
Abstract
Gene duplication is an important mechanism for acquiring new genes and creating genetic novelty in organisms. Many new gene functions have evolved through gene duplication and it has contributed tremendously to the evolution of developmental programmes in various organisms. Gene duplication can result from unequal crossing over, retroposition or chromosomal (or genome) duplication. Understanding the mechanisms that generate duplicate gene copies and the subsequent dynamics among gene duplicates is vital because these investigations shed light on localized and genomewide aspects of evolutionary forces shaping intra-specific and inter-specific genome contents, evolutionary relationships, and interactions. Based on whole-genome analysis of Arabidopsis thaliana, there is compelling evidence that angiosperms underwent two whole-genome duplication events early during their evolutionary history. Recent studies have shown that these events were crucial for creation of many important developmental and regulatory genes found in extant angiosperm genomes. Recent studies also provide strong indications that even yeast (Saccharomyces cerevisiae), with its compact genome, is in fact an ancient tetraploid. Gene duplication can provide new genetic material for mutation, drift and selection to act upon, the result of which is specialized or new gene functions. Without gene duplication the plasticity of a genome or species in adapting to changing environments would be severely limited. Whether a duplicate is retained depends upon its function, its mode of duplication, (i.e. whether it was duplicated during a whole-genome duplication event), the species in which it occurs, and its expression rate. The exaptation of preexisting secondary functions is an important feature in gene evolution, just as it is in morphological evolution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A gene duplication is an event in which one gene gives rise to two genes that cannot be operationally distinguished from each other. The duplicated genes remain in the same genome and therefore are paralogues and in different genome as orthologues. Gene duplication is believed to play an important role in evolution by providing material for evolution of new gene functions. A duplicated gene provides a greater, less-constrained chance for natural selection to shape a novel function (Long et al. 2003). An important question in the early evolution of life forms is how a single-celled bacterium evolved into multicellular complex organisms, and a few genes with thousands of base pairs evolved to several thousand genes with millions of base pairs (Zhang 2003). Bridges (1936) first identified the bar eye locus in Drosophila and its effect on eye shape when duplicated. Ohno (1970) discussed possible fates of duplicated genes in Evolution by gene duplication, and concluded that gene duplication is the only means by which a new gene can arise, and argues that in the past whole genomes have been duplicated, causing the change from invertebrates to vertebrates, which could occur only if whole genomes were duplicated (Bergman 2006). Ohno’s (1970) representation of duplication as evolutionary force opened up a major field of research into the possible evolutionary consequences of gene duplication. After the 1980s, when molecular markers were developed, and the 1990s, when genome sequencing became common, gene duplication analysis and determining the evolutionary pathways of organisms became a research field. Kellis et al. (2004) agree that ‘whole-genome duplication (WGD) followed by massive gene loss and specialization has long been postulated as a powerful mechanism of evolutionary innovation’.
Molecular mechanisms of gene duplication
Duplicated genes may be produced by unequal crossing over, retrotransposition, duplicated DNA transposition and polyploidization.
Unequal crossing over
Unequal crossing over produces tandem repeated sequences, i.e. continuous repeats of DNA sequence. Depending on the position of crossing over, the duplicated region can contain part of a gene, an entire gene, or several genes (Zhang 2003). Crossing over in a bivalent carrying a duplication in one of the two chromosomes may lead to different consequences. If the duplicated segment pairs with its homologous segment in the other chromosome in complete disregard of other homologous segments then the unequal crossing over produces duplication of other segments. If the duplicated segments are present in reverse order of the original segments or if duplication is present on the other arm then the pairing and crossing over forms dicentric along with acentric fragments. If the duplicated segments are on another, nonhomologous chromosome, crossing over with this duplicated region will produce two interchange chromosomes (Gupta 2007).
