
Abstract
The synthesis and initial spectroscopic characterization of a phosphorus(V) octaethylporphyrin-nitroxide adduct is reported. The nitroxide (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) is axially and covalently bound to the phosphorus porphyrin (PPor+) to give the studied PPor+-TEMPO conjugate. The complex allows the influence of the type of bonding and geometry of the central element in the porphyrin on the excited state dynamics to be investigated. In addition, the high oxidation potential of the PPor+ 1.35 V vs SCE means that it is a strong oxidant in its excited state and is energetically well-placed to allow charge transfer (CT) from TEMPO to PPor+ to occur. Cyclic voltammetry data and DFT computations show that the CT state is the lowest excited state, lying well below the excited sing-doublet and trip-quartet states of the complex. Time-resolved optical data show that the presence of the bound nitroxide leads to a large decrease in the excited state lifetime in acetonitrile, suggesting that rapid charge separation and recombination occurs. However, transient EPR (TREPR) data reveal that the trip-quartet state is also populated and that emissive spin polarization is generated in the ground state at room temperature, consistent with radical-quartet pair interactions analogous to the radical-triplet pair mechanism. At low temperature, the TREPR data show that the trip-quartet state is formed with strong absorptive net polarization. The shape of the spectrum provides evidence of significant dynamics in the excited state probably due to reversible transitions between the thermally accessible quartet and doublet states.
Graphical abstract

Synopsis: Spectroscopic studies of a phosphorus (V) porphyrin with an axial covalently bound TEMPO radical reveal different relaxation pathways following light excitation. Rapid deactivation of the excited sing-doublet state populates the trip-doublet and trip-quartet states as well as the CT state generated by oxidation of the radical by the excited porphyrin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Supramolecular complexes consisting of one or more chromophores appended with different electron and energy donors and acceptors or paramagnetic species have attracted a tremendous amount of attention over the past several decades for their potential use in solar energy conversion,1,2,3 spintronics,4 quantum information science,5,6 and magnetic resonance methods.7,8 In particular, there have been many studies of the effect of tethered radicals on the excited state dynamics of organic chromophores.9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 In these systems the presence of the additional spin on the radical changes the multiplicity of the states of the chromophore so that the singlet states become sing-doublets (S=1/2) and the triplet states become trip-doublets (S=1/2) and trip-quartets (S=3/2). Decay of the excited sing-doublet to the trip-doublet is spin allowed and promoted by mixing of these two states by the exchange interactions between the three unpaired electrons. In most cases, the trip-quartet state is the lowest excited state and its spin sublevels are selectively populated during an intersystem crossing from the sing-doublet and/or trip-doublet resulting in both net and multiplet polarization.12,15,25 In the majority of these systems, the chromophore has a ring structure with extended π-conjugation and the radical is attached to the chromophore via covalent bonds on the periphery of the ring. However, there are a few examples10,12,16,26 in which the radical is attached by either coordination or covalently to a metal or metalloid center via an axial bond perpendicular to the chromophore plane. The spin polarization patterns obtained with axially bound radicals differ from those obtained when the radical is in the same plane as the chromophore and generally display much stronger net polarization.
Recently, we reported the behavior of an aluminum(III) porphyrin (AlPor) complex in which a TEMPO nitroxide radical was covalently bound via an axial bond to the aluminum(III) center16 to form AlPor-TEMPO. Under illumination in frozen solution, a purely absorptive spin polarization pattern was observed that was assigned to the excited trip-doublet and trip-quartet states that were proposed to undergo rapid equilibration. Aluminum(III) porphyrins have the unusual property of being able to form both axial covalent and coordination bonds. When the Al(III) center is inserted into the free base porphyrin it assumes a five-coordinate structure with an axial halide or hydroxy group.27 The hydroxy group can then be used to couple porphyrin to substituents by ester formation. However, the five-coordinate Al(III) center is also a strong Lewis acid and can be coordinated by Lewis bases such as pyridine and imidazole to form a six-coordinate geometry. In the case of AlPor-TEMPO, coordination of pyridine to the porphyrin caused a marked change in the spin polarization pattern and a transient electron paramagnetic resonance (TREPR) spectrum typical of the lowest excited quartet state with strong multiplet polarization and weak net polarization was observed.16 It was proposed that this change in the polarization was caused by changes in the orbital energies and exchange couplings such that the equilibration between the trip-doublet and trip-quartet states becomes slower in the presence of the coordinated pyridine.
