
Abstract
Tensegrity (tensional integrity) is an emerging concept governing the structure of the body. Integrin-mediated mechanical tension is essential for connective tissue function in vivo. For example, in adult skin fibroblasts, the integrin β1 subunit mediates adhesion to collagen and fibronectin. Moreover, integrin β1, through its abilities to activate latent TGFβ1 and promote collagen production through focal adhesion kinase/rac1/nicotinamide adenine dinucleotide phosphate oxidase (NOX)/reactive oxygen species (ROS), is essential for dermal homeostasis, repair and fibrosis. The integrin β1-interacting protein CCN2, a member of the CCN family of proteins, is induced by TGFβ1; yet, CCN2 is not a simple downstream mediator of TGFβ1, but instead synergistically promote TGFβ1-induced adhesive signaling and fibrosis. Due to its selective ability to sense mechanical forces in the microenvironment, CCN2 may represent an exquisitely precise target for therapeutic intervention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
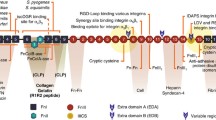
Chronic fibrotic diseases are characterized by the overproduction of scar tissue. These disorders can result in organ failure and death. The importance of mechanical tension in the cellular microenvironment is being increasingly appreciated as contributing to the pathogenesis of fibrotic conditions (Ingber 2003; Huang and Ogawa 2012). Cells such as fibroblasts sense extracellular mechanical forces through cell surface structures termed focal adhesions which contain cell surface receptors called integrins that connect the cytoskeleton to the surrounding extracellular matrix (ECM). Understanding the precise mechanism underlying how fibroblasts sense and communicate local mechanical signals is likely to be of future clinical benefit.
Myofibroblasts, the specialized form of mesenchymal cell responsible for wound repair and fibrosis, are characterized by α-smooth muscle actin (SMA)-containing stress fibers (Hinz 2009). Tissue damage results in elevated mechanical tension exerted by the surrounding microenvironment, causing differentiation of resident fibroblasts to myofibroblasts (Hinz 2009). For example, the potent fibrogenic cytokine TGF-β1 is activated in response to mechanical tension and up-regulates α-SMA in fibroblasts grown on stiff, but not on compliant, surfaces via a focal adhesion kinase (FAK)/src-dependent mechanism (Arora et al. 1999; Liu et al. 2007). In addition, α-SMA is only capable of being incorporated into stress fibers in cells subjected to significant mechanical loading (Goffin et al. 2006; Hinz 2006). Moreover, myofibroblasts inherently exert enhanced mechanical tension on their surrounding ECM through their enhanced adherent and contractile abilities, and hence directly contribute to the tensile strength of scar tissue (Chen et al. 2005; Hinz 2006; Vedrenne et al. 2012). The scar tissue itself exerts strong mechanical forces on the myofibroblast; thus, in fibrotic disease, an autocrine pro-adhesive loop exists that would be expected to be sufficient to result in persistence of the fibrotic phenotype (Hinz 2006, 2009; Leask 2011).
Integrins, the cell surface ECM receptors responsible for sensing mechanical stress, are heterodimers comprised of α and β subunits. The main integrins that actively participate in fibroblast proliferation, collagen contraction, and myofibroblast differentiation include α1β1, α2β1, and α11β1 (Pozzi et al. 1998; Schiro et al. 1991; Carracedo et al. 2010), suggesting that integrin β1 may play a central role in mechanotransduction and fibrogenic responses in dermal fibroblasts. This notion is explored extensively elsewhere in a recent review (Leask 2013); the purpose of the present review is to briefly summarize this concept, but to focus on the potential role of CCN2 in mechanotransduction and fibrosis. Integrin β1 knockout dermal fibroblasts are less able to adhere to and contract ECM, and show impaired activation of latent TGFβ1 (Liu et al. 2010a). Fibroblast-specific integrin β1 knockout mice display delayed wound healing that is rescued by addition of active recombinant TGFβ1 (Liu et al. 2010a). Fibroblast-specific integrin β1 knockout mice possess a progressive thinning of the dermis and are resistant to bleomycin-induced skin fibrosis (Liu et al. 2009; Liu and Leask 2013). Integrin β1 knockout fibroblasts also show less collagen and α−SMA production due to reduced in FAK/rac1/NOX/ROS-dependent signaling; the addition of hydrogen peroxide rescues the collagen and α−SMA expression defects of integrin β1 knockout fibroblasts (Liu and Leask 2013). These results are consistent with the hypothesis that antioxidants might be used as anti-fibrotic therapies (Demedts et al. 2005; Behr et al. 2009; Samarakoon et al. 2012).