Retroposition
Retroposition is a process when a messenger RNA (mRNA) is reverse transcribed to complementary DNA (cDNA) and then inserted into the genome. There are several molecular features of retroposition: lack of introns and regulatory sequences of a gene, presence of a poly-A sequence, and presence of flanking short direct repeats (Brosius 1991). The major difference from unequal crossing over is that introns, if present in the original genes, will also be present in the duplicated genes but absent in retrogenes. A duplicated gene generated by retroposition is usually unlinked to the original gene, because the insertion of cDNA into the genome is more or less random (Long et al. 2003). Recent studies have found that retrogenes that are integrated near other coding regions or even in introns of expressed coding sequences are much more likely to be expressed than those that are integrated far from coding sequences (Vinckenbosch et al. 2006).
Duplicative transposition
Duplicative transposition of DNA sequences can be accomplished by one of two main pathways: nonallelic homologous recombination (NAHR) or nonhomologous end joining (NHEJ). The difference between the two pathways is based on whether homologous sequences are used as a template during double-strand-break repair, and this difference can also be used to infer the mechanism by which individual genes are duplicated. Recombination between these nonallelic homologous sequences can result in the duplication of the intervening sequences, which can then lead in turn to more duplications because of pairing between the new paralogues (Bailey et al. 2003). But other studies in humans have also found multiple cases with no repetitive DNA or long stretches of homologous sequence at duplication breakpoints, suggesting the action of NHEJ (Linardopoulou et al. 2005). Because of the relatively low proportion of duplicated sequences arranged in tandem in the human genome, it has been proposed that duplicative transposition is the major mode of duplication in humans (Samonte and Eichler 2002). The number of retrogenes maintained in both mammals (Pan and Zhang 2007) and Drosophila is lower than the number maintained by DNA-based intermediates (i.e. unequal crossing over and duplicative transposition), despite the fact that the mutation rate forming new retrocopies is higher (Pan and Zhang 2007). The lack of functional regulatory DNA is likely to be the reason that very few of these paralogues are maintained for long periods; 120 functional retrotransposed gene copies have been maintained in the human genome over the past 63 million years (Vinckenbosch et al. 2006).
Polyploidization
Polyploidization is the fourth major mechanism of formation of duplicate genes. Polyploidy is an evolutionary process whereby two or more genomes are brought together into the same nucleus, usually by hybridization followed by chromosome doubling. Ohno (1970) pointed out that two rounds of genome duplication had taken place in the evolution of vertebrates. Earlier studies provide strong indications that even yeast (S. cerevisiae), with its compact genome, is in fact an ancient tetraploid where a WGD followed by massive gene loss and specialization have long been postulated as a powerful mechanism of evolutionary innovation (Kellis et al. 2004). In plants, polyploidy was proposed to have occurred in the lineage of at least 70% of angiosperms (Masterson 1994) and in 95% of pteridophytes (Grant 1981). Moreover, the first two angiosperm species whose genomes have been fully sequenced, Arabidopsis (Arabidopsis Genome Initiative 2000) and rice (Goff et al. 2002), considered classical diploids, are apparently ancient polyploids (paleopolyploids). Many more, if not all, higher plant species, considered as diploids because of their genetic and cytogenetic behaviour, are ancient polyploids that underwent a process of extensive diploidization. Thus, polyploidy appears to be one of the major processes that has driven and shaped the evolution of higher organisms (Levy and Feldman 2002).
Does gene duplication provide the engine for evolution?
How genomes evolved from a few primordial genes to the more than 20,000 genes in higher organisms is an important question in evolution. The current primary hypothesis is that it occurred via gene duplication (Hurles 2004). Shanks (2004) concluded that ‘duplication is the way of acquiring new genes by an organism. They appear as the result of duplication’. Ohno (1970) concluded that ‘gene duplication is the only means by which a new gene can arise’ and argued that not only genes but whole genomes have been duplicated in the past, causing ‘great leaps in evolution—such as the transition from invertebrates to vertebrates, which could occur only if whole genomes were duplicated’. Similarly, the most distinctive feature of angiosperm genomes is the extent of genome duplication, an evolutionary event that has been central to angiosperm evolution. The two major branches of the angiosperms (eudicots and monocots), estimated to have diverged between 125 to 140 million years ago (Mya) and 170 to 235 Mya (Davies et al. 2004), show much more rapid structural evolution than vertebrates. This difference appears to be due largely to the tendency of angiosperms for chromosomal duplication and subsequent gene loss (Coghlan et al. 2005). Earlier analyses of genome sequences suggests that genome duplication in angiosperms may be not merely episodic, but truly cyclic, imparting various fitness advantages that erode over time, favouring new polyploidizations (Chapman et al. 2006).
