
Abstract
Chronic wounds are characterized by inadequate matrix synthesis, no re-epithelialization, infection and ultimately no wound resolution. In contrast, fibrosis is characterized by overproduction of matrix and excess matrix contraction. As research in the fields of chronic wounds and fibrosis surges forward, important parallels can now be drawn between the dysfunctions in fibrotic diseases and the needs of chronic wounds. These parallels exist at both the macroscopic level and at the molecular level. Thus in finding the individual factors responsible for the progression of fibrotic diseases, we may identify new therapeutic targets for the resolution of chronic wounds. The aim of this review is to discuss how recent advances in fibrosis research have found a home in the treatment of chronic wounds and to highlight the benefits that can be obtained for chronic wound treatments by employing a translational approach to molecules identified in fibrosis research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Development of non-healing skin lesions represent an increasing burden on today’s health care systems. The burden of non-healing skin lesions, which for simplicity will be referred to as chronic wounds, is being further exacerbated with the increasing worldwide prevalence of diabetes. The need for new and alternate therapeutics for the repair of non-healing wounds is paramount to reduce patient suffering and costs associated with treatment. Chronic wounds result from failure of the natural healing process to close dermal lesions. In contrast, fibrosis results when the healing process continues, unchecked, to the point of scarring and impaired tissue function. As research in the fields of chronic wounds and fibrosis surge forward important parallels can be drawn between the dysfunctions in fibrotic diseases and the needs of chronic wounds, including; increased proliferation, increased matrix synthesis and increased matrix contraction. Curiously, elevated inflammation is a driving force in both pathologies. The aim of this review is to discuss how recent advances in fibrosis research have found a home in the treatment of chronic wounds. Additionally, a relatively new player in fibrosis, periostin, which may have potential benefit for chronic wound resolution, will be discussed. The overall goal of this review is to highlight potential new therapeutics, which have been identified as pivotal to development of fibrosis and whether such cues could kick-start chronic wound resolution. To identify the current limitations and potential for new therapeutics, it is first necessary to understand molecules that are essential for normal wound resolution (Table 1).
Acute wound repair
The process of cutaneous wound repair is very complex and dynamic, involving multiple cell types and a plethora of growth factors, cytokines and their interactions. The aim of this paper is not to review the intricate details of acute wound healing. For such information, the reader is directed to comprehensive reviews on the subject (Clark 1996; Singer and Clark 1999; Diegelmann and Evans 2004; Barrientos et al. 2008; Schultz and Wysocki 2009).
Briefly, acute (normal) wound repair consists of three overlapping phases: inflammation, proliferative and tissue remodeling (Fig. 1). Upon tissue injury, damage to blood vessels results in the aggregation of platelets and the formation of a fibrin clot. The clot is essential for restoring hemostasis but also acts as a provisional matrix for infiltrating cells (Clark 1996). Platelets secrete several soluble factors including platelet-derived growth factor (PDGF), which initiates chemotaxis of neutrophils, macrophages and fibroblasts (Clark 1996; Diegelmann and Evans 2004). Neutrophils and macrophages cleanse the wound of foreign material. Secretion of transforming growth factor (TGF) β by platelets and macrophages facilitates migration and activation of fibroblasts. Fibroblasts infiltration of the granulation tissue is essential for transition from the inflammatory stage to the proliferative/tissue-building phase (Roberts and Sporn 1993). Concurrently, keratinocytes proliferate and migrate from the wound edge, isolating the wound from the external environment (re-epithelialization).

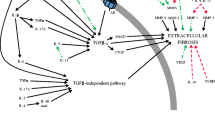
Acute wound repair consists of three overlapping phases: inflammation, proliferative and remodeling. The inflammation phase is dominated by neutrophils and macrophages, which serve to remove foreign debris, bacteria and damaged tissue. The proliferative phase includes the formation of granulation tissue and reduced inflammatory signals. The dominant cell types are fibroblasts and myofibroblasts. Matrix turnover and contraction are key features of this phase. The remodeling phase serves to rearrange and strengthen the newly formed tissue, producing a matrix-dense, relatively acellular, scar. Development of chronic wounds and fibrotic lesions are both driven by increased inflammation. However, in chronic wounds sustained inflammation and failed progression to proliferative and remodeling phases results (A). Fibrosis results from failure of the remodeling process to terminate at an appropriate point. Instead, continued matrix secretion and contraction by myofibroblasts results in excessive scarring (B)
The proliferative phase involves the formation of granulation tissue by simultaneous perfusion of the wound site with new vasculature and matrix turnover by fibroblasts (Singer and Clark 1999). Fibroblast proliferation, migration and matrix synthesis is stimulated by PDGF and TGFβ (Clark 1996). The fibrin clot is replaced by cellular fibronectin, collagen III and progressively more collagen I (Midwood et al. 2004). Differentiation of fibroblasts into the α-smooth muscle actin (α-SMA)-positive contractile, myofibroblast phenotype allows the contraction and compaction of the granulation tissue into a matrix dense scar (Tomasek et al. 2002).
The transition from granulation tissue to scar formation marks the beginning of the remodeling phase. During this phase collagen is degraded, synthesized and rearranged at a slower rate. Increasingly, collagen is organized into large bundles and cross-linked. The resulting scar tissue is relatively acellular and achieves only about 80% of the breaking strength of normal skin (Levenson et al. 1965). Therefore, acute repair does not perfectly regenerate the affected tissue, but instead strikes a favorable balance between a “good enough” repair and rapid wound closure. However, the inability of patients with existing medical conditions, such as diabetes, to resolve dermal wounds rapidly, or at all, is becoming more and more of a burden on healthcare systems.
The burden of chronic wounds
In general, chronic wounds in healthy individuals are rare (Sen et al. 2009). However, in medically compromised patients, such as those suffering from diabetes, the risk of developing non-healing skin lesions is greatly increased. The lifetime incidence of foot ulcers in diabetic patients has been estimated to be between 15% and 25% (Reiber 1996; Singh et al. 2005). The overall prevalence of pressure ulcers within Canadian healthcare institutions was estimated at 26% (Woodbury and Houghton 2004), although this estimate does not include diabetic foot ulcers, or ulcers caused by venous insufficiency. The estimated prevalence of non-healing skin lesions encompassing all etiologies within the healthcare system is closer to 35.5% (Woodbury and Houghton 2005). Unfortunately, failure of current treatment strategies to resolve chronic wounds commonly leads to amputation of the effected limb (Cavanagh et al. 2005; Wu et al. 2007). In 1993, Siitonen et al. reported the occurrence of lower extremity amputation (LEA) to be ten times higher in diabetic persons than in non-diabetics (Siitonen et al. 1993). 16 years later this trend had shown no signs of leveling off. In fact LEA occurs 19 times as often in diabetic Canadians than in the general population (Canada Diabetes Report, 2009). 60% of non-traumatic amputations in the US occurred in diabetic patients (CDC Diabetes Fact Sheet 2007), with 80% of amputations in diabetic patients being preceded by a non-healing ulcer (Driver et al. 2010). The risks associated with LEA are staggering. Perioperative mortality rates following above knee LEA have been reported as high as 18.6% in 2003 (Moxey et al. 2010) and 18% in 2005 (Ploeg et al. 2005). Recently, Aragón-Sánchez and colleagues reported 14.7% post-operative mortality following above knee amputations (Aragon-Sanchez et al. 2010). In England a significant decrease occurred in mortality rates, from 18.6% in 2003 to 15.2% in 2007, supporting a promising trend (Moxey et al. 2010). Long term mortality following LEA, however, remains unacceptably high. Several recent reports place 5-year survival rates following any LEA at approximately 50% (Hambleton et al. 2009; Papazafiropoulou et al. 2009). When major amputations (above knee) are considered alone, 5-year survival rates are as low as 11% (Morbach et al. 2009).
An estimated 2.8 million Canadians will have developed diabetes by 2012 (Canada Diabetes Report, 2009). The prevalence of diabetes in Canada has increased steadily from 4.9% to 6.2% from 2002–03 to 2006–07, representing over two million Canadians (Canada Diabetes Report 2009). Current estimates suggest that 220 million people worldwide have diabetes, and that number is expected to climb to 336 million by the year 2030 (WHO Fact Sheet No 312, 2009) (Wild et al. 2004). There is no indication that the burden of chronic wounds is shrinking. With the growing prevalence of diabetes, obesity and an aging population, the impact of chronic wounds is deserving of greater attention in healthcare research (Armstrong et al. 2007; Harding and Queen 2010). Selection of target molecules for treatment of chronic wounds based on studies of acute wound repair can be difficult and time consuming due to the complexity of acute wound repair. To expedite the search for target molecules we can take advantage of the accumulating knowledge of fibrotic skin lesions, such as keloid scars, hypertrophic scars and scleroderma. Careful dissection of these conditions of “overhealing” may offer hints towards treating non-healing chronic wounds (Table 1).
