
Abstract
Background Toll-like receptors (TLRs) may play active roles in both innate and adaptive immune responses in human intrahepatic biliary epithelial cells (HIBECs). The role of TLR3 expressed by HIBECs, however, remains unclear. Methods We determined the in vivo expression of TLRs in biopsy specimens derived from diseased livers immunohistochemically using a panel of monoclonal antibodies against human TLRs. We then examined the response of cultured HIBECs to a TLR3 ligand, polyinosinic–polycytidylic acid (polyI:C). Using siRNAs specific for Toll-IL-1R homology domain-containing adaptor molecule 1 (TICAM-1) and mitochondrial antiviral signaling protein (MAVS), we studied signaling pathways inducing IFN-β expression. Results The expression of TLR3 was markedly increased in biliary epithelial cells at sites of ductular reaction in diseased livers, including primary biliary cirrhosis (PBC), autoimmune hepatitis (AIH), and chronic viral hepatitis (CH) as compared to nondiseased livers. Although cultured HIBECs constitutively expressed TLR3 at both the protein and mRNA levels in vitro, the addition of polyI:C to culture media induced only minimal increases in IFN-β mRNA. In contrast, transfection of HIBECs with polyI:C induced a marked increase in mRNAs encoding a variety of chemokines/cytokines, including IFN-β, IL-6, and TNF-α. The induction of IFN-β mRNA was efficiently inhibited by an siRNA against MAVS but not against TICAM-1, indicating that the main signaling pathway for IFN-β induction following polyI:C transfection is via retinoic acid-inducible gene I (RIG-I)/melanoma differentiation-associated gene 5 (MDA5) in HIBECs. Conclusions TLR3 expression by biliary epithelial cells increased at sites of ductular reaction in diseased livers; further study will be necessary to characterize it’s in vivo physiological role.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epithelial cells are the first barrier against viral infection. Such cells typically express retinoic acid-inducible gene I (RIG-I)/melanoma differentiation-associated gene 5 (MDA5) and Toll-like receptor 3 (TLR3) to sense double-stranded RNAs (dsRNA), hallmarks of viral replication [1–3]. TLR3 is localized to endosomes and/or the cell surface in epithelial cells, while RIG-I/MDA5 resides in the cytoplasm [3–5]. TLR3-expressing epithelial cells are widely distributed throughout the body, with prominent expression in intestinal, cervical, uterine, endometrial, bronchial, and corneal epithelial cells, the central nervous system, and epidermal keratinocytes [6–16]. The function of TLR3 has been intensively studied in some of these epithelial cells; bronchial epithelial cells recognize dsRNA by cell-surface TLR3 and induce cellular responses, including the secretion of type 1 interferon (IFN) via the Toll-IL-1R homology domain-containing adaptor molecule 1 (TICAM-1)-interferon regulatory factor 3 (IRF3) signaling pathway [11, 12]. The intracellular RNA sensors RIG-I/MDA5 also serve as IFN inducers acting via the mitochondrial antiviral signaling protein (MAVS)-IRF3 signaling pathway, thus protecting host cells against the spread of viral invasion [2, 3].
We previously found that the expression of TLR3 and IFN-β mRNAs is significantly increased in both the portal areas and parenchyma of livers diseased with PBC [17]. There was a positive correlation between TLR3 and IFN-β mRNA levels in both areas, indicating that TLR3-type 1 IFN signaling pathway is activated in PBC; the TLR3-expressing and/or IFN-β-producing cells, however, remain unknown [17]. This prompted us to investigate TLR3 expression and IFN-β production in human intrahepatic biliary epithelial cells (HIBECs).
In this study, we used specific monoclonal antibodies against TLRs [4] to determine that intrahepatic bile ducts, but not hepatocytes, in diseased livers strongly express TLR3. TLR3 protein is found in HIBECs at low levels on the cell surface and high levels in endosomes. Our results, however, indicate that the primary signaling pathway for IFN-β induction activated by dsRNA functions via RIG-I/MDA5 in the cytoplasm but not via TLR3 expressed on the cell surface or in endosomes. This is contrary to results obtained for other types of epithelial cells, such as bronchial epithelial cells and endometrial cells, in which surface TLR3 recognizes viral dsRNA to signal the presence of infection via the TLR3-IRF3-type I interferon signaling pathway [9, 11, 12, 15]. Here we discuss dsRNA-sensing system functioning in HIBECs and the role of high expression levels of TLR3 in diseased livers.
