative disorders and aging
Many heat shock proteins are increased in tissue from elderly patients with neurodegeneration and in aged animals (Table 1) (Lee et al. 2000; Lu et al. 2004). If these increases in heat shock proteins serve to defend against proteotoxic stress, heat shock proteins should also be protective in experimental models of injury. Consistent with this hypothesis, Hsp70 has been shown to be protective in many experimental disease models, including the MPTP model of Parkinson’s disease and the β-amyloid model of Alzheimer’s disease (Dong et al. 2005; Jung et al. 2008; Kalia et al. 2010; Kumar et al. 2007; Magrane et al. 2004; Muchowski and Wacker 2005; Nagel et al. 2008; Sherman and Goldberg 2001). Furthermore, Hsp70 has been shown to enhance α-synuclein refolding or degradation, thereby decreasing α-synuclein fibrillization and toxicity (Klucken et al. 2004; Luk et al. 2008). There is an increase in the expression of HSPA1A and HSPA1B in the substantia nigra of patients with Parkinson’s disease, progressive supranuclear palsy, and frontotemporal dementia with parkinsonism (Hauser et al. 2005). In their report, Hauser and colleagues further suggested that Hsp70 induction was a “common response to mitigate the toxic effects of misfolded protein.” Hsp70 has been localized to aggregated protein deposits in Alzheimer’s brains as well as Lewy bodies in Parkinson’s brains, perhaps in an endogenous effort to reduce the toxicity of these protein aggregations (Leverenz et al. 2007; Muchowski and Wacker 2005).
In mild cognitive impairment, a potentially prodromal stage of Alzheimer’s disease, there is an increase in Hsp70 expression in the inferior parietal lobule of the telencephalic cortex (Di Domenico et al. 2010). Patients with mild cognitive impairment also exhibit increased levels of Hsp70 in plasma (Lee et al. 2008). An analysis of the hippocampal proteome in Alzheimer’s disease revealed an increase in Hsp70 expression (Sultana et al. 2007), in agreement with previous studies showing an increase in Hsp70 in the highly vulnerable temporal cortex (Yoo et al. 1999). Finally, Hsp70 expression is also increased in aged animals in the cerebral cortex, striatum, hippocampus, and cerebellum (Calabrese et al. 2004) as well as in blood cells from aged human subjects (Njemini et al. 2007). It should be noted that Hsp70 does not rise in every brain region with stress exposure, as it has been shown to decline within olfactory neurons in older human subjects and in Alzheimer’s patients (Getchell et al. 1995). Hsp70 levels are also reduced by aging in the inferior colliculus (Helfert et al. 2002). Furthermore, some authors have observed age-related reductions in Hsp70 in the striatum and cortex of older male rats and mice (Arumugam et al. 2010; Gupte et al. 2010). We have observed an increase in Hsp70 levels in the telencephalic cortex of middle-aged female rats that is reversed in the older animals, as if the response to age-related stress were biphasic (Posimo and Leak, unpublished observations). Gender is an important variable to consider as studies on males have not always translated well to women (Clayton and Collins 2014). All these Hsp70 studies reveal that no simple generalizations about heat shock proteins can be made across brain regions, cell types, species, types of stress, or gender. Furthermore, as will become evident below, the direction of the response to stress, i.e., whether there is a loss or an increase, varies greatly depending on the individual heat shock protein.
Several studies on the constitutively expressed Hsc70 protein reveal that expression is reduced in neurodegenerative conditions, unlike many of the Hsp70 studies discussed above. For example, Hsc70 levels are reduced within nigral neurons in Parkinson’s disease (Chu et al. 2009). As mentioned above, Hsc70 guides misfolded proteins to the lysosomal membrane, where LAMP2a translocates the damaged protein into the lysosomal compartment for autophagic degradation (Arias and Cuervo 2011; Massey et al. 2006; Orenstein and Cuervo 2010). Other studies have shown that Hsc70 and LAMP2a are both lowered in the substantia nigra and amygdala in Parkinson’s disease and that these markers may even be trapped within Lewy bodies (Alvarez-Erviti et al. 2010). These findings have been interpreted to suggest that the ventral midbrain suffers from significant reductions in chaperone-mediated autophagy in Parkinson’s patients. As one would expect, the changes in nigral Hsc70 protein levels in Parkinson’s disease are accompanied by a parallel loss in Hsc70 mRNA (Mandel et al. 2005). On the other hand, the mRNA levels of Hsc70 are increased in blood samples from patients with Parkinson’s disease (Molochnikov et al. 2012). These findings show that the direction of the Hsc70 stress response depends upon cell type (e.g. blood cell versus nigral neuron).
The entorhinal cortex and hippocampus in the temporal lobe both exhibit relatively low levels of Hsc70 and Hsp27 expression (Chen and Brown 2007). This finding has led to the hypothesis that these brain regions are especially vulnerable to Alzheimer’s disease because of relatively low basal expression of these two heat shock proteins (Chen and Brown 2007). Alzheimer’s disease is strongly linked with conformational changes in tau protein, which normally stabilizes microtubules in the cytoskeleton. Pathologically hyperphosphorylated tau forms an integral part of neurofibrillary tangles, one of the anatomical hallmarks of this disorder. Hsc70 is thought to rapidly engage tau under conditions of microtubule destabilization (Jinwal et al. 2010) and may regulate its proteasomal degradation by ubiquitination, perhaps in conjunction with the E3 ubiquitin ligase CHIP (Elliott et al. 2007; Shimura et al. 2004). Thus, if some brain regions and cell types exhibit low Hsc70 levels, they might be all the more vulnerable to neurofibrillary tangles, as was argued by Chen and Brown for the entorhinal cortex and hippocampus (Chen and Brown 2007).
Reports on age-related changes in Hsc70 show mixed results. We recently discovered that Hsc70 levels drop with age in the striatum of the female rat (Gleixner et al. 2014). However, Unno and colleagues have reported an increase in the basal level of Hsc70 in the pons, medulla, striatum, and thalamus in male rats (Unno et al. 2000). Others have also found an age-related increase in Hsc70 in the striatum and substantia nigra of male rats (Calabrese et al. 2004). These discrepancies could reflect gender- and/or strain-specific effects, although the tissue from male and female rats or from different strains would have to be assayed in parallel before making any conclusions. In humans, the density of Hsc70 mRNA-expressing neurons in the hippocampus is higher in older subjects (Tohgi et al. 1995).
Unlike many Hsp70 proteins, the mitochondrial member of this family, mtHsp70, is not highly induced in response to heat stress but is increased in response to other stimuli such as glucose deprivation and oxidative injury (Aridon et al. 2011). mtHsp70 is known to play a role in the mitochondrial import complex and in mitochondrial biogenesis (Brunner et al. 1995; Schneider et al. 1994). Using an unbiased quantitative proteomic approach, Jin and colleagues have reported that mtHsp70 levels are decreased in the mitochondrial fraction of the substantia nigra in Parkinson’s patients (Jin et al. 2006). Furthermore, several variants of the gene that codes for mtHsp70 (HSPA9) have been found in a small cohort of late-onset Parkinson’s patients (De Mena et al. 2009). Some of the phenotypes associated with mtHsp70 mutations are characterized by mitochondrial dysfunction, such as respiratory incompetency and increased susceptibility to oxidative stress (Burbulla et al. 2010; Goswami et al. 2012). On the other hand, mutations in HSPA9 are not common in early-onset Parkinson’s disease (Freimann et al. 2013).