Chronic wounds vs. fibrosis
Fibrosis is a general term describing pathological conditions in which the healing process has continued, unchecked, to the point where normal tissue is replaced by scars, resulting in impaired/lost function. The central features of fibrotic diseases include; increased growth factor activity, decreased protease activity and decreased fibroblast senescence (Fig. 1) (Wynn 2007). The culmination of these features is excess matrix deposition and scar formation. Although the etiology of chronic wounds is not fully understood, three major differences have been described between chronic and acute wounds; 1) reduced growth factor activity, 2) increased protease activity and 3) increased fibroblast senescence (Schultz and Mast 1998; Harding et al. 2002). Interestingly, excessive inflammation is common to both chronic wounds (Cochrane 1977; Trengove et al. 2000) and fibrosis (Abraham and Varga 2005). The key difference is that chronic wounds become stalled in an inflammatory state, but fibrotic diseases progress beyond the initial inflammation and enter an aggressive fibrotic state (Abraham and Varga 2005). In bleomycin-induced fibrosis, the initial bout of excessive inflammation is characterized by increased numbers of macrophages (Kraling et al. 1995). Being a major source of the pro-fibrotic growth factors PDGF and TGFβ, increased macrophage numbers promote an elevated fibrotic response (Yamamoto and Nishioka 2005). The elevated fibrotic response results in the increased and sustained activity of myofibroblasts, which are responsible for elevated collagen production, matrix contraction and continued secretion of TGFβ (Tomasek et al. 2002; Yamamoto and Nishioka 2005). In contrast, chronic wounds suffer from reduced expression of key growth factors, such as PDGF and TGFβ, which impedes fibroblast activation and construction of the granulation tissue (Robson 1997; Galkowska et al. 2006). Increased protease activity, originating from excessive neutrophil infiltration (Cochrane 1977) and compounded by reduced protease inhibitor levels, serves to degrade important matrix components such as collagens (Wysocki et al. 1993; Vaalamo et al. 1999; Liu et al. 2009; Yang et al. 2009) and growth factors (Chen et al. 1997). These deficiencies are further exacerbated by the senescent state of fibroblasts within the wound, which impairs the response to the already scarce growth factors (Hasan et al. 1997; Stanley et al. 1997; Hehenberger et al. 1998; Mendez et al. 1998; Loot et al. 2002; Yang et al. 2009). A question of critical importance is why, or how, do fibrotic skin lesions progress beyond the initial excessive inflammation, whereas chronic wounds become stalled (Fig. 1)?
Current targets for treatment of chronic wounds
Platelet-derived growth factor-BB
Does increased recruitment of macrophages in bleomycin-induced fibrosis provide the answer to progressing chronic wounds beyond inflammation? Fibrosis results from excessive extracellular matrix (ECM) production, primarily by a specialized type of activated fibroblast, termed myofibroblasts, so-called as they highly express the protein α-SMA (Trojanowska 2008). Macrophages release the pro-fibrotic growth factor PDGF (Yamamoto and Nishioka 2005), which plays a key role in the expansion and persistence of myofibroblast populations (Bostrom et al. 1996; Heldin and Westermark 1999; Trojanowska 2008) by modulating fibroblast migration, proliferation and activation (Barrientos et al. 2008). PDGF also contributes to TGFβ expression and thus represents a crucial initiator of granulation tissue formation (Heldin and Westermark 1999).
PDGF includes a family of homo and heterodimeric proteins (PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD), which bind to transmembrane tyrosine kinase receptors (Bennett et al. 2003; Barrientos et al. 2008). In the fibrotic disorder systemic sclerosis (SSc), expression of PDGF-BB receptors is increased within and around dermal vasculature (Klareskog et al. 1990). Bronchoalveolar lavage fluid from SSc patients contains significantly elevated levels of PDGF-AA and -BB (Ludwicka et al. 1995). Cells isolated from SSc skin biopsies express elevated PDGF-B and PDGF receptor, compared to normal patients (Zheng et al. 1998). The c-Abl inhibitors imatinib (Distler et al. 2007), dasatinib and nilotinib (Akhmetshina et al. 2008), act downstream on non-receptor tyrosine kinases of both TGFβ and PDGF to block the production of ECM proteins (Daniels et al. 2004). Bleomycin-induced dermal thickening and myofibroblast accumulation is prevented by oral administration of these inhibitors in a mouse model (Distler et al. 2007; Akhmetshina et al. 2008), suggesting that PDGF may be an excellent target for the treatment of fibrosis (Beyer et al. 2010; Leask 2010), but also highlighting PDGF as an obvious therapeutic agent for chronic wound. As a potent chemoattractant for fibroblasts (Seppa et al. 1982), PDGF is important for progression of wound repair beyond inflammation and into the proliferative phase (Gao et al. 2005). In animal models, delivery of PDGF to wounds increases fibroblast infiltration, TGFβ expression, collagen deposition, granulation tissue formation and the repaired skin’s breaking strength (Pierce et al. 1988, 1989; Mustoe et al. 1990); resulting in accelerated wound closure. Moreover, these benefits are also achieved in models of chronic wounds (Mustoe et al. 1989, 1991). It has since been shown that PDGF-BB is absent in non-healing chronic wounds, but present in actively healing pressure ulcers (Pierce et al. 1995).
PDGF is currently the only growth factor approved for use by the FDA in the treatment of diabetic foot ulcers and is marketed under the name Regranex®. Application of rhPDGF-BB is preceeded by debridement, or wound bed preparation, which is the attempt to remove the infected and necrotic components of an ulcer and establish a pseudo-acute wound environment (Kirshen et al. 2006). Delivery of rhPDGF-BB to debrided wounds improved closure of non-diabetic chronic pressure ulcers (Robson et al. 1992) and deep pressure ulcers (Mustoe et al. 1994). In neurotrophic diabetic ulcers, treatment resulted in 48% of wounds closing where as 25% of wounds closed in the placebo group (Steed 1995). In a multi-center study, administration of 100 μg/g becaplermin (rhPDGF-BB) to diabetic ulcers resulted in closure of 50% vs. 35% (treatment vs. placebo) of wounds (Wieman et al. 1998). The smaller effect size reported by Wieman in comparison with Steed possibly reflects inherent variability in the larger, multi-center study, thus more closely representing a realistic effect size with wide spread use. The addition of rhPDGF-BB to diabetic wounds has therefore produced statistically significant, although modest, increases in wound closure over placebo. At best, 50% of wounds treated with rhPDGF-BB closed, the majority of which can be attributed to standard wound care (placebo) alone. Perhaps a limitation of PDGF based treatments is its ability to recruit inflammatory cells such as macrophages and neutrophils (Deuel et al. 1982). Where as macrophages are a major source of TGFβ (Roberts and Sporn 1993) and are required for wound repair (Leibovich and Ross 1975), neutrophils are not required for wound repair in the absence of gross infection (Simpson and Ross 1972). Neutrophils are a source of inflammatory cytokines (IL-1, -6 and TNFα) (Barrientos et al. 2008), reactive oxygen species and damaging proteolytic enzymes (Diegelmann and Evans 2004). Without additional signals, which are absent in chronic wounds, it is possible that the pro-inflammatory role of PDGF may prevail. Thus complex interaction of the multitude of growth factors and matrix elements present in acute wounding may not be suitably recapitulated with rhPDGF-BB alone.
Platelet releasate
Platelet rich blood plasma contains multiple growth factors (Schultz and Grant 1991), and the components of a fibrin matrix (Mosesson 2005). Administration of autologous plasma to chronic wounds more closely mimics the complexity of the initial stages of natural wound repair, including structural matrix components. Platelet rich plasma (PRP) has been used for the treatment of chronic wounds for over 20 years. Autologel™, a PRP product, is prepared from a small sample of the patient’s blood plasma mixed with a gel base, which is immediately applied to the wound. Despite it’s long history of use, randomized controlled clinic studies for PRP are lacking. Driver and colleagues carried out the first FDA approved prospective, randomized, blinded, placebo-controlled clinical trial of PRP (Autologel™) for diabetic foot ulcers (Driver et al. 2006). Although this study showed significance (81% vs. 42% of wounds closed, treatment vs. placebo), it suffered from extensive exclusion of participants for a variety of reasons. As the vast majority of persons with chronic wounds fall outside of the numerous exclusion criteria employed in this study, the applicability of this data is limited. Margolis et al., in a retrospective, randomized, controlled study of nearly 27,000 patients (of which 21% were treated with platelet releasate), reported the overall proportion of wounds healed at 50% vs. 41% (Margolis et al. 2001). This is likely a much more realistic outcome since it reflects the effectiveness of the treatment across a very large group in the hands of many practitioners and not the efficacy within a very idealized treatment group.