Materials and methods
Liver biopsy specimen and immunohistochemical evaluation
Liver needle biopsy specimens, which were derived from seven primary biliary cirrhosis (PBC)-affected, five autoimmune hepatitis (AIH)-affected, and five chronic hepatitis C (CHC)-affected livers, were frozen in OCT compound (Sakura Finetechnical Co, Tokyo, Japan) immediately after the procedure and were stored at −80°C until use. Mouse monoclonal antibodies to human TLR1 (clone TLR1.136, IgG1, k), TLR2 (clone TLR2.45, IgG1, k), TLR3 (clone TLR3.7, IgG1, k), TLR4 (clone TLR4, IgG2a, k), and TLR6 (clone TLR6.127, IgG1, k) were generated in our laboratory [4]. Among these monoclonal antibodies, the specificity of anti-TLR3 (TLR3.7) was intensively studied. Anti-TLR3 monoclonal antibody specifically binds to the extracellular part of native TLR3 but not to denatured form of TLR3 or other TLRs, including TLR2 and TLR4. Furthermore, TLR3.7 inhibits dsRNA-induced IFN-β production by inhibiting the interassociation between dsRNA and TLR3 [4, 5]. Mouse monoclonal antibodies specific for cytokeratin (CK) 7 and CK 19 were purchased from DAKO (DAKO Japan, Kyoto, Japan). Frozen sections, 4 mm in thickness, were stained with anti-TLR and anti-CK7 or -CK19 antibodies as described elsewhere [17]. Briefly, frozen sections were first fixed in 50 and 100% acetone for 30 s and 3 min, respectively, followed by treatment with Peroxidase Blocking agent (DAKO) for 10 min. Sections were then incubated with anti-TLR monoclonal antibodies (anti-TLR1, 2, 3, 4, and 6) for 60 min at room temperature. A standard 2-step method with ENVISION+ (DAKO) was used to visualize bound antibody using 3,3′-diaminobenzidine as a chromogen (DAKO); samples were also counterstained with Mayer’s hematoxylin (DAKO). Three frozen liver biopsy specimens, which revealed normal histology, were similarly studied as nondiseased livers.
Isolation and culture of human intrahepatic biliary epithelial cells
Human intrahepatic biliary epithelial cells (HIBECs) were isolated from noncancerous liver tissues of three patients who had undergone hepatic resection for intrahepatic cholangiocarcinoma [18]. Briefly, liver specimens were digested with type IV collagenase (100 U/ml) (Sigma–Aldrich, St. Louis, MO). HIBECs were isolated immunomagnetically using Dynabeads conjugated with an epithelium-specific antibody, BerEp4 (Dynal Biotech, Norway). HIBECs were expanded in HIBEC culture medium (DMEM containing 5 μg/ml insulin, 10 ng/ml epidermal growth factor [EGF], 1.0 ng/ml hepatocyte growth factor [HGF], 4 × 10−8 M dexamethasone and 10% fetal bovine serum). All experiments were performed using HIBECs between 5 and 10 passages, which were performed using PBS containing 0.05% trypsin and 0.53 mM EDTA.
We obtained three different HIBECs (BEC3, BEC4, and BEC5) from the three different donors; each cultured HIBEC demonstrated spindle to polygonal epithelial cell morphology, with 100% positivity for CK7 and CK19 as determined by immunostaining with anti-CK7 and -CK19 monoclonal antibodies (DAKO).
Immunostaining and flow-cytometric analysis of HIBECs
HIBECs were cultured to semiconfluence in a tissue culture-treated 8-chamber glass slides (BD Biosciences, Bedford, MA) in HIBEC culture medium. Immunostaining of these cultured cells was then performed in a similar manner as that described for frozen sections of liver biopsies [17]. In brief, HIBECs were fixed with acetone, treated with peroxidase-blocking agent, and incubated with anti-TLR (anti-TLR1, -2, -3, -4, and -6) and anti-CK7 or anti-CK19 monoclonal antibodies followed by visualization of bound antibodies using a standard 2-step method with ENVISION+ (DAKO).
For flow-cytometric analysis, HIBECs were first suspended in PBS containing 0.1% sodium azide and 0.1% bovine serum albumin before incubating with 5 μg mAb (clone TLR3.7, IgG1, k) for 30 min at 4°C. Cells were washed and counterstained with FITC-conjugated goat antimouse IgG F(ab′)2 for 30 min at 4°C. We then determined fluorescence intensity and mean fluorescence shifts by flow cytometry (FACSCalibur; Becton-Dickinson).
Stimulation of HIBECs with polyI:C
Polyinosinic–polycytidylic acid (PolyI:C) was purchased from Sigma–Aldrich and reconstituted in endotoxin-free PBS. Transfection reagents, Lipofectamine 2000 and DOTAP, were purchased from Invitrogen (Carlsbad, CA) and Roche (Basel, Switzerland), respectively.