The endoplasmic reticulum Hsp70 family member, GRP78, has been shown to reduce α-synuclein toxicity in experimental models of Parkinson’s disease (Gorbatyuk et al. 2012). However, polymorphisms in the gene coding for GRP78 (HSPA5) are not thought to be strongly associated with an increased risk for developing the disorder (Chen et al. 2008). In models of Alzheimer’s disease, GRP78 has been shown to bind amyloid precursor protein and decrease β-amyloid secretion (Yang et al. 1998). Amyloid precursor protein is a ubiquitously expressed transmembrane protein from which β-amyloid peptides are cleaved. In Alzheimer’s brain tissue, β-amyloid peptides coalesce to form extracellular senile plaques or aggregations with a fibrillar, β-pleated structure (Mucke and Selkoe 2012; Walsh and Selkoe 2007). β-amyloid may also initiate cellular damage intracellularly (Hsia et al. 1999). Some studies have reported an increase in GRP78 levels in the highly vulnerable temporal cortex and hippocampus in Alzheimer’s patients (Hoozemans et al. 2005). Other studies have not reported changes in GRP78 in Alzheimer’s brain tissue (Sato et al. 2000) but have reported induction of the UPR in this condition (Lee et al. 2010b). These studies disagree with previous assessments that GRP78 and GRP94 are downregulated within the temporal cortex in Alzheimer’s patients (Katayama et al. 1999). The Katayama group further suggested that GRP78 is decreased in cells expressing mutations in presenilin, which can cause early-onset familial Alzheimer’s disease (Katayama et al. 1999). The latter authors have therefore argued that the UPR is downregulated by presenilin mutations in familial Alzheimer’s disease (Imaizumi et al. 2001; Katayama et al. 2004), although this hypothesis has been contested (Sato et al. 2000). These discrepancies may reflect differences in the stage of the illness at which tissue is harvested, the precise brain regions dissected at autopsy, and inter-laboratory technical differences in the assays. In particular, the nature of the assays must always be borne in mind while evaluating studies of postmortem tissue. For example, immunohistochemical assays allow the investigator to localize the expression of heat shock proteins within specific neuronal and glial populations, affording much higher spatial resolution than Western blotting. In Western blotting, protein changes within specific cell populations may be masked because the entire group of cells is lysed and homogenized. On the other hand, immunohistochemistry is much less quantitative and sensitive than immunoblotting, especially with the newer infrared, fluorescent methods for Westerns. The spatial/anatomical advantage of immunohistochemical studies is exemplified by reports showing that GRP78 is increased in Alzheimer’s brains within surviving neurons that appear cytologically normal in the CA3 subfield of the hippocampus and the deep layers of the entorhinal cortex (Hamos et al. 1991). Using immunohistochemical and stereological tools, GRP78 levels have also been shown to be increased in the substantia nigra of older Asian Indians, suggestive of age-related compensatory changes in the endoplasmic reticulum (Alladi et al. 2010). Contrary to the immunohistochemical findings in the human nigra, GRP78 levels are lowered in homogenates of the cortex and striatum in the aged mouse brain (Arumugam et al. 2010), suggestive of differences across brain regions, species, and/or assay techniques. Many other studies have similarly shown that GRP78 is decreased in aged tissue, such as the cerebral cortex and hippocampus (Hussain and Ramaiah 2007; Naidoo et al. 2008; Paz Gavilan et al. 2006).
As with many other heat shock proteins, the small heat shock proteins αB-crystallin, Hsp27 (Hsp25 in rodents), and Hsp32 are all stress-inducible (Krueger-Naug et al. 2002; Lanneau et al. 2010). Hsp27 and αB-crystallin have both been shown to reduce α-synuclein toxicity in experimental models of Parkinson’s disease (Outeiro et al. 2006). αB-crystallin is highly expressed in cortical neurons in Parkinson’s disease, particularly at sites predisposed to α-synuclein pathology, such as the anterior temporal and insular mesocortex (Braak et al. 2001). However, Braak and colleagues observed through a series of immunohistochemical studies that neurons expressing αB-crystallin generally do not develop Lewy pathology, and conversely, neurons with Lewy pathology fail to accumulate this heat shock protein (Braak et al. 2001). These findings suggest that those neurons that can mount a heat shock protein response are relatively protected from amyloid formation and exemplify the spatial advantages of immunohistochemical studies.
Hsp25 is known to improve the catalytic activity of the proteasome particle and to inhibit caspase-mediated apoptotic pathways (Acunzo et al. 2012; Lanneau et al. 2008, 2010). Hsp27 levels are increased within the nigrostriatal pathway of Parkinson’s victims (Zhang et al. 2005). Furthermore, Schultz and colleagues observed that ubiquitin, αB-crystallin, and Hsp27 were all increased in spheroid bodies in the globus pallidus and substantia nigra, pars reticulata of aged rhesus monkeys (Schultz et al. 2001).
Dementia may also be associated with higher Hsp27 expression. For example, Hsp27 mRNA levels are increased in dementia with Lewy bodies (Outeiro et al. 2006) and the expression of both Hsp27 and αB-crystallin is increased in reactive astrocytes of the hippocampus in Parkinson’s disease with dementia (Renkawek et al. 1994a, 1999). Recent studies have also shown that there is an increase in Hsp27 expression in the inferior parietal lobule and the hippocampus of patients suffering from mild cognitive impairment (Di Domenico et al. 2010). Furthermore, Hsp27-immunoreactive neurons and αB-crystallin-immunoreactive astrocytes are found more frequently in the temporal, parietal, and frontal lobes of patients with Alzheimer’s disease than of controls (Shinohara et al. 1993). As one might expect, Hsp27 and αB-crystallin are also present in senile plaques (Shinohara et al. 1993; Stege et al. 1999) and Hsp27 is particularly highly expressed in astrocytes of brain regions housing abundant senile plaques (Renkawek et al. 1993, 1994a; Wilhelmus et al. 2006). This may reflect a natural attempt to mitigate proteotoxic injury because Hsp27 overexpression has been shown to ameliorate symptoms in transgenic mouse models of Alzheimer’s disease (Toth et al. 2013). It is instructive that Hsp27 expression increases with the severity of the pathological changes as well as the duration of the disease (Renkawek et al. 1993, 1994a; Wilhelmus et al. 2006). We and others have reported that Hsp25 is also markedly increased by aging in the striatum, substantia nigra, globus pallidus, and cerebral cortex (Dickey et al. 2009; Gleixner et al. 2014; Gupte et al. 2010). Using immunofluorescent confocal analyses, we also determined that Hsp25 is expressed within tyrosine hydoxylase+ dopaminergic neurons in the substantia nigra, pars compacta (Gleixner et al. 2014). As one would expect age-related stress to be the most severe in the oldest group, our findings of high Hsp25 expression in the oldest animals are consistent with the highest expression of Hsp27 levels in the most severely affected Alzheimer’s patients (Renkawek et al. 1993, 1994a; Wilhelmus et al. 2006). In contrast, Hsp60, Hsp40, and Hsp32 were all found to be higher in either the striatum or nigra in middle-aged animals, not in the oldest group (Gleixner et al. 2014). Unlike the situation with Hsp25, the age-related changes in Hsp60, Hsp40, and Hsp32 that we observed in female rats are more consistent with a biphasic “aging curve.” If the duration of age-related stress leads to a biphasic aging curve in some but not all heat shock proteins, our findings suggest that the stress threshold for maximal induction of Hsp40, Hsp32, and Hsp60 is lower than for Hsp25 because they all peak earlier than Hsp25.