Use of platelet releasate for the treatment of chronic wounds offers benefits over placebo, however, similar to rhPDGF-BB, the increased incidence of wound closure is modest. The limited success of platelet releasate may be due to pro-inflammatory influences, which may undermine the pro-fibrotic effects of growth factors contained within. PDGF is potent chemoattractant for fibroblasts (Seppa et al. 1982; Lin et al. 2006), however, it does not increase expression of collagen 1, a key component of granulation tissue and scars (Tan et al. 1995; Jinnin et al. 2005). Instead, PDGF increases expression of matrix metalloproteinase (MMP)-1, which contributes to collagen degradation (Tan et al. 1995). Interestingly, where as PDGF-BB increases MMP-1 expression in human dermal fibroblasts, TGFβ1 decreases MMP-1 expression and potently induces collagen synthesis, contributing to matrix accumulation (Ignotz and Massague 1986; Edwards et al. 1987; Tan et al. 1995). Thus TGFβ is a logical alternative to PDGF as a pro-fibrotic therapeutic agent.
Transforming growth factor β
TGFβ expression during acute wound repair contributes to progression of granulation tissue formation and matrix deposition, where its ability to stimulate collagen production is so potent that is can lead to fibrosis (Barrientos et al. 2008). Platelets and macrophages are the major sources of TGFβ during the inflammatory phase (Clark 1996). Fibroblasts are the key TGFβ target cells during the proliferative phase of wound repair. PDGF and TGFβ together promote myofibroblast differentiation, however, PDGF does not stimulate α-SMA expression (Tomasek et al. 2002). TGFβ drives α-SMA expression and is required for differentiation of fibroblasts into myofibroblasts, the major contributors to collagen production and matrix remodeling (Desmouliere et al. 1993, 2005; Tomasek et al. 2002). Increased TGFβ signaling results in decreased expression of proteases, such as MMPs, and increased expression of tissue inhibitors of MMPs (TIMPs) (Edwards et al. 1987), thereby further contributing to matrix accumulation. TGFβ signaling has been extensively reviewed elsewhere (Leask and Abraham 2004; Varga and Abraham 2007). Briefly, there are three isoforms of TGFβ: TGFβ1, TGFβ2 and TGFβ3. The actions of these isoforms are largely overlapping with the possible exception of TGFβ3, which has been shown to have anti-fibrotic influences (Shah et al. 1995). Activated TGFβ binds to heteromers of the TGFβ type I and type II receptors. Type I receptors (ALK5) phosphorylate Smad2 and Smad3 which go on to bind Smad4 and translocate to the nucleus. Once in the nucleus the Smad complex binds to Smad binding elements in target genes (Fig. 2) (Massague and Wotton 2000). TGFβ has also been known to signal through focal adhesion kinase (FAK), extracellular signal-regulated kinases (ERK), c-Jun N terminal kinases (JNK) and p38 (Leask 2008). Target genes of TGFβ signaling include α-SMA, MMPs, TIMPs, periostin, connective tissue growth factor (CCN2) and collagen I (Kocher and Madri 1989; Igarashi et al. 1993; Takeshita et al. 1993; Holmes et al. 2001; Verrecchia et al. 2001).

Cannonical pro-fibrotic TGFβ signaling is initiated by the binding of active TGFβ to TGFβ receptors. Type I receptors phosphorylate receptor-Smads 2 and 3, which associate with Smad4 and translocate to the nucleus. In the nucleus the Smad complex interacts with Smad binding elements, promoting gene expression. Among the many genes influenced by TGFβ signaling are collagens I and III, α-SMA, fibronectin, TIMPs and periostin. Additionally, increased Smad7 expression creates a negative feedback loop, limiting pathway activation. The pro-fibrotic roles of periostin are summarized here. Via interactions with the ECM and BMP-1, periostin facilitates collagen crosslinking through activation of lysyl oxidase. Periostin’s influences on cell behaviours include altered proliferation, migration and adhesion. Recent evidence suggests that periostin plays a role in liberation of TGFβ from the latency-associated proteins in an MMP-2/9 dependent manner
TGFβ is central to fibrosis, where it drives excessive collagen production and myofibroblast differentiation (Wei et al. 2010), leading to matrix accumulation and contraction. The role of TGFβ in fibrosis has received a lot of attention and is the topic of several reviews (Leask and Abraham 2004; Pannu and Trojanowska 2004; Varga and Pasche 2009). Accumulating evidence implicates inappropriately elevated TGFβ signaling in the progression of SSc. Examples include: increased TGFβ receptor expression (Kawakami et al. 1998; Kubo et al. 2002; Pannu et al. 2004), increased Smad3 mRNA, protein levels and phosphorylation levels (Mori et al. 2003). Additionally, Smad3/4 nuclear localization is increased in SSc fibroblasts, both in the absence of TGFβ stimulation and in the presence of TGFβ blocking antibodies (Mori et al. 2003). The targeting of TGFβ directly in fibrosis has been successful in animal models. Injection of TGFβ neutralizing antibodies to the edges of dermal wounds in rats, results in reduced scar formation, where as addition of TGFβ increases scarring (Shah et al. 1992). Topical application of the P144 peptide, which interferes with TGFβ/receptor association, reduces bleomycin induced dermal thickening, Smad2/3 phosphorylation and α-SMA positive myofibroblast numbers (Santiago et al. 2005). Treatment strategies targeting TGFβ in fibrosis are numerous (Varga and Pasche 2009), but have so far shown limited success (Denton et al. 2007).
TGFβ’s pro-fibrotic role in acute wounds and fibrosis makes it an interesting target for treatment of chronic wounds. Indeed, TGFβ1 expression is reduced in diabetic and venous foot ulcers, compared to uninjured skin (Jude et al. 2002). In a rabbit ischemic ulcer model, topical application of rhTGFβ1 increased wound healing significantly (Beck et al. 1990; Zhao et al. 1994). These studies used young ischemic rabbits for their experiments. The combined effect of age and ischemia was address by Wu et al. They found that 60-month-old ischemic rabbits did not response to application of rhTGFβ (Wu et al. 1999). From this observation they concluded that TGFβ signaling must be defective in the aged ischemic model and topical application of TGFβ may not benefit healing of human chronic wounds. It is worth mention that, due to the cartilage within the rabbit ear, the model chosen in this series of experiments does not allow for a significant contribution of contraction in healing (Mustoe et al. 1991). Perhaps this feature of the model masks the effects of TGFβ with respect to myofibroblast differentiation and activity. Never the less, the limited benefit of TGFβ in animal models has been mirrored by the studies of Robson and colleagues, who investigated the application of TGFβ2 on human chronic venous ulcer healing. The study incorporated an open-label trial in which patients received either 0.5 μg/cm2 bovine TGFβ2 (bTGFβ2) in a lyophilized collagen matrix or placebo (matrix only). bTGFβ2 treatment resulted in a significant increase in wound closure after a 6-week regime (Robson et al. 1995). In the parallel closed-label trial, however, 2.5 μg/cm2 bTGFβ2 treatment offered no benefit over the placebo. The authors suggest that increased variation in the control groups undermined the effect of treatment in the closed-label study (Robson et al. 1995). With a small number of patients (n = 12/group) it is possible that sampling error played a significant role. We must be cautious, however, of how we interpret open-label study data since the placebo effect has been shown in earlier examples to dominate the effectiveness of treatments. At a minimum, this study stresses the requirement for large-scale randomized, blinded, multi-center, placebo-controlled clinical trials.
Recent evidence suggests that the rather disappointing results of TGFβ addition to chronic wounds may be due to dysregulation of the TGFβ signaling cascades (Hasan et al. 1997; Cowin et al. 2001; Jude et al. 2002; Pastar et al. 2010). Together these studies establish a trend towards reduced TGFβ receptor expression, although the data is somewhat conflicting when looking at the expression of specific receptors. Convincingly, TGFβ responsive genes are strikingly reduced in venous ulcers (Pastar et al. 2010). A growing body of evidence now clearly identifies additional, or accessory, signaling pathways induced by TGFβ and necessary for appropriate context-specific TGFβ signaling. As will be discussed in the coming sections, these non-canonical TGFβ signaling pathways are of particular importance to wound repair and fibrosis. Moreover, context-specific modulation of these pathways is increasingly becoming a role of matricellular proteins.
