
Abstract
It has become clear that dispersed repeat sequences have played multiple roles in eukaryotic genome evolution including increasing genetic diversity through mutation, inducing changes in gene expression, and facilitating generation of novel genes. Growing recognition of the importance of dispersed repeats has fueled development of computational tools designed to expedite discovery and classification of repeats. Here we review major existing repeat exploration tools and discuss the algorithms utilized by these tools. Special attention is devoted to ab initio programs, i.e., those tools that do not rely upon previously identified repeats to find new repeat elements. We conclude by discussing the strengths and weaknesses of current tools and highlighting additional approaches that may advance repeat discovery/characterization.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The vast majority of DNA research has focused on genes, those sequences that code for proteins or structural RNAs. However, eukaryotic genomes are characterized and often dominated by repetitive, non-genic DNA sequences [14], and indeed the vast majority of the 10,000-fold variation in eukaryotic genome sizes is due to differences in repeat sequence content [63, 82, 48]. The prevalence and evolutionary persistence of repeats in eukaryotes indicates that while repeats may have originated as “selfish DNA,” many now afford selective advantages to the genomes in which they reside. However, the mechanisms by which repeats contribute to fitness are complex and poorly understood [15, 12]. Experimental evidence has shown that repetitive regions influence expression of nearby genes [62], and in some instances it appears that insertion of even relatively short tandem repeats into an uncondensed region of chromatin can result in condensation and gene repression [25, 93, 6]. Additionally, it has long been known that mobile repetitive elements can cause insertions, deletions, and/or rearrangements that can alter gene structure and regulation, and it is possible that the tremendous increase in mobile element activity in some populations under extreme environmental stress may be a means of rapidly increasing genetic diversity through mutation [60, 10, 38]. Recent molecular evidence also suggests that some repeat elements may be instrumental in generation of new genes [37, 47, 61]. Regardless, a comprehensive understanding of gene and genome function in eukaryotes will require knowledge of repeat sequences because eukaryotic genes evolve and function within the context of a chromosomal milieu composed primarily of repetitive DNA.
The term “tandem repeat” refers to any sequence that is normally found in consecutive or nearly consecutive copies along a DNA strand. Examples of tandem repeats include the satellite, minisatellite, and microsatellite DNAs found in most eukaryotic species [17]. Microsatellites, also known as simple sequence repeats (SSRs), consist of 1–6 bases found in relatively short tandem arrays [54]. SSRs have become useful genetic markers because they are often associated with genes and because polymorphism in repeat number is common [77]. Identification of short tandem repeats such as SSRs is relatively easy and, not surprisingly, algorithms for finding SSRs are abundant [11, 81, 44]. Recognition of longer tandem repeat sequences is also fairly straightforward, although correct assembly of such arrays is difficult when there is high sequence conservation between repeat copies.
Dispersed repetitive elements are distributed throughout genomes in a non-tandem, albeit non-random, manner. The majority of dispersed repeats are transposons, sequences that can directly or indirectly move from one site to another. Those transposable elements that possess a complete set of transposition protein domains are called autonomous. However, the term autonomous does not imply that an element is active or functional. Transposons that clearly lack an intact set of mobility-associated genes are called non-autonomous. Transposition of a non-autonomous element requires participation of one or more proteins encoded by an autonomous element. Some non-autonomous transposons appear to be deletion/insertion derivatives of autonomous transposons while other non-autonomous transposons have not (yet) been linked to autonomous ancestors [91].
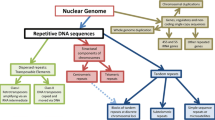
Classification of transposons has proven difficult as new types of mobile repeats are being discovered at a rapid rate, and evolutionary relationships between many dispersed repeat groups are unclear—for a recent treatment of transposon classification see Wicker et al. [91]. However, transposons can typically be divided into two classes; retrotransposons and DNA transposons. Retrotransposons are replicated and mobilized through an RNA intermediate via a copy-and-paste mechanism involving the enzyme reverse transcriptase. In contrast, DNA transposons utilize cut-and-paste or copy-and-paste methods of transposition that do not involve an RNA intermediate. An overview of common transposon groups is presented in Fig. 1.