The fate of duplicate genes
WGDs result in new gene copies of every gene in a genome and, obviously, all the flanking regulatory sequences. The birth and death of genes is a common theme in gene-family and genome evolution (Hughes and Nei 1989; Nei et al. 2000), with those genes involved in physiologies that vary greatly among species (e.g. immunity, reproduction and sensory systems) probably having high rates of gene birth and death.
Pseudogenization
It is generally not advantageous for species to carry two identical genes. Duplication of a gene produces functional redundancy. Pseudogenization, the process by which a functional gene becomes a pseudogene, usually occurs in the first few million years after duplication if the duplicated gene is not under any selection (Lynch and Conery 2000). The two major forces of pseudogenization are mutation and deletion, where changes in pseudogenization occur through promoter mutation, nonsense mutation or missense mutation in coding region, or loss of exon splicing junction. Mutations that disrupt structure and function of one of the two duplicate genes are not deleterious and are not removed by selection. Gradually, the copy of the gene that accumulates mutations becomes a pseudogene, which is either unexpressed or functionless (Zhang 2003). After a long time, pseudogenes will either be deleted from the genome or become so diverged from the parental genes such that they are no longer identifiable. Humans and mice have similar numbers of members of the olfactory receptor gene family (∼1000 genes) but the proportion of pseudogenes is >60% in humans and only 20% in mice. This may be due to reduced use of olfaction since the origin of hominoids, which can be compensated by other sensory mechanisms, such as better vision (Rouquier et al. 2000).
Occasionally pseudegenes may also serve some functions. In chicken, there is only one functional gene (VH1) encoding the heavy chain variable region of immunoglobulins, and immunoglobulin diversity is generated by gene conversion of the VH1 gene by many duplicated variable region pseudogenes that occur on its 5′ side (Ota and Nei 1995).
Conservation of gene function
The first mechanism for maintaining a duplicate copy of a gene proposed by Ohno (1970) was to simply increase the number of genes coding for functional rotein. Here both loci maintain the original functions, and this process has therefore come to be known as ‘gene conservation’. Ohno (1970) proposed two possible models, not necessarily mutually exclusive, for why these duplicates would maintain the original functions. The first model states that a second gene could provide functional redundancy if the original locus was disabled by mutation. The second possibility for why exact copies of duplicated genes are maintained is that there is an advantage to produce more of a gene product. The increased levels of protein production can be accomplished by increasing expression levels at a single locus, duplicating a gene may have an equivalent effect. The most commonly cited example of this phenomenon is the array of highly duplicated genes for histone proteins and ribosomal RNAs needed during development and other translationally active stages (Hurst and Smith 1998). How can two paralogous genes maintain the same function after duplication? One of the two possible mechanisms is concerted evolution (Li 1997) and another is purifying selection (Nei et al. 2000). Concerted evolution is a mode of gene family evolution through which members of a family remain similar in sequence and function because of frequent gene conversion and/or unequal crossing over (Hurst and Smith 1998). A strong purifying selection against mutations that modify gene function can also prevent duplicated genes from diverging.
Subfunctionalization
In general, a duplicate gene is deleterious for the genome or species, with some exceptions like histone-coding genes. Two genes with identical functions are unlikely to be stably maintained in the genome unless the presence of an extra amount of gene product is advantageous (Nowak et al. (Nowak et al. 1997)). After duplication, both daughter genes are maintained in the genome for a period of time during which they differentiate in some aspects of their functions. This can occur by subfunctionalization, in which each daughter gene adopts part of the functions of their parental gene. For example, engrailed-1 and engrailed-1b are pair of transcription factor genes in zebrafish generated by a chromosomal segmental duplication. engrailed-1 is expressed in the pectoral appendage bud, whereas engrailed-1b is expressed in a specific set of neurons in the hindbrain/spinal cord. On the other hand, the sole engrailed-1 gene of the mouse, orthologous to both genes of the zebrafish, is expressed in both pectoral appendage bud and hindbrain/spinal cord (Force et al. 1999).