Old players, but new rules in chronic wound regeneration
Non-canonical TGFβ signaling
In addition to Smad signaling, TGFβ is known to signal through Smad independent, non-canonical pathways, which have been implicated in myofibroblast differentiation, matrix contraction and fibrosis. Serini and colleagues showed that adhesion to the ED-A splice variant of fibronectin (ED-A FN) is required for myofibroblast differentiation from human subcutaneous fibroblasts (Serini et al. 1998). Although binding to ED-A FN does not cause differentiation, per se, blocking specific ED-A FN binding disrupts TGFβ-induced α-SMA expression (Serini et al. 1998); demonstrating a role for adhesive signaling in TGFβ response. Stimulation of adherent human lung fibroblasts with TGFβ results in increased FAK phosphorylation and α-SMA expression (Thannickal et al. 2003). TGFβ-induced α-SMA and collagen 1 expression has been shown to require FAK and JNK activation in mouse embryonic fibroblasts (Liu et al. 2007). When human fibroblasts are maintained in suspension or treated with the FAK/Src inhibitor, PP2, FAK phosphorylation is lost and TGFβ-induce α-SMA expression is abolished (Thannickal et al. 2003). Interestingly, Smad2 phosphorylation remains TGFβ responsive, even when cells are maintained in suspension (Thannickal et al. 2003); suggesting non-canonical signaling critically regulates myofibroblast differentiation, one of the core features of fibrosis. Fibroblasts isolated from fibrotic lesions in patients with SSc show an increased ability to contract a collagen matrix. This heightened contractility can be reduced by treatment with inhibitors of; TGFβ type 1 receptor activation, ERK activation and proteoglycan synthesis (Chen et al. 2005). Furthermore, genetic deletion of the heparan sulphate proteoglycan, syndecan 4, results in loss of TGFβ-induced ERK phosphorylation, α-SMA incorporation into stress fibers and contractility (Chen et al. 2005).
Non-cononical TGFβ signaling has been suggested to modulate, or fine tune, the response to TGFβ in a gene and context specific manner (Leask and Abraham 2004). As a result, these accessory pathways have gained the interest of fibrosis researchers since they offer potential targets for the treatment of fibrosis without disrupting the pleiotropic TGFβ/Smad pathway, which may be problematic for basal tissue functioning (Leask 2008). Modulation of TGFβ’s impact on healing via non-canonical signaling is a function that is becoming more and more prominent in a group of proteins known as matricellular proteins. Moreover, combined, TGFβ and matricellular proteins may highlight the appropriate direction for new therapeutics for chronic wounds.
Matricellular proteins in fibrosis and acute wound repair
Matricellular proteins are non-structural ECM components, which bind cell surface receptors to mediate interactions between the cell and its ECM thereby exerting control over essential events (migration, proliferation, adhesion, etc.) during wound repair (Bornstein 1995; Midwood et al. 2004; Hamilton 2008). Although genetic deletion of matricellular proteins typically results in very mild phenotypes (Hamilton 2008), wound repair can be severely compromised (Midwood et al. 2004; Hamilton 2008). Matricellular proteins have diverse roles, spanning all phases of wound repair. For example, efficient re-epithelialization is facilitated by tenascin-C, where keratinocyte migration is decreased in tenascin-C knockout mice (Matsuda et al. 1999). Thrombospondin-1 has been shown to activate latent TGFβ (Schultz-Cherry and Murphy-Ullrich 1993). Macrophage infiltration is increased in the presence of the matricellular protein osteopontin (Denhardt et al. 2001).
The role of matricellular proteins in myofibroblast behaviour has received considerable attention. Osteopontin serves as a ligand for αvβ3 integrins where binding leads to activation of FAK and numerous downstream pathways (Sodek et al. 2000). Furthermore, osteopontin is required for TGFβ-induced myofibroblast differentiation, where deletion of osteopontin eliminates TGFβ-dependent increases in α-SMA and ED-A FN expression (Lenga et al. 2008). CCN2 promotes myofibroblast differentiation in the presence of TGFβ (Leask 2008). Although, CCN2 is considered to be a cofactor in fibrosis and not a fibrotic agent itself (Leask 2010) it serves as a marker for severity of fibrosis in SSc (Takehara 2003). CCN2 is required for maximal induction of α-SMA and collagen 1 by TGFβ, where TGFβ-induced FAK and Akt activation is reduced in CCN2 null fibroblasts (Shi-wen et al. 2006). Furthermore, CCN2 can also activate ERK through a syndecan-4-dependent mechanism (Kennedy et al. 2007). Syndecan-4, which was previously discussed as being required for many facets of TGFβ-induced myofibroblast behaviour, is considered by some to be a matricellular protein (Woods 2001). Human dermal fibroblasts are unable to contract a collagen matrix in the presence of neutralizing antibodies for the matricellular protein, vitronectin (Sethi et al. 2002). Vitronectin incorporation into the ECM is inhibited by exogenous galectin-1, yet another matricellular protein (Moiseeva et al. 2003). Finally, CCN1/CYR61 acts through integrins, ERK and p38 to cause myofibroblast senescence, thus serving as a natural brake on fibrotic tissue remodeling (Jun and Lau 2010). As a group, matricellular proteins can encourage and inhibit critical events of acute and fibrotic healing. Despite mounting evidence for the importance of matricellular proteins in wound contraction and re-epithelialization, no clinical trials are currently employing these proteins for the resolution of chronic wounds. The various influences of matricellular proteins documented to date provide a multitude of avenues for exploration of potential targets for fibrosis and chronic wounds. In the following section we will use periostin as an example to illustrate the deeply rooted influence of matricellular proteins on; cellular behaviour, acute wound repair and fibrotic diseases.
Periostin
Periostin represents an, as yet, untapped resource in the treatment of fibrotic and chronic wounds (Hamilton 2008). Periostin has influences on many key aspects of acute healing and fibrosis. Collagen synthesis, fibril assembly and myofibroblast behaviour have all been increasingly linked to periostin at a functional level. Furthermore, the expression pattern of periostin following tissue injury suggests that it plays a common role in various models of acute wound repair.
Periostin is a secreted 90-kDa disulfide-linked, TGFβ inducible protein, originally designated osteoblast specific factor 2 (Takeshita et al. 1993; Horiuchi et al. 1999). Structurally, periostin consists of a typical signal sequence, an EMI domain responsible for binding to fibronectin, four tandem fasciclin-like domains that are responsible for integrin binding (Kim et al. 2002) and a C-terminal region where multiple splice variants originate (Litvin et al. 2004) (Fig. 3). Additionally, periostin has been shown to bind collagen, tenascin-C, BMP-1 and itself (Kii et al. 2006; Takayama et al. 2006; Norris et al. 2007; Kii et al. 2009; Maruhashi et al. 2010). The ability of periostin to interact with various integrin pairs (Gillan et al. 2002; Bao et al. 2004; Shao et al. 2004; Baril et al. 2007; Butcher et al. 2007) allows it to influence various biological effects including cell proliferation, cell migration, cell adhesion and epithelial to mesenchymal transformation (Horiuchi et al. 1999; Katsuragi et al. 2004; Lindner et al. 2005; Yan and Shao 2006; Vi et al. 2009; Li et al. 2010).

Domain structures of human periostin. Human full length periostin (isoform 1) consists of a typical signal peptide (SP) sequence, an EMI domain responsible for binding to fibronectin, four tandem fasciclin-like domains that are responsible for integrin binding and a C-terminal region (CTR) where multiple splice variants originate. Tryptophan 65 within the EMI domain is required for fibronectin binding. The CTR has been shown to inhibit binding of periostin to several binding partners. Numbers represent amino acid residues flanking each domain
Transient increases in periostin expression are seen in many connective tissue wound models. Nakazawa and colleagues showed an increase in periostin mRNA following induced tibial fractures in mice. Periostin expression was significantly increased by day 3, peaking at day 7, in preosteoblasts of the periosteum and undifferentiated mesenchymal cells within the soft callus (Nakazawa et al. 2004). Periostin is abundantly expressed following 8 days of balloon injury to rat carotid arteries, eventually decreasing by 4 weeks post injury (Lindner et al. 2005). Li and colleagues confirmed that periostin peaks at 7 days post injury in this model (Li et al. 2006). To determine if periostin induction is a feature common to other connective tissue injuries, Lindner and colleagues created full thickness incisional wounds in the skin of rats. Indeed, periostin expression was detected in the fibroblasts, but not keratinocytes, of the wound site (Lindner et al. 2005). The expression pattern of periostin during dermal wounding has since been further defined (Jackson-Boeters et al. 2009; Zhou et al. 2010). After incisional wounding, periostin is expressed in the dermis and basement membrane within the dermal-epidermal junction (Zhou et al. 2010). In full thickness excisional wounds, in which the dermis and epidermis are completely removed, periostin expression dominates the newly formed granulation tissue (Jackson-Boeters et al. 2009). With expression beginning at day 3, peaking by day 7 and eventually returning to basal levels by 4 weeks (Jackson-Boeters et al. 2009), the temporal expression pattern of periostin in skin mimics that in vascular balloon injury and bone fracture. These observations strongly suggest that a common role for periostin may exist in all connective tissue repairs. Interestingly, periostin expression in dermal repair coincides with the expression of α-SMA expression with the granulation tissue (Jackson-Boeters et al. 2009). Furthermore, acquisition of a smooth muscle cell phenotype, as determined by α-SMA expression, correlates with acquisition of periostin expression both in vitro and in vivo (Lindner et al. 2005).