The heat shock protein involved in antioxidant defense, Hsp32, is reportedly higher in hippocampal and cortical neurons and astrocytes in Alzheimer’s patients (Schipper 2000; Schipper et al. 2006). Furthermore, Hsp32 is increased in astrocytes in Parkinson’s disease and is found in Lewy bodies (Schipper et al. 1998). Some authors have suggested that Hsp32 might be destructive in these conditions because it breaks down heme into ferrous iron, potentially increasing the risk for Fenton chemistry and iron toxicity (Schipper 2011). Indeed, heme-derived iron and carbon monoxide may both increase the risk for oxidative stress (Desmard et al. 2007; Zhang and Piantadosi 1992). On the other hand, co-induction of apoferritin appears to limit Hsp32 toxicity (Dennery 2000; Ryter and Tyrrell 2000), and many studies have verified that Hsp32 is protective (Calabrese et al. 2009; Jazwa and Cuadrado 2010; Zhang et al. 2013). We found that Hsp32 expression is highest in the striatum at middle age, but that there is a transient drop in Hsp32 in the ventral midbrain of 16–19 months old female rats (Gleixner et al. 2014). Other groups have shown a drop in Hsp32 in the cortex and striatum of middle-aged and old male mice (Arumugam et al. 2010). However, Hsp32 levels are increased with aging in the human cerebral cortex (Hirose et al. 2003) and in human peripheral blood cells (Njemini et al. 2007).
Hsp90 exhibits high levels of expression under basal conditions because of its constitutive role in protein folding. Hsp90 is known to work in concert with Hsp70 to maintain proteins such as tau in a soluble and functional conformation (Dou et al. 2003) and to inhibit early stages of β-amyloid aggregation (Evans et al. 2006). Hsp90 can also inhibit α-synuclein aggregation by interaction with soluble oligomers (Daturpalli et al. 2013; Falsone et al. 2009). In Parkinson’s models, CHIP and Hsp90 both modulate the stability of leucine-rich repeat kinase 2 (LRRK2), a protein in which mutations increase the risk for developing Parkinson’s disease (Ding and Goldberg 2009; Hurtado-Lorenzo and Anand 2008; Ko et al. 2009; Rudenko et al. 2012; Wang et al. 2008). Furthermore, the mitochondrial Hsp90 protein, tumor necrosis factor receptor-associated protein 1 (TRAP1), is known to mitigate α-synuclein toxicity (Butler et al. 2012). Hsp90 levels are increased in the brain of Parkinson’s patients (Uryu et al. 2006) and Hsp90 is found in Lewy bodies in this condition (Auluck et al. 2002; Kalia et al. 2010; Klucken et al. 2004; McLean et al. 2002). However, serum levels of Hsp90 have been shown to be decreased in patients with Alzheimer’s disease (Gezen-Ak et al. 2013). Hsp90 has also been shown to be downregulated in the temporal lobe in Alzheimer’s disease (Yokota et al. 2006). In contrast, no changes have been observed in Hsp90 levels in the cerebellum, hippocampus, or parietal lobule in patients with mild cognitive impairment (Di Domenico et al. 2010). Similar to our recent study of the striatum and substantia nigra of the female rat, Dickey and colleagues reported no age-related change in Hsp90 in whole brain extracts (Dickey et al. 2009). However, immunohistochemical studies have shown that Hsp90 and its co-chaperone p23 increase dramatically with age in the pyramidal cells of the hippocampus proper and in polymorphic cells of the dentate gyrus (Lee et al. 2011). There is also an age-related increase in basal Hsp90 levels within human blood cells (Njemini et al. 2007). Finally, there is an increase in Hsp90 in the 20S proteasomal fraction of the aged brain and liver, suggesting increased recruitment of this protein to help rescue cells from age-related loss of proteasome activity (Dasuri et al. 2009).
Conclusions
Proteotoxic stress is a hallmark of age-related neurodegenerative disorders and heat shock proteins are one of the most important natural defenses against it. Thus, learning how to safely boost heat shock proteins at the right time and in the right location may have an immense impact on the future treatment of Alzheimer’s and Parkinson’s disease. Some of the age-related neurodegenerative disorders already elicit an endogenous increase in several heat shock proteins. This may represent a compensatory mechanism to restore homeostatic equilibrium and slow down the progression of age- or disease-related pathologies. If a particular heat shock protein is highest in the very oldest group of individuals and increased in patients with Alzheimer’s and Parkinson’s diagnoses, the threshold of proteotoxic stress at which that protein is maximally induced must be fairly high, as appears to be the case with Hsp25. However, it is evident from this review that the heat shock protein response to stress is highly variable and defies any simplistic generalizations. For example, for many heat shock proteins, there is no increase with age-related stress, but a gradual age-related decline or a biphasic response to aging. Furthermore, some heat shock proteins are not affected or decreased in postmortem tissue from Parkinson’s and Alzheimer’s victims. The patients studied in these latter reports are likely to be at advanced Braak stages of the disorder because they suffer from a clinically overt syndrome and considerable amounts of tau or α-synuclein pathology are present in the brains of patients with confirmed diagnoses (Braak and Braak 1995, 1997a, b; Braak et al. 2003). Thus, the inhibition of some heat shock proteins with the severe proteotoxic stress associated with these diagnoses is consistent with Selye’s views of the negative impact of chronic stress (Selye 1975). Further studies of heat shock protein expression are warranted as a function of earlier Braak stages, such as in patients with incidental Lewy body disease, a potentially prodromal phase of Parkinson’s disease or in patients with mild cognitive impairment, which may reflect incipient dementia. It is possible that temporal kinetic studies as a function of Braak stage will show biphasic responses of at least some heat shock proteins to disease duration and/or severity.
Given the signature topographic patterns of neurofibrillary tangle and Lewy pathology in Alzheimer’s and Parkinson’s disease, it seems likely that some brain regions suffer from greater proteotoxic stress than others. Furthermore, within specific brain regions, usually one phenotypically defined cellular population is the most vulnerable group. Further examinations of topographic heat shock protein expression as a function of brain region and cell type with immunohistochemical and Western immunoblotting tools would therefore be valuable. Furthermore, correlating heat shock protein expression within specific brain regions with their gradual development of tau+ and α-synuclein+ inclusions in Alzheimer’s or Parkinson’s disease may yield deeper insights into the effects of the dose/severity of proteotoxic stress on the nature of the heat shock protein response. As is the case with the antioxidant thiol glutathione, one might expect resilient regions/cell types to exhibit higher heat shock protein levels than more vulnerable groups (Mythri et al. 2011). On the other hand, heat shock protein induction may be higher in more vulnerable brain regions precisely because they are exposed to greater stress, as we have recently shown for the differentially vulnerable neocortex and allocortex of the telencephalon (Posimo et al. 2013). Although the allocortex is much more vulnerable to neurofibrillary and Lewy pathology than the neocortex, we discovered that it can also mount dynamic defensive responses to proteotoxic stress, such as high stress-induced increases in Hsp70 and Hsp32 (Posimo et al. 2013). If the compensatory heat shock protein response in highly susceptible brain regions were blocked, those brain regions would be expected to become all the more vulnerable to neurodegeneration. In other words, loss of heat shock protein activity in highly vulnerable brain regions is expected to be all the more catastrophic.
Given the variability in heat shock protein expression in the aged and diseased brain, we propose that the increases or decreases observed in postmortem tissue must be interpreted in the context of topographic and cell-specific neurodegeneration. If the majority of cells captured in postmortem tissue from patients with neurodegenerative disorders are those that have successfully survived proteotoxic stress, homogenized tissue from that brain region would be expected to show a net increase in at least some heat shock proteins, even in the brain regions most prone to developing tau, α-synuclein, or β-amyloid pathology. Conversely, if postmortem tissue exhibits lower levels of heat shock proteins, the majority of cells captured at that cross-sectional point may already be on the way to demise. Because of the striking heterogeneity of brain tissue, it might be useful to co-label heat shock proteins with apoptotic markers such as cleaved caspase 3 or TUNEL staining at the cellular level in order to determine whether those cells that are destined to die are indeed the ones with lowest heat shock protein expression and whether the survivors retain or increase heat shock proteins. Similarly, heat shock proteins could be co-labeled with markers for tau+ or α-synuclein+ inclusions to determine whether cells that manage to avoid Lewy or tangle pathology have higher heat shock protein expression, as shown for αB-crystallin by Braak and colleagues (Braak et al. 2001). In this manner, compensatory responses in a few resistant cell types would not be masked by loss of heat shock proteins in other, more vulnerable and numerous cell types in the usual assays of homogenized brain tissue.