Neofunctionalization
Origin of novel gene function is one of the most important outcomes of gene duplication. The evolution of a novel fruit shape in tomato (Solanum lycopersicum) appears to have been created by the chance duplication and transposition of a gene (SUN) into a new regulatory context. SUN and its progenitor (IQD12) belongs to a gene family that contains a plant-specific, 67 amino acid motif (called IQ67) that is involved in calmodulin signalling. SUN is expressed at much higher levels during the early stages of fruit development, and this upregulation is clearly correlated with an elongated fruit shape instead of the round shape governed by the gene IQD12 (Xiao et al. 2008). Ni et al. (2009) reported the involvement of polyploidy in neofunctionalization. The natural allopolyploid Arabidopsis suecica is readily resynthesized in the laboratory from its model progenitors A. thaliana and A. arenosa. An interesting feature of this allopolyploid is that it grows to a larger stature and produces more biomass than either of its parents. Among 128 genes upregulated in the allotetraploid relative to its parents, ∼67% were found to have either circadian clock associated 1 (CCA1) or evening-element binding sites in their upstream regulatory regions. Further analyses showed that the CCA1 and LHY (late elongated hypocotyl) genes were epigenetically suppressed in the allopolyploid and that this suppression strongly is correlated with increased starch synthesis and chlorophyll content, ultimately leading to greater plant biomass. The study by Ni et al. (2009) illustrates the importance of instantaneous shifts in genetic networks and their associated metabolism caused by allopolyploidy, which is likely to serve as an important source of evolutionary novelty.
Duplication and speciation
Most gene pairs formed by a WGD have only a brief evolutionary lifespan before one copy becomes deleted, leaving the other to survive as a single-copy locus. We might expect that the probability of retention is initially equal for both duplicates following WGD, but earlier results have suggested that one duplicate may be more susceptible to loss than the other. It was shown that, in A. thaliana, one paralogon (duplicated genomic region) tends to contain significantly more genes than the other (Thomas et al. 2006). There is strong evidence for one round of genome doubling after the eudicot divergence and a second polyploidization event some time following the divergence of Arabidopsis and Brassica from their common ancestor with the Malvaceae, represented by cotton (Adams and Wendel 2005).
WGD has been proposed to be a lineage splitting force because of the subsequent occurrence of gene losses independently in different populations. In particular, reciprocal gene loss (RGL) occurs when two paralogues created by WGD are retained until speciation, after which each species loses a different copy. After duplication, one of the two redundant copies of a gene should theoretically be free to degenerate and become lost from the genome without any consequence. One analysis performed just after artificial allopolyploidization in cotton found that one paralogue is silenced or downregulated in 5% of gene pairs and that silencing is often organ-specific (Adams et al. 2004).
Genome duplication and the origin of angiosperm
It has been suggested that large-scale gene duplication or WGD events can be associated with important evolutionary transitions involving the origins of higher taxa. Angiosperms appear rather suddenly in the fossil record during the Jurassic (208–145 Mya), with no obvious ancestors for a period of 80–90 million years before their appearance (Doyle and Donoghue 1993). Nevertheless, the existence during the Jurassic of all known sister taxa of the angiosperms implies that the angiosperm lineage must have been established by that time (Doyle and Donoghue 1993). The ancestral lineage is often termed ‘angiophytes’. It is presumed that angiophytes went through a period of little diversification during the Late Triassic (220 Mya) and Jurassic (Wing and Boucher 1998), either because the diversity-enhancing features, such as flowers, of the crown-group angiosperms had not yet evolved in stem angiophytes, or because the diversity among angiophytes was inhibited during the Jurassic by environmental conditions or biotic interactions (Wing and Boucher 1998).