The functional role of periostin is not fully understood, however, several lines of evidence point towards a role in collagen deposition and myofibroblast differentiation. In murine models of myocardial infraction and ventricular hypertrophy (thickening of the myocardium), increased periostin expression results in increased collagen deposition (Katsuragi et al. 2004; Oka et al. 2007; Shimazaki et al. 2008; Stansfield et al. 2009). Genetic ablation of periostin results in increased incidence of ventricular rupture following myocardial infarction due to reduced α-SMA positive cells and impaired collagen formation (Shimazaki et al. 2008). Addition of recombinant periostin to pancreatic stellate cells results in increased expression of key fibrotic proteins such as: α-SMA, collagen 1, fibronectin, TGFβ1 and periostin (Erkan et al. 2007). Collagen fibrils from the skin of periostin knockout mice are reduced in diameter and display decreased crosslinking (Norris et al. 2007). The consequences are reduced tensile strength and an alteration of the visco-elastic properties of the skin (Norris et al. 2007). Recent insights into the functional role of periostin in collagen crosslinking implicate periostin as a scaffold protein, aiding in the incorporation of BMP-1 into the ECM where it can activate lysyl oxidase (an enzyme involved in collagen fibril crosslinking) (Maruhashi et al. 2010).
Keloid scars are fibrotic dermal lesions that result from excessive production of ECM components, such as collagen, during wound repair (Abergel et al. 1985). Periostin is the single most up-regulated gene in keloid scars by cDNA microarray analysis (Naitoh et al. 2005). Wang and colleagues reported increased periostin expression in keloid and hypertrophic scars, relative to normal human skin, where periostin expression was positively correlated with TGFβ1 expression (Wang et al. 2007). Periostin is highly expressed in tissues affected by Dupuytren’s disease, a progressive disease that results in a scar-like, collagen-rich cord within the palmar fascia and permanent contracture of the hand (Vi et al. 2009). Fibroblasts isolated from diseased tissue have an increased ability to contract a collagen matrix, which is further enhanced by addition of recombinant periostin (Vi et al. 2009). Periostin-induced contractility is accompanied by an increase in α-SMA protein. In addition to skin fibrosis, periostin has been associated with bone marrow fibrosis, where it correlates with the severity of fibrosis (Oku et al. 2008). Periostin has been implicated with sub-epithelial fibrosis of bronchial asthma (Takayama et al. 2006), where, recently, it has been shown to drive TGFβ signaling (Sidhu et al. 2010). Over-expression of periostin in human bronchial epithelial cells results in increased periostin secretion, collagen synthesis and TGFβ expression/protein/activity (Sidhu et al. 2010). Addition of recombinant periostin to primary human bronchial epithelial cells increased collagen expression in a TGFβ dependent manner.
The role of periostin in acute wound repair and in fibrotic diseases has received a lot of attention in the past 10 years. Research into chronic wounds, however, is lacking and very few studies have looked at periostin in this context. Of particular importance, expression of periostin following injury coincides with fibrotic, not inflammatory, stages of repair (Jackson-Boeters et al. 2009). In fact, chronically inflamed skin contains very little periostin, with levels lower than that of normal skin (Zhou et al. 2010).
Future work must exploit the influence of matricellular proteins, like periostin, on acute wound repair and apply them to pathological healing conditions. Matricellular proteins are expressed during development, but are typically absent in the adult, except during tissue remodeling or repair (Bornstein 1995). Their tight regulation during wound repair and absence in adult tissues makes matricellular proteins an ideal localized target for therapies (Midwood et al. 2004).
Future directions
Optimization of a favorable wound environment, via debridement and possibly protease inhibitors, will certainly form the foundation of any future treatment regimes. To date, resolution of chronic wounds via administration of growth factors has produced limited benefits. Future treatment strategies must integrate the wealth of knowledge available from studies of acute and fibrotic repair, particularly the importance of cross talk between cells, structural matrix components, growth factors and matricellular proteins. The importance of the matrix on cell behaviour should not be underestimated in choosing alternative target molecules.
Historically, the introduction of physical matrices to chronic wounds has primarily focused on maintaining a moist environment to encourage healing (Queen et al. 2004). Examples of these modern dressings include hydrocolloid dressings, alginates and foam dressings (Qin and Gilding 1996; Chaby et al. 2007). Despite weak evidence of clinical efficacy, these products have obtained wide spread use (Chaby et al. 2007). One limitation of these dressings is their lack of biological activity, although hybrids containing biologically active components are now available (Donaghue et al. 1998; Murakami et al. 2010).
Introduction of exogenous ECM components, such as collagens fibronectin and fibrin, provide a scaffold on which cells can migrate into the wound area (Greiling and Clark 1997), while also greatly influencing the behaviour of cells through binding of surface receptors and activation of signaling pathways (Schultz and Wysocki 2009). One currently available ECM based product is de-cellularized porcine small intestine submucosa (SIS), which is marketed under the name OASIS® Wound Matrix (Mostow et al. 2005). In a randomized clinical trial, OASIS® Wound Matrix was shown to be at least as effective as Regranex® (rhPDGF-BB) in healing diabetic foot ulcers (Niezgoda et al. 2005). Interestingly, growth factors embedded within SIS can influence cell behaviours such as proliferation and cell morphology (Voytik-Harbin et al. 1997). We propose that an ECM based bioactive scaffold can also provide a vehicle for delivery of matricellular proteins, where the choice of matricellular proteins can be tailored to the etiology of the target wound. Realization of such a treatment, however, depends on a greater understanding of the role of matricellular proteins in the pathogenesis of chronic wounds.
Conclusions
The burden of chronic wounds on healthcare systems worldwide is enormous. With no signs that the prevalence of diabetes will decrease in the near future there is continued demand for new and effective treatments for chronic wounds. Parallels between the state of fibrotic diseases and the needs of chronic wounds (increased proliferation, increased matrix synthesis and increased matrix contraction) provide an interesting opportunity to discover new therapeutic targets relevant to both pathologies. Treatment of chronic wounds currently involves repeated debridement followed by application of moist dressings. The development of advanced treatment strategies, as discussed here, has been beneficial in complimenting current wound care practice. Efficacy of these strategies has been limited, however. This is likely due to the complexity and diversity of chronic wounds. Our limited understanding of how molecules such as TGFβ and periostin, for example, interact slows progress in designing better treatments. Therefore, in order to produce new and effective treatments we must increase our understanding of the etiology of chronic wounds, with particular attention to the role of matricellular proteins.