Transposons are traditionally classified into retrotransposons and DNA transposons [91]. Retrotransposons (A–F) are replicated and mobilized through an RNA intermediate via a copy-and-paste mechanism involving the enzyme reverse transcriptase (rt). They possess 5′ and 3′ untranslated regions (UTRs) containing minus-strand (PBS) and plus-strand (PPT) priming sites, respectively. They typically can be divided into long terminal repeat (LTR) retrotransposons (A–D) [34, 41] and non-LTR retrotransposons (E–F) [64]. (A) Gypsy elements contain an ORF with gag and pol genes. The gag gene codes for viral capsid proteins while the pol gene codes for proteinase (pr), integrase (int), reverse transcriptase (rt), and RNase H (rh) activities. (B) Copia elements are similar in overall structure to gypsy elements. However, the two groups possess distinctly different reverse transcriptase amino acid sequences. In most instances, they also exhibit variation in the relative position of int. (C) In LARDs (large retrotransposon derivatives), protein-coding regions have been replaced by a relatively long, conserved, noncoding region. (D) TRIMs, terminal repeat retrotransposons in miniature, contain short LTRs, PBS and PPT sites, and little else. (E) LINEs, long interspersed nuclear elements, are non-LTR retrotransposons that possess 1 or 2 ORFs. One ORF encodes a pol protein with rt and endonuclease (en) activities. If there is a second ORF, it encodes a nucleic acid binding protein (nabp) with chaperone and esterase activities. The 3′ UTR sometimes contains the canonical polyadenylation sequence (ATAAA) and a tract of poly-A. LINEs are transcribed by RNA polymerase II. (F) SINEs, short interspersed nuclear elements, possess a region with similarity to a tRNA (TR) or other small RNA, a tRNA-unrelated region (TU), and a region that, in some instances, appears to be LINE-derived (LD). SINEs are transcribed by RNA polymerase III. DNA transposons (G–J) can be mobilized through either a cut-and-paste mechanism (G–H) or through other mechanisms that do not involve RNA intermediates (I–J). They multiply via their host’s replication machinery. (G) TIR DNA transposons (cut-and-paste) are characterized by terminal inverted repeats (TIRs) and one ORF that encodes a transposase gene. (H) MITEs, miniature inverted-repeat transposable elements, are extremely short, TIR-flanked, cut-and-paste transposons with no coding capacity. (I) Helitrons are DNA sequences that are propagated through a rolling-circle replication mechanism [42, 30]. Autonomous Helitrons possess a helicase gene that encodes an enzyme with 5′-3′ helicase and nuclease/ligase activities. Helitrons may also contain genes for RPA-like (rpal) single-stranded DNA binding proteins. Helitrons do not create target site duplications and lack TIRs. Both autonomous Helitrons and most non-autonomous Helitron-like transposons have conserved 5′-TC and 3′-CTRR sequences at their termini. (J) Mavericks/Polintons are large elements that encode integrase (int), DNA polymerase B (dpolB), and up to 8 other proteins [43, 68]. It has been argued that Mavericks/Polintons contain all of the genes necessary for both self-transposition and self-replication [43]
In computational terms, automated dispersed repeat identification is complicated by the fact that dispersed repeats may exhibit considerable inter-copy divergence. Moreover, the replication, insertion, and excision of multiple mobile repeat elements over the course of thousands or millions of years can result in complex mosaics making identification of repeat boundaries and definition of repeat families difficult [66]. However, there is an increasing number of algorithms that have been developed for studying dispersed repeats. Some of these tools, e.g., Smit et al. [74], can also be used in detection of tandem repeats.
Here we review the major algorithmic approaches currently employed in dispersed repeat identification/classification and the tools that utilize these algorithms.Footnote 1 While we provide overviews of library- and signature-based methods, the bulk of the present review is focused on algorithms/tools that identify repeats without utilizing previously characterized repeat sequences or repeat-specific motifs. Such ab initio tools are becoming more and more important due to tremendous increases in the amount and diversity of sequences being generated in genome projects. For each ab initio tool we describe the sequence substrate utilized (shotgun reads, or assembled genomic regions), the approach used for initial identification of repeats, and the method used to extract descriptions of repeat families. We conclude with a discussion of the utility of the various tools and present some ideas regarding potential means through which automated repeat identification could be improved.
In the discussion below, repeat identification tools are introduced based upon the algorithms they use to identify and classify potential repeats. However, to permit “at a glance” comparison of tools, we provide Table 1 which lists the basic features of each of the tools, and information on how each tool can be obtained.
Library- and Signature-based Identification Techniques
Library-based Techniques
Library-based systems identify repetitive sequences by comparing input datasets against a set of reference repeat sequences, i.e., a library [39]. RepeatMasker [74] is the predominant library-based tool used in repeat identification and it has become the de facto standard for repeat identification among all methods. As the name implies, RepeatMasker was designed to discover repeats and mask (i.e., remove) them so as to prevent complications in sequence assembly and gene characterization. The tool includes a set of statistically optimal scoring matrices calculated for a range of background GC levels permitting estimation of the divergence of query sequences compared to a curated repeat library [39]. A search engine such as BLAST, WU-BLAST, or Crossmatch is utilized in the comparison process.Footnote 2 The use of WU-BLAST for matching is faster than using Crossmatch with only a slight loss in detection ability [18]. The degree of similarity required for a query sequence to be paired with a reference sequence(s) can be specified by the user. Because identification of repeats by RepeatMasker is based entirely upon shared similarity between library repeat sequences and query sequences, any region of query sequence with significant similarity to a reference sequence in the repeat library is marked as a repeat whether or not it is found multiple times in the query sequence dataset. Both the sequence information for repeat regions and the annotation reports produced by RepeatMasker are presented in a simple, user-friendly format. RepeatMasker is often used in conjunction with Repbase [39], a large curated repeat library containing data from numerous eukaryotes, but it can be used with clade-specific repeat databases [65, 90, 70] as well. RepeatMasker is the only repeat finder among those reviewed that can fully utilize multi-processor systems as it is delivered. This feature along with the simple database search approach makes it one of the fastest (when used with WU-BLAST) and most effective repeat finders available. Installation is straightforward and there are very few parameters to adjust. Despite its wide use and many advantages, RepeatMasker cannot be used to find repetitive sequences that do not share significant nucleotide similarity with previously defined repeat sequences. However, the use of RepeatMasker with Repbase or some other curated repeat database is often considered an essential first step in repeat analysis of a genome.
Several library-based repeat detection tools use visualization techniques to display data in formats that can facilitate interpretation. PLOTREP [83], for example, clusters and visually displays the variants for a reference sequence or repeat element. Censor [40] uses BLAST to identify matches between input sequences and a reference library of known repetitive sequences. The length and number of gaps in both the query and library sequences are considered along with the length of the alignment in generating similarity scores. Regions of query sequences with similarity scores exceeding a user-defined minimum threshold are recorded. This tool reports the positions of the matching regions of the query sequence along with their classification. It also produces a “masked file” similar to RepeatMasker containing the original sequence with all detected repeats visually demarcated.
Signature-based Techniques
Signature-based repeat identification tools search a query sequence(s) for nucleotide or amino acid motifs and spatial arrangements characteristic of a particular repeat group. Unlike library-based tools, all signature-based tools employ heuristics based on a priori information of particular repeat types. However, some signature-based tools also may use reference sequence libraries at some stage in the analysis process.