The recent transitional–combinational theory of angiosperm origin suggests an evolution from Jurassic seed ferns through three fundamental transitions: (i) evolution of the carpel, (ii) emergence of double fertilization, and (iii) origin of the flower. The extant (or modern) angiosperms did not appear until the early Cretaceous (145–125 Mya), when the final combination of these three angiosperm features occurred, as supported by evidence from microfossils and macrofossils (Stuessy 2004). The fossil record provides excellent evidence for this rapid diversification in floral form during the earliest phases of recorded flowering plant history. This diversification of angiosperms occurred during a period (the Aptian, 125–112 Mya) when their pollen and megafossils were rare components of terrestrial flora and species diversity was low (Crane et al. 1995). Angiosperm fossils show a dramatic increase in diversity between the Albian (112–99.6 Mya) and the Cenomanian (99.6–93.5 Mya) at a global scale (Crane et al. 2004).
In 1996, when sequencing of the flowering plant A. thaliana (Brassicaceae) genome began, this model plant, with its small genome, was not expected to be an ancient polyploid. However, five years after the release of its genome sequence (Arabidopsis Genome Initiative 2000), there is compelling evidence that the genome of Arabidopsis, or rather that of its ancestors, has been duplicated thrice (events referred as 1R, 2R and 3R) during the past 250 million years (Simillion et al. 2002; Bowers et al. 2003). Ancient polyploidy events might have directly influenced the increase in number of plant species and plant complexity observed since the early Cretaceous. However, other factors, such as expansion and functional diversification of specific gene families following a polyploidy event, are likely to have been more influential and could explain, at least in part, the origin and fast diversification of angiosperm lineages that occurred owing to biased retention of genes after duplication (De Bodt et al. 2005). Blanc and Wolfe (2004) studied the relationship between gene function and loss of duplicates after the most recent polyploidy event (3R). Maere et al. (2005) developed an evolutionary model based on the KS (number of synonymous substitutions per synonymous site) distribution of the Arabidopsis paranome where they took into account the three major genomewide duplication events (1R, 2R and 3R) and a continuous mode of small-scale gene duplications (referred to as 0R). All these studies concluded that both copies of duplicated genes involved in transcriptional regulation and signal transduction have been preferentially retained following genome duplications. It has also been observed that duplicated copies of developmental genes have been retained following genome duplications (Blanc and Wolfe 2004; Maere et al. 2005), particularly following the two older events (1R and 2R). Overall, the three polyploidy events in the ancestors of Arabidopsis might have been responsible for >90% of the transcription factor, signal transducer and developmental genes created during the past 250 million years (Maere et al. 2005).
Duplication analysis in model organisms
Since 1990, the genome sequencing projects launched in Arabidopsis and other plant species have allowed analysis of the evolutionary pattern of different species by various chromosome rearrangement. Similarity and collinearity analysis of different species or analysis within species among different chromosomes has clearly shown the process of genome duplication over time and its role in species diversification. Duplication analysis of some of the model organisms based on genome sequencing data or in comparison with other species is presented briefly below.
Duplication in Arabidopsis genome
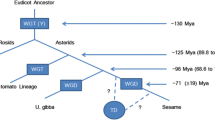
The Arabidopsis Genome Initiative published its sequence analysis in 2000. They used large-insert bacterial artificial chromosome (BAC), phage (P1) and transformation-competent artificial chromosome (TAC) libraries as the primary substrates for sequencing. The Arabidopsis genome sequence provides a complete view of chromosomal organization and clues to its evolutionary history. It revealed, through 1528 tandem arrays containing 4140 individual genes, that 17% of all genes of Arabidopsis are arranged in tandem arrays (Arabidopsis Genome Initiative 2000). After aligning all five chromosomes of Arabidopsis to each other in both orientations using MUMmer (Delcher et al. 1999), the results were filtered to identify all segments at least 1000 bp in length with at least 50% identity. It revealed 24 large duplicated segments of 100 kb or larger, comprising 65.6 Mb or 58% of the genome. But use of TBLASTX (Mayer et al. 1999) to identify collinear clusters of genes in large duplicated chromosomal segments showed that duplicated regions encompass 67.9 Mb 60% of the genome (Arabidopsis Genome Initiative 2000). As the majority of the Arabidopsis genome is represented in duplicated (but not triplicated) segments, it appears most likely that Arabidopsis, like maize, had a tetraploid ancestor (Gaut and Doebley 1997). A comparative sequence analysis of Arabidopsis and tomato estimated that a duplication occurred 112 Mya to form a tetraploid. The degrees of conservation of the duplicated segments seen at present might be due to divergence from an ancestral autotetraploid form, or might reflect differences present in an allotetraploid ancestor (Ku et al. 2000).