Finally, another variable to consider in studies of heat shock proteins is gender, as Parkinson’s disease appears to be more common in men and Alzheimer’s disease in women. For example, Hsp70 is regulated by exercise in a sex-specific manner in the myocardium, with potential implications for the prescription of exercise to men versus women (Milne and Noble 2008). For this reason, we have listed gender in Table 1. However, differences in heat shock protein expression as a function of gender can only be rigorously determined when female and male tissue is examined in parallel under precisely the same assay conditions. Although a few studies have controlled for gender dimorphisms, many studies on human tissue simply combine male and female subjects because of a low “n”. Although no obvious sex differences were noted in Table 1, the typically small sample size of many human studies may not generate sufficient statistical power to detect a subtle gender difference. The rodent studies in Table 1 do not suffer from this sample size limitation, but only examine male tissue, with the exception of our recent study on female rats (Gleixner et al. 2014). Therefore, Clayton and colleagues have recently argued that females should be examined with greater frequency (Clayton and Collins 2014).
In conclusion, it would be useful to investigate the heat shock protein response to age- and disease-related proteotoxic stress with greater temporal and spatial resolution as well as a new focus on gender. These data would help support or refute the hypothesis that early increases in heat shock proteins serve to delay the progression of age-related proteinopathies within resistant cell subpopulations whereas decreases in heat shock proteins serve to hasten cellular demise in the more vulnerable groups. Furthermore, if there are sex differences in heat shock proteins, it will be important to determine if they contribute to male/female differences in the risk of developing neurodegeneration.
Abbreviations
- ARE:
-
Antioxidant response element
- AIF:
-
Apoptosis-inducing factor
- Apaf1:
-
Apoptotic protease activation factor 1
- Ask1:
-
Apoptosis signal-regulating kinase
- ATF6:
-
Activating transcription factor 6
- BAG-1:
-
Bcl-2-associated athanogene
- CHIP:
-
Carboxy terminus of Hsp70-interacting protein
- CHOP:
-
CCAAT-enhancer-binding protein homologous protein
- JNK:
-
c-Jun N-terminal kinase
- ERAD:
-
Endoplasmic reticulum-associated protein degradation
- GRP:
-
Glucose-regulated protein
- Hsc70:
-
Heat shock cognate 70
- Hsp:
-
Heat shock protein
- HO1:
-
Heme oxygenase 1
- Hip:
-
Hsp70-interacting protein
- Hop:
-
Hsp70/90-organizing protein
- Keap1:
-
Kelch-like ECH-associated protein 1
- LRRK2:
-
Leucine-rich repeat kinase 2
- LAMP2a:
-
Lysosomal-associated membrane transporter 2a
- mtHsp70:
-
Mitochondrial Hsp70
- Nrf2:
-
Nuclear factor erythroid 2 related factor 2
- PERK:
-
PRKR-like endoplasmic reticulum kinase
- IRE1:
-
Serine/threonine-protein kinase/endoribonuclease
- TPR1:
-
Tetratrico-peptide repeat-1
- TRAP1:
-
TNF receptor-associated protein 1
- UPR:
-
Unfolded protein response
References
Acunzo J, Katsogiannou M, Rocchi P (2012) Small heat shock proteins HSP27 (HspB1), alphaB-crystallin (HspB5) and HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol 44:1622–31
Alladi PA, Mahadevan A, Vijayalakshmi K, Muthane U, Shankar SK, Raju TR (2010) Ageing enhances alpha-synuclein, ubiquitin and endoplasmic reticular stress protein expression in the nigral neurons of Asian Indians. Neurochem Int 57:530–9
Allen NJ, Barres BA (2009) Neuroscience: Glia - more than just brain glue. Nature 457:675–7
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH (2010) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 67:1464–72
Angot E, Steiner JA, Hansen C, Li JY, Brundin P (2010) Are synucleinopathies prion-like disorders? Lancet Neurol 9:1128–38
Arias E, Cuervo AM (2011) Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol 23:184–9
Aridon P, Geraci F, Turturici G, D’Amelio M, Savettieri G, Sconzo G (2011) Protective role of heat shock proteins in Parkinson’s disease. Neurodegener Dis 8:155–68
Arumugam TV, Phillips TM, Cheng A, Morrell CH, Mattson MP, Wan R (2010) Age and energy intake interact to modify cell stress pathways and stroke outcome. Ann Neurol 67:41–52
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295:865–8
Aztatzi-Santillan E, Nares-Lopez FE, Marquez-Valadez B, Aguilera P, Chanez-Cardenas ME (2010) The protective role of heme oxygenase-1 in cerebral ischemia. Cent Nerv Syst Agents Med Chem 10:310–6
Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ (2012) Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci Off J Soc Neurosci 32:13454–69
Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 19:4535–45
Barres BA (2008) The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60:430–40
Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR (2000) Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2:469–75
Blake MJ, Fargnoli J, Gershon D, Holbrook NJ (1991) Concomitant decline in heat-induced hyperthermia and HSP70 mRNA expression in aged rats. Am J Physiol 260:R663–7
Boger HA, Granholm AC, McGinty JF, Middaugh LD (2010) A dual-hit animal model for age-related parkinsonism. Prog Neurobiol 90:217–29
Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16:271–8, discussion 278–84
Braak H, Braak E (1997a) Diagnostic criteria for neuropathologic assessment of Alzheimer’s disease. Neurobiol Aging 18:S85–8
Braak H, Braak E (1997b) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–7
Braak H, Del Tredici K, Sandmann-Kiel D, Rub U, Schultz C (2001) Nerve cells expressing heat-shock proteins in Parkinson’s disease. Acta Neuropathol (Berl) 102:449–54
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Brocchieri L, de Macario EC, Macario AJ (2008) hsp70 genes in the human genome: conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol Biol 8:19
Brown MK, Naidoo N (2012) The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol 3:263
Brunner M, Schneider HC, Lill R, Neupert W (1995) Dissection of protein translocation across the mitochondrial outer and inner membranes. Cold Spring Harb Symp Quant Biol 60:619–27
Bukau B, Horwich AL (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92:351–66
Burbulla LF, Schelling C, Kato H, Rapaport D, Woitalla D, Schiesling C, Schulte C, Sharma M, Illig T, Bauer P, Jung S, Nordheim A, Schols L, Riess O, Kruger R (2010) Dissecting the role of the mitochondrial chaperone mortalin in Parkinson’s disease: functional impact of disease-related variants on mitochondrial homeostasis. Hum Mol Genet 19:4437–52
Butler EK, Voigt A, Lutz AK, Toegel JP, Gerhardt E, Karsten P, Falkenburger B, Reinartz A, Winklhofer KF, Schulz JB (2012) The mitochondrial chaperone protein TRAP1 mitigates alpha-Synuclein toxicity. PLoS Genet 8:e1002488
Calabrese EJ (2010) Hormesis is central to toxicology, pharmacology and risk assessment. Hum Exp Toxicol 29:249–61
Calabrese EJ (2013) Biphasic dose responses in biology, toxicology and medicine: accounting for their generalizability and quantitative features. Environ Pollut 182:452–60
Calabrese EJ, Blain RB (2011) The hormesis database: the occurrence of hormetic dose responses in the toxicological literature. Regul Toxicol Pharmacol: RTP 61:73–81
Calabrese V, Scapagnini G, Ravagna A, Colombrita C, Spadaro F, Butterfield DA, Giuffrida Stella AM (2004) Increased expression of heat shock proteins in rat brain during aging: relationship with mitochondrial function and glutathione redox state. Mech Ageing Dev 125:325–35