Integrin-binding matricellular proteins are present within the cellular microenvironment and contribute to tissue plasticity by their ability to enabling a rapid response to changing conditions (Kyriakides and Bornstein 2003). These ECM play minimal roles in matrix structural integrity, but regulate signaling responses through their abilities to directly bind ECM proteins, growth factors and cytokines and also through their ability to trigger integrin signaling. Hence matricellular proteins modulate cellular processes such as cell adhesion and migration, ECM deposition, cell survival, and proliferation. The actual in vivo role of each matricellular protein varies depending based on the particular context and microenvironment.
One frequently studied member of the CCN family of the matricellular proteins is CCN2 (formerly known as connective tissue growth factor). CCN2 is largely absent from normal skin but is selectively upregulated in the dermal fibroblasts during wound healing and fibrosis (Igarashi et al. 1993; Kapoor et al. 2008; Liu et al. 2010b). In fibroblasts, CCN2 supports cell adhesion by binding integrin subunits containing integrin β1 (Leu et al. 2003; Chen et al. 2004). CCN2 also promotes integrin β1-mediated adhesion in other systems (Gao and Brigstock 2005). In vitro and in vivo, CCN2 enhances and alters pro-fibrotic, adhesive signaling responses to ECM components and growth factors (Mori et al. 1999; Chen et al. 2004; Shi-wen et al. 2006; Wang et al. 2011). Although dispensible for development of connective tissue in skin and for normal tissue repair (Liu and Leask 2011; Shangxi Liu, Katherine Thompson and Andrew Leask, unpublished observations), CCN2 is required for bleomycin-induced skin fibrogenesis (Liu et al. 2011). Initially, CCN2 was shown to be upregulated in response to TGFβ, and was considered to be a downstream regulator of this cytokine. A similar situation was also reported for the related protein CCN1 (Chen et al. 2001). However, it appears that TGFβ1 and CCN2/CCN1 are responsible for different yet interacting signaling mechanisms; genes responded in a different manner to the mixture of CCN1 and TGF-β1 than either CCN1 or TGFβ1 alone (Chen et al. 2001). For example, the effects of CCN1 and TGF-β1 on Col1α1 expression were antagonistic; in integrin α5 expression, the effects of CCN1 and TGF-β1 overlapped; in MMP1 expression, TGF-β1 completely suppressed the strong inducing effect of CCN1; and in PAI-1 expression, effects of CCN1 and TGF-β1 were synergistic (Chen et al. 2011). A similar situation appears to operate with CCN2 (Shi-wen et al. 2006); CCN2 selectively enhances adhesive signaling responses to TGFβ1. Intriguingly, CCN2 appears to act not on affecting myofibroblast differentiation of resident fibroblasts but may act to recruit myofibroblasts (possibly pericytes or mesenchymal precursor cells) to the fibrotic lesion (Liu et al. 2011). In summation, CCN2 may prove in the future to be an exquisitely suitable target to affect mechanotransduction in the fibrotic milieu.
References
Arora PD, Narani N, McCulloch CA (1999) The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am J Pathol 1999(154):871–882
Behr J, Demedts M, Buhl R, Costabel U, Dekhuijzen RP, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Lankhorst I, Sardina M, Boissard G, IFIGENIA study group (2009) Lung function in idiopathic pulmonary fibrosis—extended analyses of the IFIGENIA trial. Respir Res 10:101
Carracedo S, Lu N, Popova SN, Jonsson R, Eckes B, Gullberg D (2010) The fibroblast integrin alpha11beta1 is induced in a mechanosensitive manner involving activin A and regulates myofibroblast differentiation. J Biol Chem 285:10434–10445
Chen CC, Mo FE, Lau LF (2001) The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J Biol Chem 276(50):47329–47337, Epub 2001 Oct 2
Chen Y, Abraham DJ, Shi-Wen X, Pearson JD, Black CM, Lyons KM, Leask A (2004) CCN2 (connective tissue growth factor) promotes fibroblast adhesion to fibronectin. Mol Biol Cell 15(12):5635–5646
Chen Y, Shi-Wen X, van Beek J, Kennedy L, McLeod M, Renzoni EA, Bou-Gharios G, Wilcox-Adelman S, Goetinck PF, Eastwood M, Black CM, Abraham DJ, Leask A (2005) Matrix contraction by dermal fibroblasts requires transforming growth factor-beta/activin-linked kinase 5, heparan sulfate-containing proteoglycans, and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol 167(6):1699–1711
Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Capron F, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M, IFIGENIA Study Group (2005) High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 353(21):2229–2242