Duplication in S. cerevisiae
Wolfe and Shields (1997) interpreted presence and distribution of duplicate regions in the S. cerevisiae genome as supporting a model of WGD. Kellis et al. (2004) showed that S. cerevisiae arose from complete duplication of eight ancestral chromosomes, and subsequently returned to functionally normal ploidy by massive loss of nearly 90% of duplicated genes in small deletions. They identified 145 paired regions in S. cerevisiae, tiling 88% of the genome and containing 457 duplicated gene pairs. The experiment was conducted by using Kluyveromyces waltii, a close relative of S. cerevisiae, to identify orthologous regions. The two genomes are related by a 1:2 mapping, most local regions in K. waltii mapped to two regions in S. cerevisiae, with each containing matches to only a subset of the K. waltii genes. This clearly proved that an ancient WGD had occurred in the previous lineages of yeast.
Gene and chromosome duplication in rice
The International Rice Genome Sequencing Project was organized to achieve >99.99% accurate sequence using a map-based clone-by-clone sequencing strategy (Sasaki and Burr 2000). More than 104,000 ESTs from a variety of rice tissues have been entered in an EST database (www.ncbi.nlm.nih.gov/dbEST/dbEST_summary.html). Goff et al. (2002) described a random fragment shotgun sequencing of Oryza sativa L. ssp. japonica (cv. Nipponbare) to discover rice genes, molecular markers for breeding, and mapped sequences for association of candidate genes and the traits they control. Global duplication of predicted genes was determined using BLAST by comparing all Hgenes (high predicted genes with confidence scores of >75%) and Mgenes (medium predicted genes with confidence scores from 1 to 75%). Of these, 77% were found to be homologous to at least one other predicted gene (Goff et al. 2002). Chromosomal duplications were identified by comparing (BLASTN) more than 2000 mapped rice cDNA markers (Harushima et al. 1998) to the anchored portion of Syd (Syngenta draft sequence; data access at www.tmri.org) and observed that the proportion of locally duplicated genes ranged from 15.4 to 30.4%, depending on the chromosome. The largest chromosomal duplication is on chromosomes 11 and 12 (Harushima et al. 1998; Wilson et al. 1999). The amino acid substitution rate (d A ) was used to estimate the age of genome duplications. A rice WGD is reported to have occured 40–50 Mya (Goff et al. 2002).
Conclusions
Duplicate gene evolution has most likely played a substantial role in both the rapid changes in organismal complexity apparent in deep evolutionary splits and the diversification of more closely related species. The most important contribution of gene duplication towards evolution is provision of new genetic material for different mechanisms of evolution i.e. mutation, drift and selection, to act upon, the result of which is specialized or new gene functions. Duplication increases buffering capacity of genomes or species in adapting to changing environments where only two variants (alleles) exist at any locus within a (diploid) individual. Although duplicated genes and genomes can provide raw material for evolutionary diversification and the functional divergence of duplicated genes might offer a selective advantage to polyploids over a long time scale, a beneficial effect of these duplications is assumed shortly after the duplication event. It is also posibble that differential gene duplication and pseudogenization in geographically isolated populations causes reproductive isolation and speciation, although this intriguing hypothesis awaits observational evidence. Extensive functional studies targeted at duplicated genes are required if we are to more fully understand the range of evolutionary outcomes. Collaborations between the proteomics and evolutionary-genetics communities would facilitate investigation of the potential role of gene duplication during evolution of the protein–protein and cell–cell interactions that are fundamental to the biology of multicellular organisms.
References
Adams K. L. and Wendel J. F. 2005 Allele-specific, bi-directional silencing of an alcohol dehydrogenase gene in different organs of interspecific diploid cotton hybrids. Genetics 171, 2139–2142.
Adams K. L., Percifield R. and Wendel J. F. 2004 Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid. Genetics 168, 2217–2226.
Arabidopsis Genome Initiative 2000 Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815.
Bailey J. A., Liu G. and Eichler E. E. 2003 An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 73, 823–834.