Abbreviations
- α-SMA:
-
alpha smooth muscle actin
- bTGFβ2:
-
bovine transforming growth factor beta 2
- CCN2:
-
connective tissue growth factor
- ED-A FN:
-
extra type III domain-A fibronectin
- ERK:
-
extracellular signal-regulated kinases
- FAK:
-
focal adhesion kinase
- JNK:
-
c-Jun N terminal kinases
- LEA:
-
lower extremity amputation
- MMP:
-
matrix metalloproteinase
- PDGF:
-
platelet-derived growth factor
- PRP:
-
platelet rich plasma
- rhPDGF-BB:
-
recombinant human platelet-derived growth factor B homodimer
- SIS:
-
small intestine submucosa
- SSc:
-
systemic sclerosis
- TGFβ:
-
transforming growth factor beta
- TIMP:
-
tissue inhibitors of matrix metalloproteinases
References
Abergel RP, Pizzurro D, Meeker CA et al (1985) Biochemical composition of the connective tissue in keloids and analysis of collagen metabolism in keloid fibroblast cultures. J Invest Dermatol 84:384–390
Abraham DJ, Varga J (2005) Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol 26:587–595
Akhmetshina A, Dees C, Pileckyte M et al (2008) Dual inhibition of c-abl and PDGF receptor signaling by dasatinib and nilotinib for the treatment of dermal fibrosis. FASEB J 22:2214–2222
Aragon-Sanchez J, Hernandez-Herrero MJ, Lazaro-Martinez JL et al (2010) In-hospital complications and mortality following major lower extremity amputations in a series of predominantly diabetic patients. Int J Low Extrem Wounds 9:16–23
Armstrong DG, Wrobel J, Robbins JM (2007) Guest Editorial: are diabetes-related wounds and amputations worse than cancer? Int Wound J 4:286–287
Bao S, Ouyang G, Bai X et al (2004) Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 5:329–339
Baril P, Gangeswaran R, Mahon PC et al (2007) Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene 26:2082–2094
Barrientos S, Stojadinovic O, Golinko MS et al (2008) Growth factors and cytokines in wound healing. Wound Repair Regen 16:585–601
Beck LS, Chen TL, Hirabayashi SE et al (1990) Accelerated healing of ulcer wounds in the rabbit ear by recombinant human transforming growth factor-beta 1. Growth Factors 2:273–282
Bennett SP, Griffiths GD, Schor AM et al (2003) Growth factors in the treatment of diabetic foot ulcers. Br J Surg 90:133–146
Beyer C, Distler JH, Distler O (2010) Are tyrosine kinase inhibitors promising for the treatment of systemic sclerosis and other fibrotic diseases? Swiss Med Wkly 140:w13050
Bornstein P (1995) Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol 130:503–506
Bostrom H, Willetts K, Pekny M et al (1996) PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85:863–873
Butcher JT, Norris RA, Hoffman S et al (2007) Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase. Dev Biol 302:256–266
Cavanagh PR, Lipsky BA, Bradbury AW et al (2005) Treatment for diabetic foot ulcers. Lancet 366:1725–1735
Chaby G, Senet P, Vaneau M et al (2007) Dressings for acute and chronic wounds: a systematic review. Arch Dermatol 143:1297–1304
Chen SM, Ward SI, Olutoye OO et al (1997) Ability of chronic wound fluids to degrade peptide growth factors is associated with increased levels of elastase activity and diminished levels of proteinase inhibitors. Wound Repair Regen 5:23–32
Chen Y, Shi-Wen X, van Beek J et al (2005) Matrix contraction by dermal fibroblasts requires transforming growth factor-beta/activin-linked kinase 5, heparan sulfate-containing proteoglycans, and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol 167:1699–1711
Clark R (1996) The molecular and cellular biology of wound repair, 2nd edn. Plenum Publishing Corporation, New York
Cochrane CG (1977) Role of granulocytes in immune complex-induced tissue injuries. Inflammation 2:319–333
Cowin AJ, Hatzirodos N, Holding CA et al (2001) Effect of healing on the expression of transforming growth factor beta(s) and their receptors in chronic venous leg ulcers. J Invest Dermatol 117:1282–1289
Daniels CE, Wilkes MC, Edens M et al (2004) Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest 114:1308–1316
Denhardt DT, Giachelli CM, Rittling SR (2001) Role of osteopontin in cellular signaling and toxicant injury. Annu Rev Pharmacol Toxicol 41:723–749
Denton CP, Merkel PA, Furst DE et al (2007) Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum 56:323–333
Desmouliere A, Geinoz A, Gabbiani F et al (1993) Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 122:103–111
Desmouliere A, Chaponnier C, Gabbiani G (2005) Tissue repair, contraction, and the myofibroblast. Wound Repair Regen 13:7–12
Deuel TF, Senior RM, Huang JS et al (1982) Chemotaxis of monocytes and neutrophils to platelet-derived growth factor. J Clin Invest 69:1046–1049
Diegelmann RF, Evans MC (2004) Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci 9:283–289
Distler JH, Jungel A, Huber LC et al (2007) Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum 56:311–322
Donaghue VM, Chrzan JS, Rosenblum BI et al (1998) Evaluation of a collagen-alginate wound dressing in the management of diabetic foot ulcers. Adv Wound Care 11:114–119
Driver VR, Hanft J, Fylling CP et al (2006) A prospective, randomized, controlled trial of autologous platelet-rich plasma gel for the treatment of diabetic foot ulcers. Ostomy/Wound Management 52:68–70, 2, 4 passim
Driver VR, Fabbi M, Lavery LA et al (2010) The costs of diabetic foot: the economic case for the limb salvage team. J Vasc Surg 52:17S–22S
Edwards DR, Murphy G, Reynolds JJ et al (1987) Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. EMBO J 6:1899–1904
Erkan M, Kleeff J, Gorbachevski A et al (2007) Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 132:1447–1464
Galkowska H, Wojewodzka U, Olszewski WL (2006) Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen 14:558–565
Gao Z, Sasaoka T, Fujimori T et al (2005) Deletion of the PDGFR-beta gene affects key fibroblast functions important for wound healing. J Biol Chem 280:9375–9389
Gillan L, Matei D, Fishman DA et al (2002) Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res 62:5358–5364
Greiling D, Clark RA (1997) Fibronectin provides a conduit for fibroblast transmigration from collagenous stroma into fibrin clot provisional matrix. J Cell Sci 110(Pt 7):861–870
Hambleton IR, Jonnalagadda R, Davis CR et al (2009) All-cause mortality after diabetes-related amputation in Barbados: a prospective case-control study. Diabetes Care 32:306–307
Hamilton DW (2008) Functional role of periostin in development and wound repair: implications for connective tissue disease. J Cell Commun Signal 2:9–17
Harding K, Queen D (2010) Chronic wounds and their management and prevention is a significiant public health issue. Int Wound J 7:125–126
Harding KG, Morris HL, Patel GK (2002) Science, medicine and the future: healing chronic wounds. BMJ 324:160–163
Hasan A, Murata H, Falabella A et al (1997) Dermal fibroblasts from venous ulcers are unresponsive to the action of transforming growth factor-beta 1. J Dermatol Sci 16:59–66
Hehenberger K, Heilborn JD, Brismar K et al (1998) Inhibited proliferation of fibroblasts derived from chronic diabetic wounds and normal dermal fibroblasts treated with high glucose is associated with increased formation of l-lactate. Wound Repair Regen 6:135–141
Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79:1283–1316
Holmes A, Abraham DJ, Sa S et al (2001) CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem 276:10594–10601
Horiuchi K, Amizuka N, Takeshita S et al (1999) Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res 14:1239–1249
Igarashi A, Okochi H, Bradham DM et al (1993) Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell 4:637–645
Ignotz RA, Massague J (1986) Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem 261:4337–4345
Jackson-Boeters L, Wen W, Hamilton DW (2009) Periostin localizes to cells in normal skin, but is associated with the extracellular matrix during wound repair. J Cell Commun Signal 3:125–133
Jinnin M, Ihn H, Mimura Y et al (2005) Regulation of fibrogenic/fibrolytic genes by platelet-derived growth factor C, a novel growth factor, in human dermal fibroblasts. J Cell Physiol 202:510–517
Jude EB, Blakytny R, Bulmer J et al (2002) Transforming growth factor-beta 1, 2, 3 and receptor type I and II in diabetic foot ulcers. Diabet Med 19:440–447
Jun JI, Lau LF (2010) Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2:627–631
Katsuragi N, Morishita R, Nakamura N et al (2004) Periostin as a novel factor responsible for ventricular dilation. Circulation 110:1806–1813
Kawakami T, Ihn H, Xu W et al (1998) Increased expression of TGF-beta receptors by scleroderma fibroblasts: evidence for contribution of autocrine TGF-beta signaling to scleroderma phenotype. J Invest Dermatol 110:47–51
Kennedy L, Liu S, Shi-Wen X et al (2007) CCN2 is necessary for the function of mouse embryonic fibroblasts. Exp Cell Res 313:952–964