Twenty-four hours prior to the start of polyI:C stimulation, we changed the culture medium from HIBEC culture medium to basal medium (1:1 mixture of Ham’s F12 and DMEM supplemented with 10% FBS without insulin, EGF, HGF, and dexamethasone). HIBECs were then incubated in the presence of polyI:C (40 μg/ml) or transfected with polyI:C using Lipofectamine 2000 or DOTAP according to the manufacturer’s instructions. Optimal conditions for transfection by Lipofectamine 2000 and DOTAP were 0.8 μg/well and 1.0 μg/well polyI:C, respectively, in a 12-well plate (Becton Dickinson, Franklin Lakes, NJ) (data not shown).
RNA extraction and quantitation of mRNA
Total RNA was isolated from HIBECs using an RNeasy MiniKit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Following RNase-free DNase I (QIAGEN) treatment, we synthesized first-strand complementary DNAs (cDNA) from 1.0 μg total RNA using a SuperScript First-Strand Synthesis System (Invitrogen). PCR amplification utilized FAST DNA SYBR Green I (Roche), which allows for automated quantification of amplified products in real-time using a Light-Cycler (Roche). We purchased primer sets specific for IFN-γ, IL-6, TNF-α, IL-8, and TLR3 from Roche. One microliter of each reverse-transcribed cDNA was used for real-time PCR analysis. Initial denaturation was performed at 95°C for 10 min followed by 40 amplification cycles of denaturation at 95°C for 10 s, annealing at 68°C for 10 s, and extension at 72°C for 16 s. We performed a standard melting curve analysis for every quantitation. Results were expressed as the ratio of cytokine/chemokine cDNA to GAPDH cDNA copy numbers in individual samples. Changes in mRNA levels were expressed as fold induction.
Enzyme-linked immunosorbent assay (ELISA)
HIBEC culture supernatants were assessed for cytokine/chemokine secretion using ELISA kits specific for IFN-β (PBL Biomedical Laboratories, Piscataway, NJ) and TNF-α, IL-6, and IL-8 (Beckman Coulter, Fullerton, CA), according to the manufacturers’ instructions. Absorbance at either 405 or 450 nm was measured using a microplate reader (Multiskan JX, Thermo electron corporation, Vantaa, Finland).
Effect of siRNA on IFN-β mRNA induction by polyI:C
Oligonucleotides used for siRNA knockdown analysis were purchased from Proligo (St. Louis, MO) (GFP: sense, gcagcacgacuucuucaagtt, and antisense, cuugaagaagucgugcugctt, MAVS: sense, ccaccuugaugccugugaaca, and antisense, uucacaggcaucaagguggua, TICAM-1: sense, gaccagacgccacuccaactt, and antisense, guuggaguggcgucugguctt).
BEC3 cells (2.5 × 105 per well) were plated on 12-well plates using basal medium 24 h before siRNA transfection. On day 0, we transfected each siRNA oligonucleotide into BEC3 using Lipofectamine 2000. After 24 h (on day 1), the culture medium was replaced with fresh basal medium. Forty-eight hours after siRNA transfection (on day 2), cells were stimulated with polyI:C (final concentration 40 μg/ml in phosphate-buffered saline), Lipofectamine 2000 (0.8 μg/well in 12-well plates) or DOTAP (1.0 μg/well in 12-well plates). Six hours after polyI:C stimulation, we purified total RNA from BEC3 cells using an RNeasy Mini kit (Qiagen). RT-PCR was performed using M-MLV Reverse transcriptase (Promega, Madison, WA). Quantitative PCR analyses were carried out on an iCycler iQ Real-Time detection system (Bio-Rad, Hercules, CA) using Platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen) using the following primers; β-actin: forward, cctggcacccagcacaat, and reverse, gccgatccacacggagtact; IFN-β: forward, caacttgcttggattcctacaaag, and reverse, tattcaagcctcccattcaattg; MAVS: forward, ggtacccgagtctcgtttcct, and reverse, ttgtcttcagcaaacggcatt; TICAM-1: forward, agcgccttcgacattctaggt, and reverse, aggagaaccatggcatgca [19]. Quantitative PCR data was analyzed by the 2−ΔΔCT methods [20].
Ethics board
This study was approved by the Ethics Board of our institute. We obtained written informed consent from each subject for use of their biopsy and operation samples to advance knowledge on the cause of PBC.
Results
HIBECs strongly express TLR3 molecule in vitro
Previous analysis of the expression of TLRs mRNA in HIBECs by RT-PCR revealed that mRNAs encoding TLR1, -2, -3, -4, -5, -6, and -9, but not TLR7 and -8, are constitutively expressed in HIBECs [18]. The expression of TLR proteins, however, has not been examined. In this study, we studied the expression of TLR proteins in HIBECs using mouse monoclonal antibodies against TLR1, -2, -3, -4, and -6 [4]. TLR3 was strongly expressed on HIBECs (Fig. 1c) at an intensity comparable to that of CK19, a marker of biliary epithelial cells in the liver (Fig. 1f). In contrast, TLR1, -2, and -4 were weakly expressed on HIBECs (Fig. 1a, b, d). Flow-cytometric analysis revealed that while TLR3 was strongly expressed in the cytoplasm (Fig. 2a), it was only weakly expressed at the cell surface of HIBECs (Fig. 2b).