Bergman J. 2006 Does gene duplication provide the engine for evolution? J. Creation 20, 99–104.
Blanc G. and Wolfe K. H. 2004 Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16, 1679–1691.
Bowers J. E., Chapman B. A., Rong J. and Paterson A. H. 2003 Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438.
Bridges C. B. 1936 The Bar gene a duplication. Science 83, 210–211.
Brosius J. 1991 Retroposons – seeds of evolution. Science 251, 753.
Chapman B. A., Bowers J. E., Feltus F. A. and Paterson A. H. 2006 Buffering crucial functions by paleologous duplicated genes may impart cyclicality to angiosperm genome duplication. Proc. Natl. Acad. Sci. USA 103, 2730–2735.
Coghlan A., Eichler E. E., Oliver S. G., Paterson A. H. and Stein L. 2005 Chromosome evolution in eukaryotes: a multi-kingdom perspective. Trends Genet. 21, 673–682.
Crane P. R., Friis E. M. and Pedersen K. R. 1995 The origin and early diversification of angiosperms. Nature 374, 27–33.
Crane P. R., Herendeen P. and Friis E. M. 2004 Fossils and plant phylogeny. Am. J. Bot. 91, 1683–1699.
Davies T. J., Barraclough T. G., Chase M. W., Soltis P. S., Soltis D. E. and Savolainen V. 2004 Darwin’s abominable mystery: Insights from a super tree of the angiosperms. Proc. Natl. Acad. Sci. USA 101, 1904–1909.
De Bodt S., Maere S. and Van de Peer Y. 2005 Genome duplication and the origin of angiosperms. Trends Ecol. Evol. 20, 591–597.
Delcher A. L., Kasif S., Fleischmann R. D., Peterson J., White O. and Salzberg S. L. 1999 Alignment of whole genomes. Nucleic Acids Res. 27, 2369–2376.
Doyle J. A. and Donoghue M. J. 1993 Phylogenies and angiosperm diversification. Paleobiology 19, 141–167.
Force A., Cresko W. A., Pickett F. B., Proulx S. R., Amemiya C. and Lynch M. 1999 The origin of subfunctions and modular gene regulation. Genetics 170, 433–446.
Gaut B. S. and Doebley J. F. 1997 DNA sequence evidence for the segmental allotetraploid origin of maize. Proc. Natl. Acad. Sci. USA 94, 6809–6814.
Goff S. A., Ricke D., Lan T. H., Presting G., Wang R., Dunn M. et al. 2002 A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100.
Grant V. 1981 Plant speciation, 2nd edition. Columbia University Press, New York, USA
Gupta P. K. 2007 Duplication and deficiencies. In Cytogenetics, 7th edition, pp. 19–43. Rastogi Publication, Meerut, India.
Harushima Y., Yano M., Shomura A., Sato M., Shimano T., Kuboki Y. et al. 1998 A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 148, 479–494.
Hughes A. L. and Nei M. 1989 Evolution of the major histocompatibility complex: independent origin of nonclassical class I genes in different groups of mammals. Mol. Biol. Evol. 6, 559–579.
Hurles M. 2004 Gene duplication: the genomic trade in spare parts. PloS Biol. 2, 900–904.
Hurst L. D. and Smith N. G. C. 1998 The evolution of concerted evolution. Proc. R. Soc. London. Ser. B 265, 121–127.
Kellis M., Birren B. W. and Lander E. S. 2004 Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cereviseae. Nature 428, 617–624.
Ku H. M., Vision T., Liu J. and Tanksley S. D. 2000 Comparing sequenced segments of the tomato and Arabidopsis genomes: Large-scale duplication followed by selective gene loss creates a network of synteny. Proc. Natl. Acad. Sci. USA 97, 9121–9126.
Levy A. A. and Feldman M. 2002 The impact of polyploidy on grass genome evolution. Plant Physiol. 130, 1587–1593.
Li W. H. 1997 Molecular evolution, 1st edition. Sinauer Associates, Sunderland, Massachusetts.
Linardopoulou E. V., Williams E. M., Fan Y., Friedman C., Young J. M. and Trask B. J. 2005 Human sub-telomeres are hot spots of inter chromosomal recombination and segmental duplication. Nature 437, 94–100.