Signature-based tools used to identify long terminal repeat (LTR) retrotransposons (Fig. 1) include LTR_STRUC and RetroTector©. LTR_STRUC [59] searches a query sequence for pairs of similar LTRs separated from each other by a physical distance expected for this retrotransposon group. The authors report that LTR_STRUC can successfully locate retrotransposons when LTR regions have > 75% sequence identity. RetroTector© [76] uses a variety of techniques to identify potential LTR retroelements. The program identifies putative LTR pairs based on comparison with conserved LTR sequence motifs and analysis of spatial relationships among closely associated LTRs. A variety of procedures are then used to search surrounding areas for conserved retrotransposon protein motifs and other LTR retroelement features.
Miniature inverted-repeat transposable elements (MITEs) have also been identified using signature-based tools. FINDMITE [84] uses a string matching technique adapted from the Knuth–Morris–Pratt algorithm [20] to search for potential pairs of terminal inverted repeat (TIR)/target site duplication sequences separated from each other by a distance characteristic of MITEs (Fig. 1). MAK [92], another MITE identification tool, assumes that all members of a MITE family are homologous. When provided an input sequence of a MITE element, MAK uses BLAST, implemented as part of sub-pipelines, to (a) identify and retrieve other members of the family that share similarity along their complete length or at both the terminal regions, (b) generate a consensus sequence that serves as the anchor element for the family [40], and (c) locate the genes, if any, that exist in close proximity to the MITE family elements. This tool is particularly useful in comparative genomics studies as a characterized MITE element from one species can be used to “seed” searches for similar elements in related species.
Inverted repeats are features of several DNA transposon groups including TIR transposons, MITEs, and Mavericks/Polintons (Fig. 1). Inverted Repeats Finder (IRF), a tool developed by Warburton et al [87] searches for pairs of non-overlapping short, identical sequences in reverse complement orientations. For each identified pair, the halfway point between the two members (i.e., a center position) is recorded. After this process is complete for the query sequence, IRF searches for “clusters” of pairs that roughly share the same center position, and aligns and extends these to produce candidate inverted repeats. A “narrowband” technique [11] is used to eliminate those candidates with alignment scores beneath a user-defined threshold. According to the authors, searching for short reverse complement sequence pairs facilitates identification of short inverted repeats separated by relatively small “spacers,” while searching for longer reverse complement pairs aids in identification of long inverted repeats with larger spacers.
Andrieu et al. [5] base their signature-based tool for identifying transposable elements, TE-HMM, on the observation that transposons have compositional (nucleotide) biases compared to genes. For a species, TE-HMM builds three different hidden Markov models using training sets of retrotransposons, DNA transposons, and gene sequences. TE-HMM can then be used to place a particular query sequence or section of a query sequence into one of these three categories based upon the compositional model it most closely resembles. Using TE-HMM on several test species, the authors report high specificity values (with somewhat lower sensitivity values) in identification of retrotransposons and DNA transposons. Because compositional bias varies among species, it is usually necessary to build new hidden Markov models for new species.
Ab initio Repeat Identification
Ab initio algorithms identify repetitive elements without using reference sequences or known repeat motifs in the repeat identification process. To facilitate comparison of ab initio tools, we use the following definitions:
-
Assembled genomic region: a relatively long (Mb to Gb) region of continuous DNA sequence, e.g., a whole chromosome or an assembled chromosomal region.
-
Family: a group of repetitive sequences inferred to have a common ancestor based upon sequence similarity. Note that in the context of this paper, family is not meant to imply a taxonomic level in a formal hierarchical classification scheme.
-
Element: a broad term for an individual member of a repeat family. If an assembled genomic region is used as the substrate for repeat identification, then each identified element will be traceable back to a specific location on the query sequence. If sequence reads are used as the starting substrate, then it is likely that the exact physical locale of an identified element will not be known without additional research/information.
-
Consensus sequence: a “pseudomolecule” composite of all the members in a repeat family in which each place in the nucleotide chain is occupied by the base most commonly found at that position.
We have divided the process of detecting repeats into two stages. The first stage deals with initial identification of repetitive sequences. The second stage, repeat family definition, is focused on identifying the boundaries of the repeats and extraction of the consensus sequence for each family. Below we discuss major ab initio repeat finding algorithms/tools within the framework of these two stages. Note that some tools perform repeat identification (stage 1) without generating a family definition (stage 2).
Stage 1: Initial Identification of Repetitive Sequences
All ab initio discovery of repeat families begins with identification of relatively short sequences that are found multiple times in a sequence or sequence set. Four basic (but not entirely exclusive) groups of approaches have been utilized in initial identification and clustering of repeats.
-
Self-comparison: compares the uncharacterized DNA sequence with itself to identify clusters of similar sequences.
-
k-mer: involves explicit enumeration of all frequently occurring exact substrings (called k-mers or “words”) in the query sequence(s). Two substrings of length k are not matched unless their sequences are identical.
-
Spaced seed: similar to k-mer approaches except that the “seeds” used in the matching process possess a predefined level of tolerance for mismatch or indels.
-
Dot matrix: plots the input sequence against itself.
-
Periodicity: transforms sequence data from the sequence (time) domain into the frequency domain and performs analysis on the frequency data.
Self-Comparison Approaches
One of the first attempts to build a system for automated detection of repetitive elements was the Repeat Pattern Toolkit [2]. It uses the nucleotide–nucleotide BLAST module, i.e., BLASTN (http://www.ncbi.nlm.nih.gov/blast/), with the overlap option applied to an assembled genomic region. Local alignments are calculated separately for word sizes of 8 and 12 bp, and the results are merged. The product is a set of words and their pairwise similarity scores. A graph-based single link clustering algorithm is then used to group sequences. Single link clustering regards two sequences as belonging to the same cluster if they share a similar nucleotide stretch(es) longer than a certain proportion of one of the two elements. Each sequence is considered to be a vertex in a graph, and two vertices are linked if they overlap beyond some threshold. Connected components form groups of related repeat elements [7].