TLRs immunostaining in cultured human intrahepatic biliary epithelial cells (HIBECs). BEC3 cells were stained with mouse monoclonal antibodies: (a) TLR1.136 (diluted 1/80); (b) TLR2.45 (diluted 1/100); (c) TLR3.7 (diluted 1/100); (d) HTA125 (diluted 1/70); (e) TLR6.127 (diluted 1/80); (f) anti-CK19 (diluted 1/200) for TLR1, TLR2, TLR3, TLR4, TLR6, and CK19, respectively, as described in the text. BEC3 cells stained strongly with TLR3.7 but only weakly with TLR1.136, TLR2.45, and HTA125

Flow-cytometeric analysis of TLR3 in cultured human intrahepatic biliary epithelial cells (HIBECs). BEC3 cells were stained with TLR3.7 monoclonal antibody intracellularly (a) or extracellularly (b). BEC3 cells exhibited strong intracellular staining with TLR3.7 but only weak cell surface staining
Biliary epithelial cells strongly express TLR3 at sites of ductular reaction in vivo
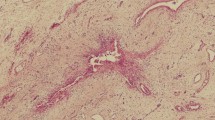
We examined the expression of TLRs in vivo using frozen sections of liver needle biopsy specimens using the monoclonal antibodies to TLR1, -2, -3, -4, and -6. Again, TLR3 was strongly expressed by intrahepatic biliary epithelial cells, especially at sites of ductular reaction in all diseased livers affected by PBC (Fig. 3b), AIH (Fig. 3c), and CHC (Fig. 3d). TLR3 was only weakly expressed on small bile ducts in normal livers (Fig. 3a). TLR3 was not expressed in hepatocytes of either diseased or normal livers. Minimal expression of TLR1, -2, -4, and -6 was observed in either diseased or normal livers (data not shown).

In vivo expression of TLR3 in intrahepatic biliary epithelial cells. TLR3 was strongly expressed on intrahepatic biliary epithelial cells, especially at sites of ductular reactions, in livers from patients with PBC (b), AIH (c), and CHC (d). In contrast, TLR3 was weakly expressed on small bile ducts in normal liver (a)
Induction of IFN-β by polyI:C stimulation in HIBECs
We examined the induction of IFN-β by HIBECs cultured for 24 h in basal medium containing 0–160 μg/ml of polyI:C, a ligand for TLR3. IFN-β secretion was consistently undetectable in culture supernatants, as determined by ELISA (data not shown). IFN-β mRNA copy numbers were calculated as 0.00024 ± 0.00017/GAPDH (mean value ± SD, n = 3) in the basal state, which increased by 2.7- to 5.5-fold (0.0012 ± 0.0010/GAPDH, n = 3) after 4–8 h of polyI:C stimulation (Fig. 4a). The increase in IFN-β mRNAs, however, was not statistically significant (P = 0.195). In contrast, IFN-β mRNA expression was markedly increased by polyI:C transfection using Lipofectamine 2000 (Fig. 4b). Maximal induction reached approximately 150-fold 4–8 h after transfection. Approximately 250 pg/ml IFN-β was secreted into culture medium over a 24-h period after polyI:C transfection. These results suggest that TLR3, even when expressed on the cell surface of HIBECs, does not recognize extracellular polyI:C; instead, TLR3 localized to endosomes and/or RIG-I/MDA5 in the cytoplasm sense polyI:C in HIBECs.

Induction of IFN-β mRNA by polyI:C in HIBECs. BEC3 cells were either cultured in basal medium containing polyI:C (a) or transfected with polyI:C using Lipofectamine 2000 (b). mRNA encoding IFN-β was strongly induced by polyI:C-transfection, while IFN-β mRNA induction was minimal following the addition of polyI:C to culture medium
We also examined the induction of other chemokine/cytokine mRNAs. We observed the potent induction of mRNAs encoding for IL-6 (basal level 0.019–0.052/GAPDH) and TNF-α (basal level 0.00072–0.00081/GAPDH) to levels 50- and 120-fold greater than baseline, respectively (Fig. 5b, c). We detected 4- to 6-fold induction of mRNA for IFN-α (basal level 0.0059–0.0128/GAPDH), IL-8 (basal level 0.241–0.859/GAPDH), and TLR3 (basal level 0.0064–0.0081/GAPDH) (Fig. 5d–f). This upregulation in gene expression is also attributable to intracytoplasmic polyI:C recognition, since addition of polyI:C to culture medium did not induce any increase of mRNA levels for IL-6, TNF-α, IFN-α, IL-8, and TLR3 (data not shown).