Long M., Betran E., Thornton K. and Wang W. 2003 The origin of new genes: glimpses from the young and old. Nat. Rev. Genet. 4, 865–875.
Lynch M. and Conery J. S. 2000 The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155.
Maere S., De Bodt S., Raes J., Casneuf T., Van Montagu M., Kuiper M. and Van de Peer Y. 2005 Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA 102, 5454–5459.
Masterson J. 1994 Stomatal size in fossil plants: evidence for polyploidy in majority of angiosperms. Science 264, 421–423.
Mayer K., Schüller C., Wambutt R., Murphy G., Volckaert G., Pohl T. et al. 1999 Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana. Nature 402, 769–777.
Nei M., Rogozin I. B. and Piontkivska H. 2000 Purifying selection and birth-and-death evolution in the ubiquitin gene family. Proc. Natl. Acad. Sci. USA 97, 10866–10871.
Ni Z., Kim E. D. , Ha M., Lackey E., Liu J., Zhang Y. et al. 2009 Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 457, 327–331.
Nowak M. A., Boerlijst M. C. and Smith J. M. 1997 Evolution of genetic redundancy. Nature 388, 167–171.
Ohno S. 1970 Evolution by gene duplication. Springer-Verlag, New York, USA.
Ota T. and Nei M. 1995 Evolution of immunoglobulin VH pseudogenes in chickens. Mol. Biol. Evol. 12, 94–102.
Pan D. and Zhang L. 2007 Quantifying the major mechanisms of recent gene duplications in the human and mouse genomes: a novel strategy to estimate gene duplication rates. Genome Biol. 8, R158.
Rouquier S., Blancher A. and Giorgi D. 2000 The olfactory receptor gene repertoire in primates and mouse: evidence for reduction of the functional fraction in primates. Proc. Natl. Acad. Sci. USA 97, 2870–2874.
Samonte R. V. and Eichler E. E. 2002 Segmental duplications and the evolution of the primate genome. Nat. Rev. Genet. 3, 65–72.
Sasaki T. and Burr B. 2000 International rice genome sequencing project. The effort to completely sequence the rice genome. Curr. Opin. Plant Biol. 3, 138–141.
Shanks N. 2004 God, the devil, and darwin. Oxford University Press, New York, USA.
Simillion C., Vandepoele K., Van Montagu M. C., Zabeau M. and Van de Peer Y. 2002 The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 99, 13627–13632.
Stuessy T. F. 2004 A transitional–combinatorial theory for the origin of angiosperms. Taxon 53, 3–16.
Thomas B. C., Pedersen B. and Freeling M. 2006 Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homologue leaving clusters enriched in the sensitive genes. Genome Res. 16, 934–946.
Vinckenbosch N., Dupanloup I. and Kaessamann H. 2006 Evolutionary fate of retroposed gene copies in the human genome. Proc. Natl. Acad. Sci. USA 103, 3220–3225.
Wilson W. A., Harrington S. E., Woodman W. L., Lee M., Sorrells M. E. and McCouch S. R. 1999 Inferences on the genome structure of progenitor maize through comparative analysis of rice, maize and the domesticated panicoids. Genetics 153, 453–473.
Wing S. L. and Boucher L. D. 1998 Ecological aspects of the cretaceous flowering plant radiation. Annu. Rev. Earth Planet Sci. 26, 379–421.
Wolfe K. H. and Shields D. C. 1997 Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387, 708–713.
Xiao H., Jiang N., Schaffner E., Stockinger E. J. and van der Knaap E. 2008 A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319, 1527–1530.
Zhang J. 2003 Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 192–198.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Magadum S., Banerjee U., Murugan P., Gangapur D. and Ravikesavan R. 2013 Gene duplication as a major force in evolution. J. Genet. 92, xx–xx]
Rights and permissions
About this article
Cite this article
MAGADUM, S., BANERJEE, U., MURUGAN, P. et al. Gene duplication as a major force in evolution. J Genet 92, 155–161 (2013). https://doi.org/10.1007/s12041-013-0212-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-013-0212-8