Kii I, Amizuka N, Minqi L et al (2006) Periostin is an extracellular matrix protein required for eruption of incisors in mice. Biochem Biophys Res Commun 342:766–772
Kii I, Nishiyama T, Li M et al (2009) Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem 285:2028–2039
Kim JE, Jeong HW, Nam JO et al (2002) Identification of motifs in the fasciclin domains of the transforming growth factor-beta-induced matrix protein betaig-h3 that interact with the alphavbeta5 integrin. J Biol Chem 277:46159–46165
Kirshen C, Woo K, Ayello EA et al (2006) Debridement: a vital component of wound bed preparation. Adv Skin Wound Care 19:506–517, quiz 17–9
Klareskog L, Gustafsson R, Scheynius A et al (1990) Increased expression of platelet-derived growth factor type B receptors in the skin of patients with systemic sclerosis. Arthritis Rheum 33:1534–1541
Kocher O, Madri JA (1989) Modulation of actin mRNAs in cultured vascular cells by matrix components and TGF-beta 1. In Vitro Cell Dev Biol 25:424–434
Kraling BM, Maul GG, Jimenez SA (1995) Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology 63:48–56
Kubo M, Ihn H, Yamane K et al (2002) Upregulated expression of transforming growth factor-beta receptors in dermal fibroblasts of skin sections from patients with systemic sclerosis. J Rheumatol 29:2558–2564
Leask A (2008) Targeting the TGFbeta, endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell Signal 20:1409–1414
Leask A (2010) Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res 106:1675–1680
Leask A, Abraham DJ (2004) TGF-beta signaling and the fibrotic response. FASEB J 18:816–827
Leibovich SJ, Ross R (1975) The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol 78:71–100
Lenga Y, Koh A, Perera AS et al (2008) Osteopontin expression is required for myofibroblast differentiation. Circ Res 102:319–327
Levenson SM, Geever EF, Crowley LV et al (1965) The healing of rat skin wounds. Ann Surg 161:293–308
Li G, Oparil S, Sanders JM et al (2006) Phosphatidylinositol-3-kinase signaling mediates vascular smooth muscle cell expression of periostin in vivo and in vitro. Atherosclerosis 188:292–300
Li G, Jin R, Norris RA et al (2010) Periostin mediates vascular smooth muscle cell migration through the integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase (FAK) pathway. Atherosclerosis 208:358–365
Lin H, Chen B, Sun W et al (2006) The effect of collagen-targeting platelet-derived growth factor on cellularization and vascularization of collagen scaffolds. Biomaterials 27:5708–5714
Lindner V, Wang Q, Conley BA et al (2005) Vascular injury induces expression of periostin: implications for vascular cell differentiation and migration. Arterioscler Thromb Vasc Biol 25:77–83
Litvin J, Selim AH, Montgomery MO et al (2004) Expression and function of periostin-isoforms in bone. J Cell Biochem 92:1044–1061
Liu S, Xu SW, Kennedy L et al (2007) FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell 18:2169–2178
Liu Y, Min D, Bolton T et al (2009) Increased matrix metalloproteinase-9 predicts poor wound healing in diabetic foot ulcers. Diabetes Care 32:117–119
Loot MA, Kenter SB, Au FL et al (2002) Fibroblasts derived from chronic diabetic ulcers differ in their response to stimulation with EGF, IGF-I, bFGF and PDGF-AB compared to controls. Eur J Cell Biol 81:153–160
Ludwicka A, Ohba T, Trojanowska M et al (1995) Elevated levels of platelet derived growth factor and transforming growth factor-beta 1 in bronchoalveolar lavage fluid from patients with scleroderma. J Rheumatol 22:1876–1883
Margolis DJ, Kantor J, Santanna J et al (2001) Effectiveness of platelet releasate for the treatment of diabetic neuropathic foot ulcers. Diabetes Care 24:483–488
Maruhashi T, Kii I, Saito M et al (2010) Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem 285:13294–13303
Massague J, Wotton D (2000) Transcriptional control by the TGF-beta/Smad signaling system. EMBO J 19:1745–1754
Matsuda A, Yoshiki A, Tagawa Y et al (1999) Corneal wound healing in tenascin knockout mouse. Invest Ophthalmol Vis Sci 40:1071–1080
Mendez MV, Stanley A, Phillips T et al (1998) Fibroblasts cultured from distal lower extremities in patients with venous reflux display cellular characteristics of senescence. J Vasc Surg 28:1040–1050
Midwood KS, Williams LV, Schwarzbauer JE (2004) Tissue repair and the dynamics of the extracellular matrix. Int J Biochem Cell Biol 36:1031–1037
Moiseeva EP, Williams B, Samani NJ (2003) Galectin 1 inhibits incorporation of vitronectin and chondroitin sulfate B into the extracellular matrix of human vascular smooth muscle cells. Biochim Biophys Acta 1619:125–132
Morbach S, Groblinghoff U, Schulze H et al (2009) All-cause mortality after diabetes-related amputation in Barbados: a prospective case-control study: response to Hambleton et al. Diabetes Care 32:e100, author reply e1
Mori Y, Chen SJ, Varga J (2003) Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum 48:1964–1978
Mosesson MW (2005) Fibrinogen and fibrin structure and functions. J Thromb Haemost 3:1894–1904
Mostow EN, Haraway GD, Dalsing M et al (2005) Effectiveness of an extracellular matrix graft (OASIS Wound Matrix) in the treatment of chronic leg ulcers: a randomized clinical trial. J Vasc Surg 41:837–843
Moxey PW, Hofman D, Hinchliffe RJ et al (2010) Epidemiological study of lower limb amputation in England between 2003 and 2008. Br J Surg 97:1348–1353
Murakami K, Aoki H, Nakamura S et al (2010) Hydrogel blends of chitin/chitosan, fucoidan and alginate as healing-impaired wound dressings. Biomaterials 31:83–90
Mustoe TA, Purdy J, Gramates P et al (1989) Reversal of impaired wound healing in irradiated rats by platelet-derived growth factor-BB. Am J Surg 158:345–350
Mustoe TA, Landes A, Cromack DT et al (1990) Differential acceleration of healing of surgical incisions in the rabbit gastrointestinal tract by platelet-derived growth factor and transforming growth factor, type beta. Surgery 108:324–329, discussion 9–30
Mustoe TA, Pierce GF, Morishima C et al (1991) Growth factor-induced acceleration of tissue repair through direct and inductive activities in a rabbit dermal ulcer model. J Clin Invest 87:694–703
Mustoe TA, Cutler NR, Allman RM et al (1994) A phase II study to evaluate recombinant platelet-derived growth factor-BB in the treatment of stage 3 and 4 pressure ulcers. Arch Surg 129:213–219
Naitoh M, Kubota H, Ikeda M et al (2005) Gene expression in human keloids is altered from dermal to chondrocytic and osteogenic lineage. Genes Cells 10:1081–1091
Nakazawa T, Nakajima A, Seki N et al (2004) Gene expression of periostin in the early stage of fracture healing detected by cDNA microarray analysis. J Orthop Res 22:520–525
Niezgoda JA, Van Gils CC, Frykberg RG et al (2005) Randomized clinical trial comparing OASIS Wound Matrix to Regranex Gel for diabetic ulcers. Adv Skin Wound Care 18:258–266
Norris RA, Damon B, Mironov V et al (2007) Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem 101:695–711
Oka T, Xu J, Kaiser RA et al (2007) Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res 101:313–321
Oku E, Kanaji T, Takata Y et al (2008) Periostin and bone marrow fibrosis. Int J Hematol 88:57–63
Pannu J, Trojanowska M (2004) Recent advances in fibroblast signaling and biology in scleroderma. Curr Opin Rheumatol 16:739–745
Pannu J, Gore-Hyer E, Yamanaka M et al (2004) An increased transforming growth factor beta receptor type I:type II ratio contributes to elevated collagen protein synthesis that is resistant to inhibition via a kinase-deficient transforming growth factor beta receptor type II in scleroderma. Arthritis Rheum 50:1566–1577
Papazafiropoulou A, Tentolouris N, Soldatos RP et al (2009) Mortality in diabetic and nondiabetic patients after amputations performed from 1996 to 2005 in a tertiary hospital population: a 3-year follow-up study. J Diab Complications 23:7–11
Pastar I, Stojadinovic O, Krzyzanowska A et al (2010) Attenuation of the transforming growth factor beta-signaling pathway in chronic venous ulcers. Mol Med 16:92–101
Pierce GF, Mustoe TA, Senior RM et al (1988) In vivo incisional wound healing augmented by platelet-derived growth factor and recombinant c-sis gene homodimeric proteins. J Exp Med 167:974–987
Pierce GF, Mustoe TA, Lingelbach J et al (1989) Platelet-derived growth factor and transforming growth factor-beta enhance tissue repair activities by unique mechanisms. J Cell Biol 109:429–440
Pierce GF, Tarpley JE, Tseng J et al (1995) Detection of platelet-derived growth factor (PDGF)-AA in actively healing human wounds treated with recombinant PDGF-BB and absence of PDGF in chronic nonhealing wounds. J Clin Invest 96:1336–1350
Ploeg AJ, Lardenoye J-W, Vrancken Peeters M-PFM et al (2005) Contemporary series of morbidity and mortality after lower limb amputation. In: Eur J Vasc Endovasc Surg 633–637