Induction of chemokine/cytokine mRNAs in HIBECs following polyI:C-transfection. We observed strong induction of mRNAs encoding IFN-β (a), IL-6 (b), and TNF-α (c), but only weak induction of mRNAs for IFN-α (d), IL-8 (e), and TLR3 (f)
Induction of IFN-β mRNA by polyI:C transfection depend on MAVS pathway but not on TICAM-1 pathway in HIBECs
To further confirm the functional role of TLR3 in the induction of IFN-β mRNA in HIBECs, we performed knockdown experiments using siRNA specific for TICAM-1 or MAVS. We first evaluated the efficiency of knockdown. Knockdown significantly reduced the mRNA levels of MAVS and TICAM-1 in HIBECs to approximately 30% of baseline using the corresponding siRNA (Fig. 6a). We then examined the effect of MAVS or TICAM-1 knockdown on the induction of IFN-β mRNA. As more efficient targeting of nucleotides to the endosomal compartment was reported by using DOTAP in comparison to Lipofectamine 2000 [21], we utilized DOTAP for the induction of IFN-β mRNA in knockdown experiments.

Effect of MAVS or TICAM-1 knockdown on the induction of IFN-β mRNA following polyI:C transfection. mRNA levels of MAVS and TICAM-1 in HIBECs significantly decreased to 30% of baseline levels by knockdown using an appropriate siRNAs in HIBECs (a). The induction of IFN-β mRNA in HIBECs after polyI:C transfection was efficiently inhibited by MAVS but not by TICAM-1 knockdown (b)
Interferon-β induction following polyI:C stimulation using Lipofectamine 2000 was largely dependent on MAVS/IPS-1, but not on TICAM-1 (Fig. 6b left side). Unexpectedly, similar results were obtained following polyI:C stimulation using DOTAP (Fig. 6b right side). These results suggested that the RIG-I/MDA5 (sensors of dsRNA in the cytosol)-MAVS signaling pathway plays a major role in the induction of IFN-β mRNA in HIBECs. Abundant expression of TLR3 in endosomes does not appear to participate significantly in polyI:C-mediated IFN-β induction in HIBECs.
Discussion
In this study, we provide the first data demonstrating that TLR3 is expressed in vitro in the cultured HIBECs; in these cells, IFN-β mRNA is strongly induced by polyI:C transfection, but only weakly induced by extrinsic polyI:C. Antibody blocking of TLR3 on HIBECs did not result in abrogation of IFN-β promoter activity, suggesting that cell-surface TLR3 participates only minimally in IFN-β promoter activation on dsRNA recognition (data not shown). These results suggested that endosomal, not cell surface, TLR3 is actively involved in type I IFN production by HIBECs. The results obtained by siRNA knockdown of TICAM-1 or MAVS, however, indicated that cytoplasmic RNA sensors like RIG-I/MDA5, not endosomal TLR3, are the major receptors initiating type I IFN induction in HIBECs.
To limit the growth of commensal organisms on their surface and to defend underlying tissues from invading pathogens, epithelial cells have both innate immune antimicrobial functions and the ability to modulate the recruitment and activity of innate and adaptive immune system [1, 3]. Human fibroblasts, colon epithelial cells, lung epithelial cells, corneal epithelial cells and keratinocytes, as well as the respective cell lines, express TLR3 on their cell surfaces [4, 5, 7, 12, 14, 16]. Recent analyses of TLR3 subcellular localization, however, have suggested that TLR3 is localized to endoplasmic reticulum (ER) and early endosomes in most human epithelial cell types [5]. A similar localization of TLR3 was observed in HIBECs in the present study; the HIBECs express TLR3 on both the cell surface and within intracellular organelles.
Unexpectedly, surface TLR3 in HIBECs exerted only a weak ability to induce type I IFN in response to polyI:C. As polyI:C must be internalized and delivered to the ER or early endosomes, in which TLR3 is abundant, to activate TLR3, it was speculated that the capacity of HIBECs to internalize polyI:C is weak. Intracellular polyI:C that was internalized into cells by lipofection, however, did not play a major role in activating the type I IFN promoter via TLR3. These results indicate that even if the bile fluid contains dsRNA that may be derived from the gastrointestinal tract via the portal vein, hepatocytes or cholangiocytes infected with virus, or apoptotic cell debris, bile fluid only minimally stimulates TLR3 on the surface or in endosomes to induce type I IFN, although it is also possible that bile fluid may contain as yet unknown TLR3-ligand to induce type I IFN. Further studies of TLR3 in HIBECs will be needed to identify the functional specificities of the surface-expressed and endosome-expressed TLR3.