RECON [7], currently one of the most widely used ab initio repeat identification tools, is also based on BLAST searches. RECON begins with an all-to-all BLAST analysis of multiple sequence reads using WU-BLASTN. This is followed by application of single link clustering to alignment results. An undirected graph G is constructed with each image (i.e., overlapping regions of assembled reads) as a vertex, and two images are connected by an edge if they overlap beyond a threshold. The shortest sequence that contains all images in a connected component in this graph is deemed an element. However, since this procedure can result in elements that are composite, the element and all images used to construct it are aligned together. The element is split up at every point with a significant aggregation of image ends. Note that this process will collapse all identical elements for a repeat family located at different sites in the genome into a single element.
PILER [29] uses a local alignment procedure called pairwise alignment of long sequences (PALS) that is tailored for repeat identification in assembled genomic regions. To improve efficiency, only location coordinates (end points) of hits are recorded. PALS uses banded searching (local alignment of sequences that are located within a certain range of each other) to optimize identification of repeat families with profiles characteristic of known repeat types.
k-mer Approaches
k-mer or “word counting” approaches view a repeat as a substring w of length k that occurs more than once in a sequence S of length n. A repetitive subsequence w that cannot be extended without introducing mismatches is called a maximal repeat. Since there are 4 k possible words of length k, these approaches usually require that k be at least log 4(n) where n is the length of the genome or sequence set being studied. For example, the k-mer-based tools ReAS, RepeatScout, and RAP all recommend a value of k that is greater than log 4(n). The value of k required for indexing assembled plant genomes is roughly 12 to 19 based on plant genome size estimates [9]. Because direct indexing of all sequences of this length is impractical, a key issue that must be addressed by algorithms that use the k-mer approach for repeat finding is compact and efficient representation of substrings. Increasing the value of k decreases the sensitivity of the repeat searching procedure while decreasing the seed size increases the computational complexity of the search and the probability of matches occurring at random.
REPuter [45] was one of the first tools to implement a k-mer search algorithm for repeat finding. Its search engine component, REPfind, uses the efficient suffix tree data structure developed by Weiner [88] for storing all repeated exact k-mers in a sequence that have lengths greater than or equal to a user-specified size. Suffix trees can be used to search for strings in linear space and time with a complexity of O(n + z) where z is the number of maximal repeats. This representation allows the algorithm to scale when handling large sequences from eukaryotic genomes. The REPuter k-mer approach has also been effectively used by other tools. For example, RepeatFinder [85] and RepeatGluer [66] both use the REPuter engine to generate an initial list of maximal repeats. Alternatively, RepeatFinder can also use the output from another suffix-tree-based tool, RepeatMatch, which is based on MUMmer [23].
While REPuter builds initial clusters by finding all repetitive sequences longer than a threshold value, other k-mer tools group sequences based upon shared high frequency k-mers of a pre-defined length. Aligned sequences identified by a specific k-mer are then extended by a variety of mechanisms. Using a fixed-length k-mer reduces the time and space complexity of the search process.
The authors of ReAS [52], an approach based on fixed-length k-mers, have adapted major components of their RePS [86] sequence assembly tool for identification of transposable elements in shotgun sequence reads. ReAS utilizes sequence reads as a substrate to avoid errors introduced by incorrect sequence assemblies. The ReAS algorithm employs a randomly selected, high frequency k-mer as “bait” to retrieve sequence reads containing that k-mer. The value of k is determined based on genome size (n) using the formula k ≥ log 4(n) as described above. The “captured” sequence reads for a given k-mer are processed by ClustalW [18] to generate an initial 100 bp consensus sequence centered on the k-mer. If another high copy k-mer exists near either end of the initial consensus sequence, it is used to capture additional sequences from the input dataset. The newly retrieved sequences are then utilized to extend the initial consensus sequence if there is 95% identity in the region of overlap. Consensus sequence extension is repeated up to five times.
RepeatScout also builds a library of high frequency fixed length k-mers and uses these as seeds for a greedy search during the family definition stage [67]. RepeatScout implements a modified version of the classical local alignment algorithm by incorporating a penalty-based scoring system in screening the k-mers.
Spaced Seed Approaches
An extension of k-mer approaches is the concept of spaced seeds. Instead of searching for perfectly identical matches of length k, spaced seed algorithms conduct searches using “seeds” containing a defined level of tolerance for variation in sequence identity and/or length. The first spaced seed tool, PatternHunter [56], allowed mismatches in fixed positions while requiring perfect matching in others. This increased the sensitivity (and somewhat surprisingly) the speed of searches compared to standard k-mer approaches. Multiple spaced seed techniques [56, 51, 36] take this idea even further by using several optimal spaced seed patterns in searches. The multiple/optimal spaced seed concept was utilized in development of PatternHunter II [51] and also has been incorporated into some recent versions of BLAST and other alignment programs [36]. “Indel seeds” proposed by Mak et al. [57] use a spaced seed strategy except that the so-called “don′t care” positions in the spaced seeds not only tolerate single base mismatches but indels (i.e., short insertions and deletions) as well. The authors use a modified version of Inverted Repeat Finder [87] with indel seeds to increase sensitivity compared to standard spaced seeds. While indel seeds are arguably not essential when searching for conserved regions in genes, they are likely to be of considerable utility when evaluating repeats and non-coding gene regions where indels are more likely to be present.
Another spaced seed-like approach, RAP [16], implements a complex indexing strategy that allows space efficient counting of words of a specific size in which a predefined amount of degeneracy is permitted. Word counters are created for each position in the sequence, and all potential words of size k beginning from each sequence position are enumerated using a multi-array data structure.