Calabrese V, Cornelius C, Mancuso C, Barone E, Calafato S, Bates T, Rizzarelli E, Kostova AT (2009) Vitagenes, dietary antioxidants and neuroprotection in neurodegenerative diseases. Front Biosci 14:376–97
Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP (2010) Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid Redox Signal 13:1763–811
Calabrese V, Cornelius C, Cuzzocrea S, Iavicoli I, Rizzarelli E, Calabrese EJ (2011) Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol Aspects Me 32:279–304
Calabrese V, Cornelius C, Dinkova-Kostova AT, Iavicoli I, Di Paola R, Koverech A, Cuzzocrea S, Rizzarelli E, Calabrese EJ (2012) Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim Biophys Acta 1822:753–83
Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G (2002) Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie 84:215–22
Carvey PM, Punati A, Newman MB (2006) Progressive dopamine neuron loss in Parkinson’s disease: the multiple hit hypothesis. Cell Transplant 15:239–50
Cecarini V, Ding Q, Keller JN (2007) Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic Res 41:673–80
Chen S, Brown IR (2007) Neuronal expression of constitutive heat shock proteins: implications for neurodegenerative diseases. Cell Stress Chaperones 12:51–8
Chen CM, Wu YR, Hu FJ, Chen YC, Chuang TJ, Cheng YF, Lee-Chen GJ (2008) HSPA5 promoter polymorphisms and risk of Parkinson’s disease in Taiwan. Neurosci Lett 435:219–22
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH (2009) Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35:385–98
Clayton JA, Collins FS (2014) Policy: NIH to balance sex in cell and animal studies. Nature 509:282–3
Conconi M, Friguet B (1997) Proteasome inactivation upon aging and on oxidation-effect of HSP 90. Mol Biol Rep 24:45–50
Conconi M, Szweda LI, Levine RL, Stadtman ER, Friguet B (1996) Age-related decline of rat liver multicatalytic proteinase activity and protection from oxidative inactivation by heat-shock protein 90. Arch Biochem Biophys 331:232–40
Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol 3:93–6
Cornelius C, Perrotta R, Graziano A, Calabrese EJ, Calabrese V (2013) Stress responses, vitagenes and hormesis as critical determinants in aging and longevity: Mitochondria as a “chi”. Immun Ageing: I & A 10:15
Cuervo AM (2004) Autophagy: in sickness and in health. Trends Cell Biol 14:70–7
Cuervo AM (2008) Autophagy and aging: keeping that old broom working. Trends Genet: TIG 24:604–12
Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A (2005) Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 1:131–40
Dabir DV, Trojanowski JQ, Richter-Landsberg C, Lee VM, Forman MS (2004) Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. Am J Pathol 164:155–66
Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B (2009) Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J Neurochem 111:192–203
Dasuri K, Zhang L, Ebenezer P, Liu Y, Fernandez-Kim SO, Keller JN (2009) Aging and dietary restriction alter proteasome biogenesis and composition in the brain and liver. Mech Ageing Dev 130:777–83
Dasuri K, Zhang L, Keller JN (2013) Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med 62:170–85
Daturpalli S, Waudby CA, Meehan S, Jackson SE (2013) Hsp90 inhibits alpha-synuclein aggregation by interacting with soluble oligomers. J Mol Biol 425:4614–28
Daugaard M, Rohde M, Jaattela M (2007) The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett 581:3702–10
De Mena L, Coto E, Sanchez-Ferrero E, Ribacoba R, Guisasola LM, Salvador C, Blazquez M, Alvarez V (2009) Mutational screening of the mortalin gene (HSPA9) in Parkinson’s disease. J Neural Transm 116:1289–93
Dennery PA (2000) Regulation and role of heme oxygenase in oxidative injury. Curr Top Cell Regul 36:181–99
Desmard M, Boczkowski J, Poderoso J, Motterlini R (2007) Mitochondrial and cellular heme-dependent proteins as targets for the bioactive function of the heme oxygenase/carbon monoxide system. Antioxid Redox Signal 9:2139–55
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106:13010–5
Di Domenico F, Sultana R, Tiu GF, Scheff NN, Perluigi M, Cini C, Butterfield DA (2010) Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: an investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Res Protocol 1333:72–81
Dickey C, Kraft C, Jinwal U, Koren J, Johnson A, Anderson L, Lebson L, Lee D, Dickson D, de Silva R, Binder LI, Morgan D, Lewis J (2009) Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am J Pathol 174:228–38
Dickson DW (2009) Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol 3:1–23
Ding X, Goldberg MS (2009) Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PLoS One 4:e5949
Ding Q, Dimayuga E, Keller JN (2006) Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid Redox Signal 8:163–72
Dobson CM (2003) Protein folding and misfolding. Nature 426:884–90
Dong Z, Wolfer DP, Lipp HP, Bueler H (2005) Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol Ther: J Am Soc Gene Ther 11:80–8
Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H (2003) Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A 100:721–6
Durrenberger PF, Filiou MD, Moran LB, Michael GJ, Novoselov S, Cheetham ME, Clark P, Pearce RK, Graeber MB (2009) DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in parkinsonian astrocytes. J Neurosci Res 87:238–45
Elliott E, Tsvetkov P, Ginzburg I (2007) BAG-1 associates with Hsc70. Tau complex and regulates the proteasomal degradation of Tau protein. J Biol Chem 282:37276–84
Esser C, Alberti S, Hohfeld J (2004) Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim Biophys Acta 1695:171–88
Evans CG, Wisen S, Gestwicki JE (2006) Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J Biol Chem 281:33182–91
Falsone SF, Kungl AJ, Rek A, Cappai R, Zangger K (2009) The molecular chaperone Hsp90 modulates intermediate steps of amyloid assembly of the Parkinson-related protein alpha-synuclein. J Biol Chem 284:31190–9
Freimann K, Zschiedrich K, Bruggemann N, Grunewald A, Pawlack H, Hagenah J, Lohmann K, Klein C, Westenberger A (2013) Mortalin mutations are not a frequent cause of early-onset Parkinson disease. Neurobiol Aging 34(2694):e19–20
Friguet B, Bulteau AL, Chondrogianni N, Conconi M, Petropoulos I (2000) Protein degradation by the proteasome and its implications in aging. Ann N Y Acad Sci 908:143–54
Gabai VL, Yaglom JA, Volloch V, Meriin AB, Force T, Koutroumanis M, Massie B, Mosser DD, Sherman MY (2000) Hsp72-mediated suppression of c-Jun N-terminal kinase is implicated in development of tolerance to caspase-independent cell death. Mol Cell Biol 20:6826–36
Gao HM, Hong JS (2011) Gene-environment interactions: key to unraveling the mystery of Parkinson’s disease. Prog Neurobiol 94:1–19
Getchell TV, Krishna NS, Dhooper N, Sparks DL, Getchell ML (1995) Human olfactory receptor neurons express heat shock protein 70: age-related trends. Ann Otol Rhinol Laryngol 104:47–56
Gezen-Ak D, Dursun E, Hanagasi H, Bilgic B, Lohman E, Araz OS, Atasoy IL, Alaylioglu M, Onal B, Gurvit H, Yilmazer S (2013) BDNF, TNFalpha, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J Alzheimers Dis: JAD 37:185–95
Gleixner AM, Pulugulla SH, Pant DB, Posimo JM, Crum TS, Leak RK (2014) Impact of aging on heat shock protein expression in the substantia nigra and striatum of the female rat. Cell Tissue Res 357:43–54
Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426:895–9
Goldberg AL (2007) Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans 35:12–7
Gorbatyuk MS, Shabashvili A, Chen W, Meyers C, Sullivan LF, Salganik M, Lin JH, Lewin AS, Muzyczka N, Gorbatyuk OS (2012) Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol Ther: J Am Soc Gene Ther 20:1327–37