In this study, we also provide the first evidence that the expression of TLR3 by intrahepatic biliary epithelial cells is markedly increased at sites of ductular reaction in diseased livers, including those affected by PBC, AIH, and CHC. TLR3 protein expression increased in synovial tissues from patients with RA. In addition, cultured RA synovial fibroblasts were activated by the TLR3 ligand polyI:C and by RNA released from necrotic synovial fluid cells, suggesting that necrotic cells may act as an endogenous TLR3 ligand leading to the stimulation of proinflammatory gene expression and autoimmunity [22–24]. The overexpression of TLR3 in thyrocytes is associated with the development of Hashimoto’s autoimmune thyroiditis [25]. TLR3 activation can drastically enhance susceptibility to immune destruction of solid organs, as seen in autoimmune hepatitis [26]. Exposure of pancreatic β cells to the combination of dsRNA and IFN-α, -β, or -γ significantly increases apoptosis [27, 28]. TLR3 can directly trigger apoptosis in human umbilical vein endothelial cells and cancer cells [29, 30]. TLR3 plays a role in the development of hepatitis C-associated glomerulonephritis through the induction of mesangial cell apoptosis [31]. Thus, enhanced TLR3 expression in intrahepatic biliary epithelial cells may play a critical role in the induction and maintenance of inflammation, immune destruction, and/or biliary epithelial cell apoptosis in vivo in diseased liver such as PBC, whereas enhanced TLR3 expression in biliary epithelial cells in CHC may play a critical role for protecting them from hepatitis virus infection.
TLR3 in the nervous system induces the expression of a range of neuroprotective mediators and angiogenic factors, chemokines, and anti-inflammatory cytokines that regulate astrocyte cellular growth, differentiation, and migration [32]. Activation of TLR3 protects against DSS-induced acute colitis [33]. Thus, it is possible that high TLR3 expression in HIBECs at sites of ductular reaction may protect against cell death or stimulate tissue repair and regeneration by inducing the production of as yet unknown protective and/or growth factors. The strong expression of TLR3 at ductal plate in human fetal liver indicates the importance of TLR3 in the regeneration and/or development of biliary epithelial cells (data not shown). Therefore, it is also considered possible that as yet unknown TLR3-ligand is involved in the development of ductular reaction in diseased livers including PBC, AIH, and CHC.
Enhanced expression of various molecules, including MHC-class I and class II antigen, adhesion molecules (ICAM-1, VCAM-1, LFA-3, etc.), chemokines (MCP-1, SDF-1, Fractalkine, etc.), cytokines (IL-6, IL-8, TNF-α, etc.), costimulatory molecules (B7, PD-L1, PD-L2, etc.), and TLR4, have also been reported in biliary epithelial cells in livers affected by PBC [34–38]. In addition to these molecules involved in innate and acquired immune response, we here demonstrated for the first time that RIG-I/MDA5–MAVS signaling pathway is operative in the strong induction of IFN-β by dsRNA stimulation in HIBECs. TLR3 and RIG-I/MDA5 expression increase in the presence of IFN-α, IFN-β, IFN-γ, and TNF-α in vitro [39–43]. These results may indicate that intrahepatic biliary epithelial cells are involved as an immunoregulatory organ in various liver diseases, including PBC, AIH, and CHC. In addition, the portal inflammation is closely associated with ductular reaction in periportal areas. As hepatic stem cells are speculated to reside alongside biliary epithelial cells in canal of Hering [44, 45], the existence of multiple IFN-inducing pathways, including TLR3 and RIG-I/MDA5, may suggest the importance of this innate immune effector pathway in the protection of putative hepatic stem cells from viral infection.
In conclusion, we demonstrated for the first time the increased expression of TLR3 at sites of ductular reaction in diseased livers including PBC, AIH, and CHC. Since cytoplasmic RNA sensors like RIG-I/MDA5, not TLR3, seem to be the major receptors initiating strong type I IFN induction in biliary epithelial cells, we speculate that there is another important role in TLR3 that is highly expressed in biliary epithelial cells. Further study will be necessary to characterize its in vivo physiological role.