Dot Matrix Approaches
One of the earliest and simplest repeat finding techniques was the dot-plot [75] in which a sequence is plotted against itself. Auto Dot PLOT or Adplot [80] is an adaptation of the dot-plot principle applied to a single assembled genomic region. Similar k-mer elements located within a user-specified range are detected in the first step. This information is recorded along with the inter-element distance. A sliding window based filtering method is applied to repeat families whose sum of element lengths is below a threshold. The focus of this tool is visualization of the distribution of repetitive regions over the sequence. The authors claim that Adplot is more effective than dot-plot for analyzing repeats families dispersed over the genome.
Periodicity Approaches
Periodicity-based approaches are fundamentally different than the aforementioned techniques. The Spectral Repeat Finder of Sharma et al. [72] uses Fourier transforms to analyze DNA sequence in the frequency domain rather than the commonly used time domain (where an alphabetic sequence is viewed as a time series). The power spectrum of the sequence generated from the Fourier transforms is used to identify both short term and long term autocorrelations of the sequence with itself. High intensity peaks in the power spectrum of the sequence represent candidate repetitive regions or elements. These candidate repeats are used to seed a local alignment search to detect similar elements and to determine the consensus sequence for the family. Since the signal strength deteriorates with the dispersion of repeats, this method is most effective for exact tandem repeats, although the authors indicate that it can also be used to locate some dispersed repeats. The time complexity of the algorithm is O(n 2).
Stage 2: Defining Repeat Families
The methods described in the preceding section are used to generate sets of similar elements whereas the following section discusses techniques used to extend and combine elements into families, where possible, and to extract descriptions of the consensus (or prototype) sequence for each repeat family.
Clustering
Some tools implement repeat family identification by further clustering to derive the final family definition. This process may be guided by biological heuristics.
RepeatFinder [85] begins with the initial set of exact repeats identified using one of two suffix tree approaches and then merges different exact repeats that are close together (merging using gaps) or that overlap (merging using overlap) to generate a set of “merged repeats.” The merged repeats are then grouped into categories; two merged repeats are placed in the same category if they contain at least one exact repeat. A final round of clustering is performed in which BLAST is used to compare each category against all other categories. After a final round of merging, an element is selected from each resulting category as the representative sequence (prototype) for the repeat family; an objective function is defined for each clustering protocol (merging using overlaps and merging using gaps) and a category prototype is derived that minimizes the appropriate objective function. Clustering operations are performed using only element location coordinates affording a more compact data structure and therefore improved time and memory efficiency. The running time of RepeatFinder is dominated by the all versus all comparison in the first step and the memory requirement is dominated by the REPuter algorithm [85]. The memory requirements for the underlying suffix tree data structure can grow up to many gigabytes for moderate to large eukaryotic genomes [46].
PILER [29] adopts a novel heuristic-based approach for repeat identification and characterization in assembled genomic regions. The PILER algorithm is designed to analyze an assembled genomic region(s) and find only repeat families whose structure is characteristic of known subclasses of repetitive sequences. PILER works on the premise that the entire DNA sequence is assembled with a reasonably low number of errors because the algorithm is completely dependent on the position of repeats in the genome for all classification. The output of the clustering step is recorded in terms of start and end coordinates. Similar elements are then clustered into “piles.” Piles are actually sets of overlapping hits similar to the categories in RepeatFinder. The time required to create the piles increases linearly with the length of the sequence. The characteristics of elements clustered in a pile are matched against four author-defined profiles (tandem arrays, dispersed families, pseudosatellites, and terminal repeats). The MUSCLE [27] alignment program is used to generate the consensus sequence for each family detected in the classification step. These consensus sequences can be used to create RepeatMasker or BLAST libraries to search for complete or partial members in the genome [29].
Graph Representation with Heuristics
Repeat Pattern Toolkit [2] builds a repeat graph G = (V,E) using only ungapped local alignments from the clustering step. Vertices V represent the repetitive sequences or elements. Weighted edges E represent the relationship among similar elements. Connected components from the graph are converted into minimum spanning trees using Kruskal′s algorithm and Binsort [20] in \({\text{O}}\left( {\left| {\text{E}} \right|{\text{ }} + {\text{ }}\left| {\text{V}} \right|{\text{log}}\left| {\text{V}} \right|} \right)\) time. Each minimum spanning tree represents a repeat family. Each tree is reduced to a single vertex to deduce the consensus sequence for the family. This vertex is the weighted midpoint of all the other vertices in the graph. A limitation of this technique is its inability to address repeat families that have elements with indels since only ungapped alignments are analyzed.
Bao and Eddy [7] extended and improved upon the work of Agarwal and States [2] with RECON. The algorithm refines the elements derived from the results of local alignment of multiple sequence reads. The final set of elements is represented as a repeat graph H where each element is a vertex and two types of edges represent relationships among elements. Elements with an overlap ratio above a specified threshold are connected by edges designated as primary while those with significant alignment but with overlap below the ratio threshold are connected by edges designated as secondary. Primary edges are interpreted as denoting elements that belong to the same family and secondary edges as denoting elements from similar but distinct families.
RepeatGluer takes a novel approach for extracting the descriptions of repeat families [66]. The focus is on deciphering the mosaic of sub-repeats nested within repetitive regions in the genome using A-Bruijn graphs, an extension of the de Bruijn graphs [22]. The original de Bruijn graph represents repeat families as a mosaic of perfect repeats. The concept has been generalized by Pevzner et al. [66] to A-Bruijn graphs to enable approximate matches or imperfect repeats to be represented within this framework. The algorithm constructs an adjacency matrix from the supplied assembled genomic region(s). This matrix is used to construct a weighted A-Bruijn graph G where the weight of the edge between two vertices is the number of edges joining them. The graph G can model all relationships accurately but can become extremely complex to interpret. A number of biologically derived heuristics are used to simplify the graph. Finally, each set of connected components or “tangle” is resolved to a consensus sequence. The consensus sequence for a repeat family is constructed using consensus sequences from all similar elements for each sub-repeat within the repeat family.