Goswami AV, Samaddar M, Sinha D, Purushotham J, D’Silva P (2012) Enhanced J-protein interaction and compromised protein stability of mtHsp70 variants lead to mitochondrial dysfunction in Parkinson’s disease. Hum Mol Genet 21:3317–32
Grochot-Przeczek A, Dulak J, Jozkowicz A (2012) Haem oxygenase-1: non-canonical roles in physiology and pathology. Clin Sci 122:93–103
Gupte AA, Morris JK, Zhang H, Bomhoff GL, Geiger PC, Stanford JA (2010) Age-related changes in HSP25 expression in basal ganglia and cortex of F344/BN rats. Neurosci Lett 472:90–3
Hamos JE, Oblas B, Pulaski-Salo D, Welch WJ, Bole DG, Drachman DA (1991) Expression of heat shock proteins in Alzheimer’s disease. Neurology 41:345–50
Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715–25
Hauser MA, Li YJ, Xu H, Noureddine MA, Shao YS, Gullans SR, Scherzer CR, Jensen RV, McLaurin AC, Gibson JR, Scott BL, Jewett RM, Stenger JE, Schmechel DE, Hulette CM, Vance JM (2005) Expression profiling of substantia nigra in Parkinson disease, progressive supranuclear palsy, and frontotemporal dementia with parkinsonism. Arch Neurol 62:917–21
Helfert RH, Glatz FR 3rd, Wilson TS, Ramkumar V, Hughes LF (2002) Hsp70 in the inferior colliculus of Fischer-344 rats: effects of age and acoustic stress. Hear Res 170:155–65
Hirose W, Ikematsu K, Tsuda R (2003) Age-associated increases in heme oxygenase-1 and ferritin immunoreactivity in the autopsied brain. Legal Med 5(Suppl 1):S360–6
Hohfeld J, Jentsch S (1997) GrpE-like regulation of the hsc70 chaperone by the anti-apoptotic protein BAG-1. EMBO J 16:6209–16
Hohfeld J, Minami Y, Hartl FU (1995) Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell 83:589–98
Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W (2005) The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110:165–72
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A 96:3228–33
Hurtado-Lorenzo A, Anand VS (2008) Heat shock protein 90 modulates LRRK2 stability: potential implications for Parkinson’s disease treatment. J Neurosci Off J Soc Neurosci 28:6757–9
Hussain SG, Ramaiah KV (2007) Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun 355:365–70
Imaizumi K, Miyoshi K, Katayama T, Yoneda T, Taniguchi M, Kudo T, Tohyama M (2001) The unfolded protein response and Alzheimer’s disease. Biochim Biophys Acta 1536:85–96
Jazwa A, Cuadrado A (2010) Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr Drug Targets 11:1517–31
Jellinger KA (2008) Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neurodegener Dis 5:118–21
Jellinger KA (2009) Recent advances in our understanding of neurodegeneration. J Neural Transm 116:1111–62
Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Hohfeld J, Patterson C (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem 276:42938–44
Jin J, Hulette C, Wang Y, Zhang T, Pan C, Wadhwa R, Zhang J (2006) Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol Cell Proteomics: MCP 5:1193–204
Jinwal UK, O’Leary JC 3rd, Borysov SI, Jones JR, Li Q, Koren J 3rd, Abisambra JF, Vestal GD, Lawson LY, Johnson AG, Blair LJ, Jin Y, Miyata Y, Gestwicki JE, Dickey CA (2010) Hsc70 rapidly engages tau after microtubule destabilization. J Biol Chem 285:16798–805
Johnson JL, Brown C (2009) Plasticity of the Hsp90 chaperone machine in divergent eukaryotic organisms. Cell Stress Chaperones 14:83–94
Jung AE, Fitzsimons HL, Bland RJ, During MJ, Young D (2008) HSP70 and constitutively active HSF1 mediate protection against CDCrel-1-mediated toxicity. Mol Ther: J Am Soc Gene Ther 16:1048–55
Kabani M, Martineau CN (2008) Multiple hsp70 isoforms in the eukaryotic cytosol: mere redundancy or functional specificity? Curr Genom 9:338–248
Kalia SK, Kalia LV, McLean PJ (2010) Molecular chaperones as rational drug targets for Parkinson’s disease therapeutics. CNS Neurol Disord Drug Targets 9:741–53
Kalia LV, Kalia SK, Chau H, Lozano AM, Hyman BT, McLean PJ (2011) Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS One 6:e14695
Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M (1999) Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol 1:479–85
Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M (2004) Induction of neuronal death by ER stress in Alzheimer’s disease. J Chem Neuroanat 28:67–78
Kaushik S, Cuervo AM (2006) Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med 27:444–54
Kaushik S, Cuervo AM (2009) Methods to monitor chaperone-mediated autophagy. Methods Enzymol 452:297–324
Kawamoto Y, Akiguchi I, Shirakashi Y, Honjo Y, Tomimoto H, Takahashi R, Budka H (2007) Accumulation of Hsc70 and Hsp70 in glial cytoplasmic inclusions in patients with multiple system atrophy. Brain Res Protocol 1136:219–27
Keller JN, Hanni KB, Markesbery WR (2000a) Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev 113:61–70
Keller JN, Huang FF, Markesbery WR (2000b) Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 98:149–56
Keller JN, Gee J, Ding Q (2002) The proteasome in brain aging. Ageing Res Rev 1:279–93
Keller JN, Dimayuga E, Chen Q, Thorpe J, Gee J, Ding Q (2004) Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol 36:2376–91
Kennedy D, Jager R, Mosser DD, Samali A (2014) Regulation of apoptosis by heat shock proteins. IUBMB Life
Kisselev AF, Goldberg AL (2005) Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol 398:364–78
Kisselev AF, Garcia-Calvo M, Overkleeft HS, Peterson E, Pennington MW, Ploegh HL, Thornberry NA, Goldberg AL (2003) The caspase-like sites of proteasomes, their substrate specificity, new inhibitors and substrates, and allosteric interactions with the trypsin-like sites. J Biol Chem 278:35869–77
Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ (2004) Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem 279:25497–502
Ko HS, Bailey R, Smith WW, Liu Z, Shin JH, Lee YI, Zhang YJ, Jiang H, Ross CA, Moore DJ, Patterson C, Petrucelli L, Dawson TM, Dawson VL (2009) CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc Natl Acad Sci U S A 106:2897–902
Koga H, Kaushik S, Cuervo AM (2011) Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev 10:205–15
Kroemer G (2001) Heat shock protein 70 neutralizes apoptosis-inducing factor. Sci World J 1:590–2
Krueger-Naug AM, Plumier JC, Hopkins DA, Currie RW (2002) Hsp27 in the nervous system: expression in pathophysiology and in the aging brain. Prog Mol Subcell Biol 28:235–51
Kumar P, Ambasta RK, Veereshwarayya V, Rosen KM, Kosik KS, Band H, Mestril R, Patterson C, Querfurth HW (2007) CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum Mol Genet 16:848–64
Labbadia J, Morimoto RI (2014) Proteostasis and longevity: when does aging really begin? F1000 prime reports 6: 7
Lanneau D, Brunet M, Frisan E, Solary E, Fontenay M, Garrido C (2008) Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med 12:743–61
Lanneau D, Wettstein G, Bonniaud P, Garrido C (2010) Heat shock proteins: cell protection through protein triage. Sci World J 10:1543–52
Leak RK (2014) Adaptation and sensitization to proteotoxic stress. Dose–response: Publ Int Hormesis Soc 12:24–56
Lecker SH, Goldberg AL, Mitch WE (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol: JASN 17:1807–19
Lee CK, Weindruch R, Prolla TA (2000) Gene-expression profile of the ageing brain in mice. Nat Genet 25:294–7
Lee KS, Chung JH, Oh BH, Hong CH (2008) Increased plasma levels of heat shock protein 70 in patients with vascular mild cognitive impairment. Neurosci Lett 436:223–6
Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ (2010a) Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285:9262–72
Lee JH, Won SM, Suh J, Son SJ, Moon GJ, Park UJ, Gwag BJ (2010b) Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp Mol Med 42:386–94