Abbreviations
- BEC:
-
Biliary epithelial cell
- CK:
-
Cytokeratin
- dsRNA:
-
Double stranded RNA
- ER:
-
Endoplasmic reticulum
- ELISA:
-
Enzyme-linked immunosorbent assay
- GAPDH:
-
Glyceraldehydes-3-phosphate dehydrogenase
- HIBEC:
-
Human intrahepatic biliary epithelial cell
- HRP:
-
Horseradish peroxidase
- IFN:
-
Interferon
- IL:
-
Interleukin
- IRF:
-
Interferon regulatory factor
- MAVS:
-
Mitochondrial anti-viral signaling protein
- MDA5:
-
Melanoma differentiation associated gene-5
- MyD88:
-
Myeloid differentiation factor 88
- PBC:
-
Primary biliary cirrhosis
- PBMC:
-
Peripheral blood mononuclear cells
- PolyI:C:
-
Polyinosinic–polycytidylic acid
- PRR:
-
Pattern-recognition receptor
- RIG-I:
-
Retinoic acid-inducible gene I
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- siRNA:
-
Small interfering RNA
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- TICAM-1:
-
Toll-IL-1R homology domain containing adaptor molecule 1
References
Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol 2004;4:499–511.
Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagoshi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 2004;5:730–7.
Lee MS, Kim Y-J. Pattern-recognition receptor signaling initiated from extracellular, membrane, and cytoplasmic space. Mol Cells 2007;23:1–10.
Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T. Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun 2002;293:1364–9.
Matsumoto M, Funami K, Tanabe M, Hiroyuki O, Shingai M, Seto Y, et al. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol 2003;171:3154–62.
Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of Toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 2000;68:7010–17.
Furrie E, Macfarlane S, Thomson G, Macfarlane GT. Toll-like receptors-2, -3 and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology 2005;115:565–74.
Schaefer TM, Desouza K, Fahey JV, Beagley KW, Wira CR. Toll-like receptor (TLR) expression and TLR-mediated cytokine/chemokine production by human uterine epithelial cells. Immunology 2004;112:428–36.
Jorgenson RL, Young SL, Lesmeister MJ, Lyddon TD, Misfeldt ML. Human endometrial epithelial cells cyclically express Toll-like receptor 3(TLR3) and exhibit TLR3-dependent responses to dsRNA. Human Immunol 2004;66:469–82.
Schaefer TM, Fahey JV, Wright JA, Wira CR. Innate immunity in the human female reproductive tract: antiviral response of uterine epithelial cells to the TLR3 agonist poly(I:C). J Immunol 2005;174:992–1002.
Guillot L, Goffic RL, Bloch S, Escriou N, Akira S, Chignard M, et al. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 2005;280:5571–80.
Ritter M, Mennerich D, Weith A, Seither P. Characterization of Toll-like receptors in primary lung epithelial cells: strong impact of the TLR3 ligand poly(I:C) on the regulation of Toll-like receptors, adaptor proteins and inflammatory response. J Inflamm 2005;2:16.
Kumar A, Zhang J, Yu FSX. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology 2005;117:11–21.
Ueta M, Hamuro J, Kiyono H, Kinoshita S. Triggering of TLR3 by polyI:C in human corneal epithelial cells to induce inflammatory cytokines. Biochem Biophys Res Commun 2005;331:285–94.
Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol 2002;61:1013–21.
Kollisch G, Kalali BN, Voelcker V, Wallich R, Behrendt H, Ring J, et al. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 2005;114:531–41.
Takii Y, Nakamura M, Ito M, Yokoyama T, Komori A, Shimizu-Yoshida Y, et al. Enhanced expression of type I interferon and Toll-like receptor-3 in primary biliary cirrhosis. Lab Invest 2005;85:908–20.
Yokoyama T, Komori A, Nakamura M, Takii Y, Kamihira T, Shimoda S, et al. Human intrahepatic biliary epithelial cells function in innate immunity by producing IL-6 and IL-8 via the TLR4-NF-γB and -MAPK signaling pathways. Liver Int 2006;26:467–76.
Sasai M, Shingai M, Funami K, Yoneyama M, Fujita T, Matsumoto M, et al. NAK-associated protein 1 participates in both the TLR3 and the cytoplasmic pathways in type I IFN induction. J Immunol 2006;177:8676–83.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) method. Methods 2001;25:402.
Dalpke A, Frank J, Peter M, Heeg K. Activation of Toll-like receptor 9 by DNA from different bacterial species. Infect Immunol 2006;74:940–6.
Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 2004;279:12542–50.
Roelofs MF, Joosten LAB, Abdollahi-Roodsaz S, van Lieshout AWT, Sprong T, van den Hoogen FH, et al. The expression of Toll-like receptor 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of Toll-like receptor 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arth Rheum 2005;52:2313–22.
Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arth Rheum 2005;52:2656–65.
Harii N, Lewis CJ, Vasko V, McCall K, Benavides-Peralta U, Sun X, et al. Thyrocytes express a functional Toll-like receptor 3: overexpression can be induced by viral infection and reversed by phenylmethimazole and is associated with Hashimoto’s autoimmune thyroiditis. Mol Endocrinol 2005;19:1231–50.