String Extension
Algorithms covered in this review that cluster high frequency k-mers as a first step often employ string extension techniques for the second step of family definition. REPuter was one of the first repeat finders to use the string extension method [45]. The authors employ a strictly non-heuristic and purely mathematical approach for detecting repeats. The output of REPfind, REPuter′s search module, is processed further for finding degenerate repeats using either a Hamming distance model or an edit distance model [32]. The edit (or Levenshtein) distance approach has an overall time efficiency of O(n + zk 3) where n is the size of the sequence and z is the number of k-mers extended. REPuter extends the maximal repeats in both directions with the number of mismatches as a threshold. Each isolated consensus sequence is assigned an E value [4] based on the expected number of consensus sequences with similar lengths and number of errors based on the assumption that random DNA conforms to a uniform Bernoulli model. REPuter also includes a visualization module called Repvis to enable manual inspection of the detected elements in a genome context. Of note, the REPuter package has been subsumed by Vmatch [1]. Vmatch uses suffix arrays [58] that have a reduced space requirement instead of a suffix tree for indexing substrings.
RepeatScout [67] generates consensus sequences by first detecting a set of highly repetitive fixed length k-mers in an assembled genomic region as described above. The algorithm extracts a copy of each k-mer and its surrounding region and then greedily extends the boundaries on both ends yielding a consensus sequence for the repeat family representing the k-mer. RepeatScout processes each repeat element using RepeatMasker to find all similar elements for the repeat family and adjusts frequencies of other k-mers in case of overlaps. The final set of consensus sequences found by RepeatScout can be compared against gene annotation coordinate files for the organism to screen out the repeat families located in genic regions or areas of segmental duplication using scripts provided by the authors.
ReAS is focused on reconstruction of ancestral sequences of transposable elements from multiple sequence reads [52]. Consensus sequence boundaries are determined from clustered elements using the “aggregation of end points” technique derived from RECON [7]. Li et al. [52], the creators of ReAS, have used simulations to empirically determine parameters for different steps and guide their repeat element discovery process. The ancestral or consensus sequence set constructed by ReAS can be used as a library for RepeatMasker.
Conclusion
It is becoming increasingly clear that repeats are one of the principal factors responsible for the evolutionary success of eukaryotes [13, 89] Specifically, (a) repetitive DNA influences gene expression and recombination [6, 25, 73], (b) some repeat sequences have become critical in maintenance of chromosome structure [49, 55], (c) mobile repeat sequences increase genetic diversity through mutation [78, 8, 31], and (d) multiple transposition of some DNA transposons can produce chimeric molecules composed of segments of a variety of cellular genes, an observation which suggests that mobile element transposition may represent a means by which novel genes can be generated [37, 47, 61]. However, the algorithms and computational tools for identifying and studying repeat sequences are relatively primitive compared to those being utilized to explore genes. The complex structure of repetitive elements, the lack of knowledge concerning their function, and their limited conservation make the problem of identifying repeats and extracting meaningful family descriptions computationally challenging.
Ab initio tools hold the promise of enabling researchers to identify repeats in newly sequenced genomes and to discover new repetitive elements in well-studied genomes. Five approaches that have been used for initial identification of repetitive DNA are similarity based searches (e.g., BLAST), enumeration of k-mers, spaced seeds, dot-matrix approaches, and periodicity approaches. A variety of techniques are then used to extract refined descriptions of repeat families. Some ab initio tools are designed to be used with assembled genomic regions while others are targeted for shotgun reads (Table 1). The effectiveness and accuracy of six widely used ab initio repeat finders is evaluated and discussed in a companion paper [71].
There are many ways in which computational identification and characterization of repeat sequences could be improved. In addition to better, faster algorithms for repeat finding, the use of ensemble approaches that combine results from several different algorithms via voting mechanisms holds promise, and such strategies have been successfully applied to gene expression data [79, 53]. Another approach is to build pipelines that sequentially apply tools targeting different types of repeats or tools based on different algorithms. This is the approach taken by Quesneville et al. [69] for annotation of transposable elements in Drosophila and Chouvarine et al. [19] for classification of higher plant repeats. Although there are a number of tools for ab initio repeat identification, there has been little work in the development of computational tools for subsequent characterization of the discovered repeat families.
New types of repetitive elements are being discovered at a rapid rate as more genome sequences become accessible. The availability of sequenced genomes as well as the increasing recognition of the biological importance of repetitive elements will motivate the development of more sensitive and selective algorithms for ab initio repeat discovery and automated methods for classification and characterization of newly discovered repetitive elements.
Notes
Haas and Salzberg [33] have recently reviewed a subset of the repeat finders that we discuss. The focus of much of their review is mechanisms for handling the complications presented by repeats during genome assembly. The focus of our review is the use of these tools for identification of novel dispersed repeats in genomes.
BLAST is an acronym for the “Basic Local Alignment and Search Tool” developed by Altschul et al. [3]. There are currently several different BLAST modules specially designed for comparisons between different data types (see http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). WU-BLAST is a powerful alternative implementation of BLAST available from Washington University (http://blast.wustl.edu/). Crossmatch is a similarity search tool traditionally packaged with Phrap (www.phrap.org).