Lee CH, Park JH, Choi JH, Yoo KY, Ryu PD, Won MH (2011) Heat shock protein 90 and its cochaperone, p23, are markedly increased in the aged gerbil hippocampus. Exp Gerontol 46:768–72
Leverenz JB, Umar I, Wang Q, Montine TJ, McMillan PJ, Tsuang DW, Jin J, Pan C, Shin J, Zhu D, Zhang J (2007) Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol 17:139–45
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–217
Lowe J, McDermott H, Pike I, Spendlove I, Landon M, Mayer RJ (1992) alpha B crystallin expression in non-lenticular tissues and selective presence in ubiquitinated inclusion bodies in human disease. J Pathol 166:61–8
Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429:883–91
Luk KC, Mills IP, Trojanowski JQ, Lee VM (2008) Interactions between Hsp70 and the hydrophobic core of alpha-synuclein inhibit fibril assembly. Biochemistry 47:12614–25
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012a) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–53
Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012b) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209:975–86
Magrane J, Smith RC, Walsh K, Querfurth HW (2004) Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci Off J Soc Neurosci 24:1700–6
Mandel S, Grunblatt E, Riederer P, Amariglio N, Jacob-Hirsch J, Rechavi G, Youdim MB (2005) Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci 1053:356–75
Manning-Bog AB, Langston JW (2007) Model fusion, the next phase in developing animal models for Parkinson’s disease. Neurotox Res 11:219–40
Massey A, Kiffin R, Cuervo AM (2004) Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol 36:2420–34
Massey AC, Zhang C, Cuervo AM (2006) Chaperone-mediated autophagy in aging and disease. Curr Top Dev Biol 73:205–35
Mattson MP (2008) Hormesis defined. Ageing Res Rev 7:1–7
Mayer MP, Bukau B (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci: CMLS 62:670–84
McLean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT (2002) TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J Neurochem 83:846–54
McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD (2006) Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem 281:24756–68
Milne KJ, Noble EG (2008) Response of the myocardium to exercise: sex-specific regulation of hsp70. Med Sci Sports Exerc 40:655–63
Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C (2008) CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol 28:4018–25
Minami Y, Hohfeld J, Ohtsuka K, Hartl FU (1996) Regulation of the heat-shock protein 70 reaction cycle by the mammalian DnaJ homolog, Hsp40. J Biol Chem 271:19617–24
Molochnikov L, Rabey JM, Dobronevsky E, Bonucelli U, Ceravolo R, Frosini D, Grunblatt E, Riederer P, Jacob C, Aharon-Peretz J, Bashenko Y, Youdim MB, Mandel SA (2012) A molecular signature in blood identifies early Parkinson’s disease. Mol Neurodegener 7:26
Morimoto RI (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev 22:1427–38
Morimoto RI (2011) The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol 76:91–9
Morimoto RI, Cuervo AM (2014) Proteostasis and the aging proteome in health and disease. J Gerontol A: Biol Med Sci 69(Suppl 1):S33–8
Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22
Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2:a006338
Mythri RB, Venkateshappa C, Harish G, Mahadevan A, Muthane UB, Yasha TC, Srinivas Bharath MM, Shankar SK (2011) Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochem Res 36:1452–63
Nagel F, Falkenburger BH, Tonges L, Kowsky S, Poppelmeyer C, Schulz JB, Bahr M, Dietz GP (2008) Tat-Hsp70 protects dopaminergic neurons in midbrain cultures and in the substantia nigra in models of Parkinson’s disease. J Neurochem 105:853–64
Naidoo N, Ferber M, Master M, Zhu Y, Pack AI (2008) Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci Off J Soc Neurosci 28:6539–48
Njemini R, Lambert M, Demanet C, Kooijman R, Mets T (2007) Basal and infection-induced levels of heat shock proteins in human aging. Biogerontology 8:353–64
Nowotny K, Jung T, Grune T, Hohn A (2014) Accumulation of modified proteins and aggregate formation in aging. Exp Gerontol 57:122–131
Orenstein SJ, Cuervo AM (2010) Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol 21:719–26
Outeiro TF, Klucken J, Strathearn KE, Liu F, Nguyen P, Rochet JC, Hyman BT, McLean PJ (2006) Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem Biophys Res Commun 351:631–8
Pardue S, Groshan K, Raese JD, Morrison-Bogorad M (1992) Hsp70 mRNA induction is reduced in neurons of aged rat hippocampus after thermal stress. Neurobiol Aging 13:661–72
Pardue S, Wang S, Miller MM, Morrison-Bogorad M (2007) Elevated levels of inducible heat shock 70 proteins in human brain. Neurobiol Aging 28:314–24
Park HS, Lee JS, Huh SH, Seo JS, Choi EJ (2001) Hsp72 functions as a natural inhibitory protein of c-Jun N-terminal kinase. EMBO J 20:446–56
Park HS, Cho SG, Kim CK, Hwang HS, Noh KT, Kim MS, Huh SH, Kim MJ, Ryoo K, Kim EK, Kang WJ, Lee JS, Seo JS, Ko YG, Kim S, Choi EJ (2002) Heat shock protein hsp72 is a negative regulator of apoptosis signal-regulating kinase 1. Mol Cell Biol 22:7721–30
Paz Gavilan M, Vela J, Castano A, Ramos B, del Rio JC, Vitorica J, Ruano D (2006) Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging 27:973–82
Pearl LH, Prodromou C (2006) Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem 75:271–94
Posimo JM, Titler AM, Choi HJ, Unnithan AS, Leak RK (2013) Neocortex and allocortex respond differentially to cellular stress in vitro and aging in vivo. PLoS One 8:e58596
Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C, Kroemer G (2001) Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol 3:839–43
Renkawek K, Bosman GJ, Gaestel M (1993) Increased expression of heat-shock protein 27 kDa in Alzheimer disease: a preliminary study. Neuroreport 5:14–6
Renkawek K, Bosman GJ, de Jong WW (1994a) Expression of small heat-shock protein hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol 87:511–9
Renkawek K, Voorter CE, Bosman GJ, van Workum FP, de Jong WW (1994b) Expression of alpha B-crystallin in Alzheimer’s disease. Acta Neuropathol 87:155–60
Renkawek K, Stege GJ, Bosman GJ (1999) Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B-crystallin in Parkinson’s disease. Neuroreport 10:2273–6
Roodveldt C, Bertoncini CW, Andersson A, van der Goot AT, Hsu ST, Fernandez-Montesinos R, de Jong J, van Ham TJ, Nollen EA, Pozo D, Christodoulou J, Dobson CM (2009) Chaperone proteostasis in Parkinson’s disease: stabilization of the Hsp70/alpha-synuclein complex by Hip. EMBO J 28:3758–70
Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10(Suppl):S10–7
Rudenko IN, Kaganovich A, Hauser DN, Beylina A, Chia R, Ding J, Maric D, Jaffe H, Cookson MR (2012) The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem J 446:99–111
Ryter SW, Tyrrell RM (2000) The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med 28:289–309
Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES (2000) Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2:476–83
Salminen A, Ojala J, Kaarniranta K, Hiltunen M, Soininen H (2011) Hsp90 regulates tau pathology through co-chaperone complexes in Alzheimer’s disease. Prog Neurobiol 93:99–110
Salway KD, Gallagher EJ, Page MM, Stuart JA (2011) Higher levels of heat shock proteins in longer-lived mammals and birds. Mech Ageing Dev 132:287–97
Sato N, Urano F, Yoon Leem J, Kim SH, Li M, Donoviel D, Bernstein A, Lee AS, Ron D, Veselits ML, Sisodia SS, Thinakaran G (2000) Upregulation of BiP and CHOP by the unfolded-protein response is independent of presenilin expression. Nat Cell Biol 2:863–70