Qin Y, Gilding DK (1996) Alginate fibres and wound dressings. Med Device Technol 7:32–41
Queen D, Orsted H, Sanada H et al (2004) A dressing history. Int Wound J 1:59–77
Reiber GE (1996) The epidemiology of diabetic foot problems. Diabet Med 13(Suppl 1):S6–S11
Roberts AB, Sporn MB (1993) Physiological actions and clinical applications of transforming growth factor-beta (TGF-beta). Growth Factors 8:1–9
Robson MC (1997) The role of growth factors in the healing of chronic wounds. Wound Repair Regen 5:12–17
Robson MC, Phillips LG, Thomason A et al (1992) Platelet-derived growth factor BB for the treatment of chronic pressure ulcers. Lancet 339:23–25
Robson MC, Phillip LG, Cooper DM et al (1995) Safety and effect of transforming growth factor-beta(2) for treatment of venous stasis ulcers. Wound Repair Regen 3:157–167
Santiago B, Gutierrez-Canas I, Dotor J et al (2005) Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J Invest Dermatol 125:450–455
Schultz GS, Grant MB (1991) Neovascular growth factors. Eye (Lond) 5(Pt 2):170–180
Schultz GS, Mast BA (1998) Molecular analysis of the environments of healing and chronic wounds: cytokines, proteases and growth factors. Wounds 10(suppl F):F1–F9
Schultz GS, Wysocki A (2009) Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen 17:153–162
Schultz-Cherry S, Murphy-Ullrich JE (1993) Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol 122:923–932
Sen CK, Gordillo GM, Roy S et al (2009) Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen 17:763–771
Seppa H, Grotendorst G, Seppa S et al (1982) Platelet-derived growth factor in chemotactic for fibroblasts. J Cell Biol 92:584–588
Serini G, Bochaton-Piallat ML, Ropraz P et al (1998) The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol 142:873–881
Sethi KK, Yannas IV, Mudera V et al (2002) Evidence for sequential utilization of fibronectin, vitronectin, and collagen during fibroblast-mediated collagen contraction. Wound Repair Regen 10:397–408
Shah M, Foreman DM, Ferguson MW (1992) Control of scarring in adult wounds by neutralising antibody to transforming growth factor beta. Lancet 339:213–214
Shah M, Foreman DM, Ferguson MW (1995) Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J Cell Sci 108(Pt 3):985–1002
Shao R, Bao S, Bai X et al (2004) Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol Cell Biol 24:3992–4003
Shimazaki M, Nakamura K, Kii I et al (2008) Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med 205:295–303
Shi-wen X, Stanton LA, Kennedy L et al (2006) CCN2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. J Biol Chem 281:10715–10726
Sidhu SS, Yuan S, Innes AL et al (2010) Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc Natl Acad Sci USA 107:14170–14175
Siitonen OI, Niskanen LK, Laakso M et al (1993) Lower-extremity amputations in diabetic and nondiabetic patients. A population-based study in eastern Finland. Diabetes Care 16:16–20
Simpson DM, Ross R (1972) The neutrophilic leukocyte in wound repair a study with antineutrophil serum. J Clin Invest 51:2009–2023
Singer AJ, Clark RA (1999) Cutaneous wound healing. N Engl J Med 341:738–746
Singh N, Armstrong DG, Lipsky BA (2005) Preventing foot ulcers in patients with diabetes. JAMA 293:217–228
Sodek J, Ganss B, McKee MD (2000) Osteopontin. Crit Rev Oral Biol Med 11:279–303
Stanley AC, Park HY, Phillips TJ et al (1997) Reduced growth of dermal fibroblasts from chronic venous ulcers can be stimulated with growth factors. J Vasc Surg 26:994–999, discussion 9–1001
Stansfield WE, Andersen NM, Tang RH et al (2009) Periostin is a novel factor in cardiac remodeling after experimental and clinical unloading of the failing heart. Ann Thorac Surg 88:1916–1921
Steed DL (1995) Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity diabetic ulcers. Diabetic Ulcer Study Group. J Vasc Surg 21:71–78, discussion 9–81
Takayama G, Arima K, Kanaji T et al (2006) Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol 118:98–104
Takehara K (2003) Hypothesis: pathogenesis of systemic sclerosis. J Rheumatol 30:755–759
Takeshita S, Kikuno R, Tezuka K et al (1993) Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J 294(Pt 1):271–278
Tan EM, Qin H, Kennedy SH et al (1995) Platelet-derived growth factors-AA and -BB regulate collagen and collagenase gene expression differentially in human fibroblasts. Biochem J 310(Pt 2):585–588
Thannickal VJ, Lee DY, White ES et al (2003) Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 278:12384–12389
Tomasek JJ, Gabbiani G, Hinz B et al (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3:349–363
Trengove NJ, Bielefeldt-Ohmann H, Stacey MC (2000) Mitogenic activity and cytokine levels in non-healing and healing chronic leg ulcers. Wound Repair Regen 8:13–25
Trojanowska M (2008) Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology (Oxford) 47(Suppl 5):V2–V4
Vaalamo M, Leivo T, Saarialho-Kere U (1999) Differential expression of tissue inhibitors of metalloproteinases (TIMP-1, -2, -3, and -4) in normal and aberrant wound healing. Hum Pathol 30:795–802
Varga J, Abraham D (2007) Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 117:557–567
Varga J, Pasche B (2009) Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol 5:200–206
Verrecchia F, Chu ML, Mauviel A (2001) Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem 276:17058–17062
Vi L, Feng L, Zhu RD et al (2009) Periostin differentially induces proliferation, contraction and apoptosis of primary Dupuytren’s disease and adjacent palmar fascia cells. Exp Cell Res 315:3574–3586
Voytik-Harbin SL, Brightman AO, Kraine MR et al (1997) Identification of extractable growth factors from small intestinal submucosa. J Cell Biochem 67:478–491
Wang Q, Nie FF, Zhao X et al (2007) The expression of periostin in hyperplasic scars and the relations to TGF-beta1 and its receptors. Zhonghua Zheng Xing Wai Ke Za Zhi 23:229–232
Wei J, Bhattacharyya S, Tourtellotte WG et al (2010) Fibrosis in systemic sclerosis: Emerging concepts and implications for targeted therapy. Autoimmun Rev
Wieman TJ, Smiell JM, Su Y (1998) Efficacy and safety of a topical gel formulation of recombinant human platelet-derived growth factor-BB (becaplermin) in patients with chronic neuropathic diabetic ulcers. A phase III randomized placebo-controlled double-blind study. Diabetes Care 21:822–827
Wild S, Roglic G, Green A et al (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–1053
Woodbury MG, Houghton PE (2004) Prevalence of pressure ulcers in Canadian healthcare settings. Ostomy/Wound Management 50:22–38
Woodbury MG, Houghton PE (2005) The extent of chronic wounds in Canada. Wound Care Canada 3:18–22
Woods A (2001) Syndecans: transmembrane modulators of adhesion and matrix assembly. J Clin Invest 107:935–941
Wu L, Xia YP, Roth SI et al (1999) Transforming growth factor-beta1 fails to stimulate wound healing and impairs its signal transduction in an aged ischemic ulcer model: importance of oxygen and age. Am J Pathol 154:301–309
Wu SC, Driver VR, Wrobel JS et al (2007) Foot ulcers in the diabetic patient, prevention and treatment. Vasc Health Risk Manag 3:65–76
Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 117:524–529
Wysocki AB, Staiano-Coico L, Grinnell F (1993) Wound fluid from chronic leg ulcers contains elevated levels of metalloproteinases MMP-2 and MMP-9. J Invest Dermatol 101:64–68
Yamamoto T, Nishioka K (2005) Cellular and molecular mechanisms of bleomycin-induced murine scleroderma: current update and future perspective. Exp Dermatol 14:81–95
Yan W, Shao R (2006) Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial-mesenchymal transformation. J Biol Chem 281:19700–19708
Yang C, Zhu P, Yan L et al (2009) Dynamic changes in matrix metalloproteinase 9 and tissue inhibitor of metalloproteinase 1 levels during wound healing in diabetic rats. J Am Podiatr Med Assoc 99:489–496
Zhao LL, Davidson JD, Wee SC et al (1994) Effect of hyperbaric oxygen and growth factors on rabbit ear ischemic ulcers. Arch Surg 129:1043–1049
Zheng XY, Zhang JZ, Tu P et al (1998) Expression of platelet-derived growth factor B-chain and platelet-derived growth factor beta-receptor in fibroblasts of scleroderma. J Dermatol Sci 18:90–97
Zhou HM, Wang J, Elliott C et al (2010) Spatiotemporal expression of periostin during skin development and incisional wound healing: lessons for human fibrotic scar formation. J Cell Commun Signal 4:99–107
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Elliott, C.G., Hamilton, D.W. Deconstructing fibrosis research: do pro-fibrotic signals point the way for chronic dermal wound regeneration?. J. Cell Commun. Signal. 5, 301–315 (2011). https://doi.org/10.1007/s12079-011-0131-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-011-0131-5