Abbreviations
- BLAST:
-
Basic Local Alignment and Search Tool
- bp:
-
base pair
- Mb:
-
megabase
- Gb:
-
gigabase
- MITE:
-
miniature inverted-repeat transposable element
- PALS:
-
Pairwise Alignment of Long Sequences
- SSR:
-
simple sequence repeat
References
Abouelhoda MI, Kurtz S, Ohlebusch E (2004) Replacing suffix trees with enhanced suffix arrays. J Discrete Algorithm 2:53–86
Agarwal P, States DJ (1994) The Repeat Pattern Toolkit (RPT): analyzing the structure and evolution of the C. elegans genome. Proc Int Conf Intell Syst Mol Biol 2:1–9
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Altschul SF, Madden TL, Zhang J et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Andrieu O, Fiston AS, Anxolabehere D et al (2004) Detection of transposable elements by their compositional bias. BMC Bioinformatics 5:94
Assaad FF, Tucker KL, Signer ER (1993) Epigenetic repeat-induced gene silencing (RIGS) in Arabidopsis. Plant Mol Biol 22:1067–1085
Bao Z, Eddy SR (2002) Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12:1269–1276
Batzer MA, Deininger PL (2002) ALU repeats and human genomic diversity. Nature 3:370–380
Bennett MD, Leitch IJ (2004) Plant DNA C-values database (release 3.0, Jan. 2004). http://www.rbgkew.org.uk/cval/homepage.html
Bennetzen JL (2000) Transposable element contributions to plant gene and genome evolution. Plant Mol Biol 42:251–269
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580
Biemont C, Vieira C (2006) Genetics: junk DNA as an evolutionary force. Nature 443:521–524
Britten RJ (1996) Cases of ancient mobile element DNA insertions that now affect gene regulation. Mol Phylogenet Evol 5:13–17
Britten RJ, Kohne DE (1968) Repeated sequences in DNA. Science 161:529–540
Brosius J (2003) How significant is 98.5% ‘junk’ in mammalian genomes. Bioinformatics 19(suppl. 2):ii35
Campagna D, Romualdi C, Vitulo N et al (2005) RAP: a new computer program for de novo identification of repeated sequences in whole genomes. Bioinformatics 21:582–588
Charlesworth B, Sniegowski P, Stephan W (1994) The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371:215–220
Chenna R, Sugawara H, Koike T et al (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500
Chouvarine P, Saha S, Peterson DG (2008) An automated, high-throughput sequence read classification pipeline for preliminary genome characterization. Anal Biochem 373:78–87
Cormen TH, Leiserson CE, Rivest RL et al (2001) Introduction to Algorithms, 2nd Edition. MIT Press and McGraw-Hill, Cambridge, MA
Coward E, Drablos F (1998) Detecting periodic patterns in biological sequences. Bioinformatics 14:498–507
de Bruijn NG (1946) A combinatorial problem. Proc Koninklijke Nederlandse Akademie v Wetenschappen 49:758–764
Delcher AL, Kasif S, Fleischmann RD et al (1999) Alignment of whole genomes. Nucleic Acids Res 27:2369–2376
Delcher AL, Phillippy A, Carlton J et al (2002) Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 30:2478–2483
Dorer DR, Henikoff S (1994) Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell 77:993–1002
Du L, Zhou H, Yan H (2007) OMWSA: detection of DNA repeats using moving window spectral analysis. Bioinformatics 23:631–633
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Edgar RC (2007) PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8:18
Edgar RC, Myers EW (2005) PILER: identification and classification of genomic repeats. Bioinformatics 21(Suppl 1):i152–i158
Feschotte C, Wessler SR (2001) Treasures in the attic: rolling circle transposons discovered in eukaryotic genomes. Proc Natl Acad Sci USA 98:8923–8924
Frost LS, Leplae R, Summers AO et al (2005) Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol 3:722–732
Gusfield D (1999) Algorithms on Strings, Trees and Sequences: Computer Science and Computational Biology. Cambridge University Press, New York
Haas BJ, Salzberg SL (2007) Finding repeats in genome sequences. In: Lengauer T (ed) Bioinformatics—From Genomes to Therapies, 1 edn. Wiley-VCH, Weinheim, pp 197–234
Havecker ER, Gao X, Voytas DF (2004) The diversity of LTR retrotransposons. Genome Biol 5:225
Hou M, Berman P, Hsu CH et al (2007) HomologMiner: looking for homologous genomic groups in whole genomes. Bioinformatics 23:917–925
Ilie L, Ilie S (2007) Multiple spaced seeds for homology search. Bioinformatics 23:2969–2977
Jiang N, Bao Z, Zhang X et al (2004) Pack-MULE transposable elements mediate gene evolution in plants. Nature 431:569–573
Jiang N, Bao Z, Zhang X et al (2003) An active DNA transposon family in rice. Nature 421:163–167
Jurka J, Kapitonov VV, Pavlicek A et al (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110:462–467
Jurka J, Klonowski P, Dagman V et al (1996) CENSOR—a program for identification and elimination of repetitive elements from DNA sequences. Comput Chem 20:119–121
Kalendar R, Vicient CM, Peleg O et al (2004) Large retrotransposon derivatives: abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics 166:1437–1450
Kapitonov VV, Jurka J (2001) Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci U S A 98:8714–8719
Kapitonov VV, Jurka J (2006) Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci U S A 103:4540–4545
Kolpakov R, Bana G, Kucherov G (2003) mreps: Efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res 31:3672–3678
Kurtz S, Choudhuri JV, Ohlebusch E et al (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29:4633–4642
Kurtz S, Schleiermacher C (1999) REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics 15:426–427
Lai J, Li Y, Messing J et al (2005) Gene movement by Helitron transposons contributes to the haplotype variability of maize. Proc Natl Acad Sci USA 102:9068–9073
Lapitan NLV (1992) Organization and evolution of higher plant nuclear genomes. Genome 35:171–181
Lee C, Ritchie DBC, Lin CC (1994) A tandemly repetitive, centromeric DNA sequence from the Canadian woodland caribou (Rangifer tarandus caribou): its conservation and evolution in several deer species. Chromosome Res 2:293–306