Gao R, Brigstock DR (2005) Connective tissue growth factor (CCN2) in rat pancreatic stellate cell function: integrin alpha5beta1 as a novel CCN2 receptor. Gastroenterology 129(3):1019–1030
Goffin JM, Pittet P, Csucs G et al (2006) Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol 172:259–268
Hinz B (2006) Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol 85:175–181
Hinz B (2009) Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep 11:120–126
Huang C, Ogawa R (2012) Fibroproliferative disorders and their mechanobiology. Connect Tissue Res 53(3):187–196, Epub 2012 Feb 13
Igarashi A, Okochi H, Bradham DM, Grotendorst GR (1993) Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell 4(6):637–645
Ingber DE (2003) Mechanobiology and diseases of mechanotransduction. Ann Med 35:564–577
Kapoor M, Liu S, Huh K, Parapuram S, Kennedy L, Leask A (2008) Connective tissue growth factor promoter activity in normal and wounded skin. Fibrogenesis Tissue Repair 1(1):3
Kyriakides TR, Bornstein P (2003) Matricellular proteins as modulators of wound healing and the foreign body response. Thromb Haemost 90(6):986–992
Leask A (2011) Possible strategies for anti-fibrotic drug intervention in scleroderma. J Cell Commun Signal 5(2):125–129
Leask A (2013) Integrin β1: a mechanosignaling sensor essential for connective tissue deposition by fibroblasts advances in wound care. doi:10.1089/wound.2012.0365
Leu SJ, Liu Y, Chen N, Chen CC, Lam SC, Lau LF (2003) Identification of a novel integrin alpha 6 beta 1 binding site in the angiogenic inducer CCN1 (CYR61). J Biol Chem 278(36):33801–33808
Liu S, Leask A (2011) CCN2 is not required for skin development. J Cell Commun Signal 5(3):179–182
Liu S, Leask A (2013) Integrin β1 is required for dermal homeostasis. J Invest Dermatol, in revision
Liu S, Xu SW, Kennedy L, Pala D, Chen Y, Eastwood M, Carter DE, Black CM, Abraham DJ, Leask A (2007) FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell 18(6):2169–2178
Liu S, Kapoor M, Denton CP, Abraham DJ, Leask A (2009) Loss of beta1 integrin in mouse fibroblasts results in resistance to skin scleroderma in a mouse model. Arthritis Rheum 60(9):2817–2821
Liu S, Xu SW, Blumbach K, Eastwood M, Denton CP, Eckes B, Krieg T, Abraham DJ, Leask A (2010a) Expression of integrin beta1 by fibroblasts is required for tissue repair in vivo. J Cell Sci 123(Pt 21):3674–3682
Liu S, Taghavi R, Leask A (2010b) Connective tissue growth factor is induced in bleomycin-induced skin scleroderma. J Cell Commun Signal 4(1):25–30
Liu S, Shi-wen X, Abraham DJ, Leask A (2011) CCN2 is required for bleomycin-induced skin fibrosis in mice. Arthritis Rheum 63(1):239–246
Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T, Takehara K (1999) Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: a mouse fibrosis model. J Cell Physiol 181(1):153–159
Pozzi A, Wary KK, Giancotti FG, Gardner HA (1998) Integrin alpha1beta1 mediates a unique collagen-dependent proliferation pathway in vivo. J Cell Biol 142:587–594
Samarakoon R, Overstreet JM, Higgins PJ (2012) TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal 25(1):264–268. doi:10.1016/j.cellsig.2012.10.003 [Epub ahead of print]
Schiro JA, Chan BM, Roswit WT, Kassner PD, Pentland AP, Hemler ME, Eisen AZ, Kupper TS (1991) Integrin alpha 2 beta 1 (VLA-2) mediates reorganization and contraction of collagen matrices by human cells. Cell 67:403–410
Shi-wen X, Stanton LA, Kennedy L, Pala D, Chen Y, Howat SL, Renzoni EA, Carter DE, Bou-Gharios G, Stratton RJ, Pearson JD, Beier F, Lyons KM, Black CM, Abraham DJ, Leask A (2006) CCN2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. J Biol Chem 281(16):10715–10726
Vedrenne N, Coulomb B, Danigo A, Bonté F, Desmoulière A (2012) The complex dialogue between (myo)fibroblasts and the extracellular matrix during skin repair processes and ageing. Pathol Biol (Paris) 60(1):20–27
Wang Q, Usinger W, Nichols B, Gray J, Xu L, Seeley TW, Brenner M, Guo G, Zhang W, Oliver N, Lin A, Yeowell D (2011) Cooperative interaction of CTGF and TGF-β in animal models of fibrotic disease. Fibrogenesis Tissue Repair 4(1):4
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is based on a presentation given at the University of Sydney, Australia at the ICCNS-sponsored CCN/IGFBP workshop.
Rights and permissions
About this article
Cite this article
Leask, A. CCN2: a mechanosignaling sensor modulating integrin-dependent connective tissue remodeling in fibroblasts?. J. Cell Commun. Signal. 7, 203–205 (2013). https://doi.org/10.1007/s12079-013-0205-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-013-0205-7