Scherzer CR, Eklund AC, Morse LJ, Liao Z, Locascio JJ, Fefer D, Schwarzschild MA, Schlossmacher MG, Hauser MA, Vance JM, Sudarsky LR, Standaert DG, Growdon JH, Jensen RV, Gullans SR (2007) Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc Natl Acad Sci U S A 104:955–60
Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101:199–210
Schipper HM (2000) Heme oxygenase-1: role in brain aging and neurodegeneration. Exp Gerontol 35:821–30
Schipper HM (2011) Heme oxygenase-1 in Alzheimer disease: a tribute to Moussa Youdim. J Neural Transm 118:381–7
Schipper HM, Liberman A, Stopa EG (1998) Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp Neurol 150:60–8
Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, Kelly J, Arvanitakis Z (2006) Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging 27:252–61
Schipper HM, Song W, Zukor H, Hascalovici JR, Zeligman D (2009) Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J Neurochem 110:469–85
Schneider HC, Berthold J, Bauer MF, Dietmeier K, Guiard B, Brunner M, Neupert W (1994) Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature 371:768–74
Schroder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569:29–63
Schultz C, Dick EJ, Cox AB, Hubbard GB, Braak E, Braak H (2001) Expression of stress proteins alpha B-crystallin, ubiquitin, and hsp27 in pallido-nigral spheroids of aged rhesus monkeys. Neurobiol Aging 22:677–82
Seidel K, Vinet J, Dunnen WF, Brunt ER, Meister M, Boncoraglio A, Zijlstra MP, Boddeke HW, Rub U, Kampinga HH, Carra S (2012) The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol Appl Neurobiol 38:39–53
Selye H (1975) Stress without distress. Signet, Philadelphia, Vol
Sherman MY, Goldberg AL (2001) Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29:15–32
Shimura H, Schwartz D, Gygi SP, Kosik KS (2004) CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 279:4869–76
Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ (2005) The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 280:23727–34
Shinohara H, Inaguma Y, Goto S, Inagaki T, Kato K (1993) Alpha B crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer’s disease. J Neurol Sci 119:203–8
Soti C, Csermely P (2000) Molecular chaperones and the aging process. Biogerontology 1:225–33
Stege GJ, Renkawek K, Overkamp PS, Verschuure P, van Rijk AF, Reijnen-Aalbers A, Boelens WC, Bosman GJ, de Jong WW (1999) The molecular chaperone alphaB-crystallin enhances amyloid beta neurotoxicity. Biochem Biophys Res Commun 262:152–6
Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA (2007) Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis: JAD 11:153–64
Sulzer D (2007) Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci 30:244–50
Tetzlaff JE, Putcha P, Outeiro TF, Ivanov A, Berezovska O, Hyman BT, McLean PJ (2008) CHIP targets toxic alpha-Synuclein oligomers for degradation. J Biol Chem 283:17962–8
Titler AM, Posimo JM, Leak RK (2013) Astrocyte plasticity revealed by adaptations to severe proteotoxic stress. Cell Tissue Res 352:427–43
Tohgi H, Utsugisawa K, Yoshimura M, Yamagata M, Nagane Y (1995) Heat-shock cognate 70 messenger RNA expression in postmortem human hippocampus: regional differences and age-related changes. Neurosci Lett 196:89–92
Toth ME, Szegedi V, Varga E, Juhasz G, Horvath J, Borbely E, Csibrany B, Alfoldi R, Lenart N, Penke B, Santha M (2013) Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones 18:759–71
Unnithan AS, Choi HJ, Titler AM, Posimo JM, Leak RK (2012) Rescue from a two hit, high-throughput model of neurodegeneration with N-acetyl cysteine. Neurochem Int 61:356–368
Unnithan AS, Jiang Y, Rumble JL, Pulugulla SH, Posimo JM, Gleixner AM, Leak RK (2013) N-acetyl cysteine prevents synergistic, severe toxicity from two hits of oxidative stress. Neurosci Lett 560:71–6
Unno K, Asakura H, Shibuya Y, Kaiho M, Okada S, Oku N (2000) Increase in basal level of Hsp70, consisting chiefly of constitutively expressed Hsp70 (Hsc70) in aged rat brain. J Gerontol A: Biol Med Sci 55:B329–35
Uryu K, Richter-Landsberg C, Welch W, Sun E, Goldbaum O, Norris EH, Pham CT, Yazawa I, Hilburger K, Micsenyi M, Giasson BI, Bonini NM, Lee VM, Trojanowski JQ (2006) Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am J Pathol 168:947–61
Uversky VN (2009) Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci: J Virtual Libr 14:5188–238
Viana RJ, Nunes AF, Rodrigues CM (2012) Endoplasmic reticulum enrollment in Alzheimer’s disease. Mol Neurobiol 46:522–34
Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM (2011) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71
Walsh DM, Selkoe DJ (2007) A beta oligomers - a decade of discovery. J Neurochem 101:1172–84
Walters TJ, Ryan KL, Mason PA (2001) Regional distribution of Hsp70 in the CNS of young and old food-restricted rats following hyperthermia. Brain Res Bull 55:367–74
Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, Chandran J, Lin X, Lai C, Yang WJ, Moore DJ, Dawson TM, Dawson VL, Chiosis G, Cookson MR, Cai H (2008) The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci Off J Soc Neurosci 28:3384–91
Weidong L, Shen C, Jankovic J (2009) Etiopathogenesis of Parkinson disease: a new beginning? Neuroscientist: Rev J Bringing Neurobiol, Neurol Psychiatry 15:28–35
Wilhelmus MM, Otte-Holler I, Wesseling P, de Waal RM, Boelens WC, Verbeek MM (2006) Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol Appl Neurobiol 32:119–30
Wong E, Cuervo AM (2010) Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol 2:a006734
Wu ML, Ho YC, Yet SF (2011) A central role of heme oxygenase-1 in cardiovascular protection. Antioxid Redox Signal 15:1835–46
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–7
Xilouri M, Stefanis L (2010) Autophagy in the central nervous system: implications for neurodegenerative disorders. CNS Neurol Disord Drug Targets 9:701–19
Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, Yamamoto M (2008) Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol 28:2758–70
Yang Y, Turner RS, Gaut JR (1998) The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem 273:25552–5
Yokota T, Mishra M, Akatsu H, Tani Y, Miyauchi T, Yamamoto T, Kosaka K, Nagai Y, Sawada T, Heese K (2006) Brain site-specific gene expression analysis in Alzheimer’s disease patients. Eur J Clin Investig 36:820–30
Yoo BC, Seidl R, Cairns N, Lubec G (1999) Heat-shock protein 70 levels in brain of patients with Down syndrome and Alzheimer’s disease. J Neural Transm Suppl 57:315–22
Zhang Y, Barres BA (2010) Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol 20:588–94
Zhang J, Piantadosi CA (1992) Mitochondrial oxidative stress after carbon monoxide hypoxia in the rat brain. J Clin Invest 90:1193–9
Zhang Y, James M, Middleton FA, Davis RL (2005) Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet: Off Publ Int Soc Psychiatr Genet 137B:5–16
Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F (2013) Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol 100:30–47
Zhu X, Castellani RJ, Takeda A, Nunomura A, Atwood CS, Perry G, Smith MA (2001) Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mech Ageing Dev 123:39–46
Zhu X, Lee HG, Perry G, Smith MA (2007) Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta 1772:494–502
Acknowledgments
We apologize that we were not able to include all the many references on heat shock protein expression in the brain. RKL has no conflicts to declare. We are grateful to Mary Caruso, Deb Willson, and Jackie Farrer for outstanding administrative support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Leak, R.K. Heat shock proteins in neurodegenerative disorders and aging. J. Cell Commun. Signal. 8, 293–310 (2014). https://doi.org/10.1007/s12079-014-0243-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-014-0243-9