Lefebvre A, Lecroq T, Dauchel H et al (2003) FORRepeats: detects repeats on entire chromosomes and between genomes. Bioinformatics 19:319–326
Li M, Ma B, Kisman D et al (2004a) Patternhunter II: highly sensitive and fast homology search. J Bioinform Comput Biol 2:417–439
Li R, Ye J, Li S et al (2005) ReAS: Recovery of ancestral sequences for transposable elements from the unassembled reads of a whole genome shotgun. PLoS Comput Biol 1:e43
Li X, Rao S, Wang Y et al (2004b) Gene mining: a novel and powerful ensemble decision approach to hunting for disease genes using microarray expression profiling. Nucleic Acids Res 32:2685–2694
Li YC, Korol AB, Fahima T et al (2002) Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol 11:2453–2465
Lundblad V, Wright WE (1996) Telomeres and telomerase: A simple picture becomes complex. Cell 87:369–375
Ma B, Tromp J, Li M (2002) PatternHunter: faster and more sensitive homology search. Bioinformatics 18:440–445
Mak D, Gelfand Y, Benson G (2006) Indel seeds for homology search. Bioinformatics 22:e341–e349
Manber U, Myers G (1993) Suffix arrays: a new method for on-line string searches. SIAM J Comput 22:935–948
McCarthy EM, McDonald JF (2003) LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics 19:362–367
McClintock B (1984) The significance of responses of the genome to challenge. Science 226:792–801
Morgante M, Brunner S, Pea G et al (2005) Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat Genet 37:997–1002
Müller HJ (1930) Types of viable variations induced by X-rays in Drosophila. Genetics 22:299–337
Nagl W (1976) DNA endoreduplication and polyteny understood as evolutionary strategies. Nature 261:614–615
Ohshima K, Okada N (2005) SINEs and LINEs: symbionts of eukaryotic genomes with a common tail. Cytogenet Genome Res 110:475–490
Ouyang S, Buell CR (2004) The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res 32:D360–D363
Pevzner PA, Tang H, Tesler G (2004) De novo repeat classification and fragment assembly. Genome Res 14:1786–1796
Price AL, Jones NC, Pevzner PA (2005) De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1):i351–i358
Pritham EJ, Putliwala T, Feschotte C (2007) Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene 390:3–17
Quesneville H, Bergman CM, Andrieu O et al (2005) Combined evidence annotation of transposable elements in genome sequences. PLoS Comput Biol 1:166–175
Ruitberg CM, Reeder DJ, Butler JM (2001) STRBase: a short tandem repeat DNA database for the human identity testing community. Nucleic Acids Res 29:320–322
Saha S, Bridges S, Magbanua ZV et al. (2008) Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res (in press)
Sharma D, Issac B, Raghava GP et al (2004) Spectral Repeat Finder (SRF): identification of repetitive sequences using Fourier transformation. Bioinformatics 20:1405–1412
Sherman JD, Stack SM (1995) Two-dimensional spreads of synaptonemal complexes from solanaceous plants. VI. High-resolution recombination nodule map for tomato (Lycopersicon esculentum). Genetics 141:683–708
Smit AFA, Hubley R, Green P (1996–2004) RepeatMasker Open-3.0. http://www.repeatmasker.org
Sonnhammer ELL, Durbin R (1995) A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:1–10
Sperber GO, Airola T, Jern P et al (2007) Automated recognition of retroviral sequences in genomic data—RetroTector©. Nucleic Acids Res 35:4964–4976
Strachan T, Read AP (1999) Human molecular genetics, 2nd edn. Wiley & Sons, New York
Syvanen M (1984) The evolutionary implications of mobile genetic elements. Annual Rev Genet 18:271–293
Tan AC, Gilbert D (2003) Ensemble machine learning on gene expression data for cancer classification. Appl Bioinformatics 2:S75–S83
Taneda A (2004) Adplot: detection and visualization of repetitive patterns in complete genomes. Bioinformatics 20:701–708
Temnykh S, DeClerck G, Lukashova A et al (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Timberlake WE (1978) Low repetitive DNA content in Aspergillus nidulans. Science 202:973–975
Toth G, Deak G, Barta E et al (2006) PLOTREP: a web tool for defragmentation and visual analysis of dispersed genomic repeats. Nucleic Acids Res 34:W708–W713
Tu Z (2001) Eight novel families of miniature inverted repeat transposable elements in the African malaria mosquito, Anopheles gambiae. Proc Natl Acad Sci U S A 98:1699–1704
Volfovsky N, Haas BJ, Salzberg SL (2001) A clustering method for repeat analysis in DNA sequences. Genome Biol 2:research0027.1–0027.11
Wang J, Wong GK, Ni P et al (2002) RePS: a sequence assembler that masks exact repeats identified from the shotgun data. Genome Res 12:824–831
Warburton PE, Giordano J, Cheung F et al (2004) Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res 14:1861–1869
Weiner P (1973) Linear pattern matching algorithm. In: Proceedings of the 14th annual IEEE symposium on switching and automata theory, University of Iowa, Iowa City, 15–17 Oct 1973
Wessler SR (1997) Transposable elements and the evolution of gene expression. Exp Biol 1039:115–122
Wicker T, Matthews DE, Keller B (2002) TREP: a database for Triticeae repetitive elements. Trends Plant Sci 7:561–562
Wicker T, Sabot F, Hua-Van A et al (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Yang G, Hall TC (2003) MAK, a computational tool kit for automated MITE analysis. Nucleic Acids Res 31:3659–3665
Zuckerkandl E, Hennig W (1995) Tracking heterochromatin. Chromosoma 104:75–83
Acknowledgements
This research was supported, in part, by the National Science Foundation (DBI-0421717 to D.G.P. and EPS-0556308 to S.M.B.), the United States Department of Agriculture (CSREES-2006-34506-17290 and ARS-58-6402-7-241 to D.G.P.), and the Mississippi Corn Promotion Board (to D.G.P.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Dr. Ray Ming
Rights and permissions
About this article
Cite this article
Saha, S., Bridges, S., Magbanua, Z.V. et al. Computational Approaches and Tools Used in Identification of Dispersed Repetitive DNA Sequences. Tropical Plant Biol. 1, 85–96 (2008). https://doi.org/10.1007/s12042-007-9007-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-007-9007-5