Lang KS, Georgiev P, Recher M, Navarini AA, Bergthaler A, Heikenwalder M, et al. Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J Clin Invest 2006;116:2456–63.
Wen L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J Immunol 2004;172:3173–80.
Rasschaert J, Ladriere L, Urbain M, Dogusan Z, Katabua B, Sato S, et al. Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-γ-induced apoptosis in primary pancreatic β-cells. J Biol Chem 2005;280:33984–91.
Kaiser WJ, Kaufman JL, Offermann MK. IFN-γ sensitizes human umbilical vein endothelial cells to apoptosis induced by double-stranded RNA. J Immunol 2004;172:1699–710.
Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol 2006;176:4894–901.
Wornle M, Schmid H, Banas B, Merkle M, Henger A, Roeder M, et al. Novel role of Toll-like receptor 3 in hepatitis C-associated glomerulonephritis. Am J Pathol 2006;168:370–86.
Bsibsi M, Persoon-Deen C, Verwer RWH, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 2006;53:688–95.
Vijay-Kumar M, Wu H, Aitken J, Kolachala VL, Neish AS, Sitaraman SV, et al. Activation of Toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm Bowel Dis 2007;13:856–64.
Harada K, Van de Water J, Leung PS, Coppel RL, Ansari A, Nakanuma Y, et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the TH1 subset. Hepatology 1997;25:791–6.
Harada K, Nakanuma Y. Molecular mechanisms of cholangiopathy in primary biliary cirrhosis. Med Mol Morphol 2006;39:55–61.
Gershwin ME, Nishio A, Ishibashi H, Lindor K. Primary biliary cirrhosis. In: Gershwin ME, Vierling JM, Manns MP, editors. Liver immunology, Chapter 20. Philadelphia, PA: Hanley & Belfus, Inc.; 2003. p. 311–27.
Kamihira T, Shimoda S, Nakamura M, Yokoyama T, Tkii Y, Kawano A, et al. Biliary epithelial cells regulate autoreactive T cells: implications for biliary-specific diseases. Hepatology 2005;41:151–9.
Wang AP, Migita K, Ito M, Takii Y, Daikoku M, Yokoyama T, et al. Hepatic expression of Toll-like receptor 4 in primary biliary cirrhosis. J Autoimmun 2005;25:85–91.
Heinz S, Haehnel V, Karaghiosoff M, Schwarzfischer L, Muller M, Krause SW, et al. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J Biol Chem 2003;278:21502–9.
Tanabe M, Kurita-Taniguchi M, Takeuchi K, Takeda M, Ayata M, Ogura H, et al. Mechanism of up-regulation of human Toll-like receptor 3 secondary to infection of measles virus-attenuated strains. Biochem Biophys Res Commun 2003;311:39–48.
Tissari J, Siren J, Meri S, Julkunen I, Matikainen S. IFN-γ enhances TLR3-mediated antiviral cytokine expression in human endothelial and epithelial cells by up-regulating TLR3 expression. J Immunol 2005;174:4289–94.
Siren J, Imaizumi T, Sarkar D, Pietila T, Noah DL, Lin R, et al. Retinoic acid inducible gene-I and mda-5 are involved in influenza A virus-induced expression of anti-viral cytokines. Microbes Infect 2006;8:2013–20.
Liu P, Jamaluddin M, Li K, Garofalo RP, Casola A, Brasier AR. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol 2007;81:1401–11.
Theise ND, Saxena R, Portmann BC, Thung SN, Yee Y, Chiriboga L, et al. The canals of Hering and hepatic stem cells in humans. Hepatology 1999;30:1425–33.
Zhou H, Rogler LE, Teperman L, Morgan G, Rogler C. Identification of hepatocytic and bile ductular cell lineages and candidate stem cells in bipolar ductular reactions in cirrhotic human liver. Hepatology 2007;45:716–24.
Acknowledgments
This study was supported by Grants-in-Aid for Scientific Research from the Ministry of Health, Labour and Welfare of Japan and Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nakamura, M., Funami, K., Komori, A. et al. Increased expression of Toll-like receptor 3 in intrahepatic biliary epithelial cells at sites of ductular reaction in diseased livers. Hepatol Int 2, 222–230 (2008). https://doi.org/10.1007/s12072-008-9055-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-008-9055-4
Keywords
- Primary biliary cirrhosis (PBC)
- Human intrahepatic biliary epithelial cells (HIBECs)
- Interferon beta (IFN-β)
- Toll-like receptor 3 (TLR3) Toll-IL-1R homology domain-containing adaptor molecule 1 (TICAM-1)
- Mitochondrial antiviral signaling protein (MAVS)
- Retinoic acid inducible gene I (RIG-I)
- Melanoma differentiation-associated gene 5 (MDA5)