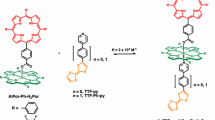
Here, we extend our investigation of the role of axial ligands on the excited state dynamics by studying an analogous phosphorus (V) porphyrin complex. In contrast to Al(III) porphyrins, the phosphorus center forms six-coordinate complexes with two axial covalent bonds.28 As shown in Scheme 1, insertion of trichloromethylphosophorus into octaethylporphyrin (OEP) results in phosphorus(V)octaethylporphyrin (PPor+) with two covalent axial bonds. The chlorine group can then be displaced to generate PPor+-TEMPO. The presence of the covalently bound axial methyl group in PPor+-TEMPO compared to coordinated pyridine in the case of AlPor-TEMPO allows the influence of the bonding of the axial ligand on the spin polarization to be studied. However, an important additional feature of PPor+ is its high oxidation potential compared to other porphyrins.28 This makes the excited state of PPor+ a very strong oxidant29,30,31 and means that excited state decay via electron transfer from TEMPO to PPor+ is also possible as has been proposed to occur in fullerene - nitroxide conjugates.32 We will show that the presence of the bound nitroxide leads to a very short excited-state lifetime in polar acetonitrile. However, relaxation of the molecule into its lowest trip-quartet state is observed in a liquid crystalline solvent at room temperature and in an ethanol glass at low temperature. Similar to the AlPor-TEMPO case, the spin polarization patterns show evidence of spin dynamics in the trip-quartet state that leads to net absorptive polarization and partial averaging of the zero-field splitting.

Synthesis of the investigated PPor+ derivatives compounds. Reaction conditions: (i) CH3PCl3, 2,6-lutidine, dichloromethane, reflux under N2 for 36 h, (ii) Dry methanol, stirred for 2 h under N2, (iii) 4-Hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl, (OH-TEMPO), dry CH2Cl2, stirred for 18 h under N2 at room temperature, and (iv) saturated solution of NH4PF6 in water.
2 Experimental
2.1 General
All chemicals and solvents used in this study were purchased from Sigma-Aldrich or Alfa-Aesar. The OEP and chromatography material (Al2O3 neutral AW 3) were obtained from Sigma-Aldrich. The precursor porphyrin (PPor-Cl.Cl) and reference porphyrin (PPor-OMe.PF6) were synthesized from OEP by established methods.30 Care was taken to avoid the entry of direct, ambient light into the samples in all the spectroscopic and electrochemical experiments. Unless otherwise specified, all the experiments were carried out at 293 ± 3 K.
Synthesis of PPor-TEMPO.PF6: A solution of OEP (100 mg, 0.187 mmol), CH3PCl2 (50 μL, 0.565 mmol) and 2,6-lutidine (100 μL, 0.859 mmol) in dry dichloromethane (10 mL) was refluxed for 36 h under nitrogen. The solvent was evaporated, and the solute was dried under a vacuum. The obtained crude PPor-Cl.Cl was added a solution of OH-TEMPO (100 mg, 0.580 mmol) in dry dichloromethane (10 mL). The resulting solution was stirred at room temperature for 18 h under a nitrogen atmosphere. The solvent was evaporated, and the residue was chromatographed on neutral alumina. The crude product was loaded onto the column using a benzene: acetonitrile (95:5) solvent mixture and eluted with benzene: methanol (95:5) to remove the low-polarity impurities. The polarity was then increased by eluting with benzene: methanol (90:10) to collect the desired compound as the chloride salt. To exchange the counterion, the chloride salt was dissolved in 10 mL of ethanol and precipitated with a saturated aqueous solution of NH4PF6. The precipitate was collected by filtration and washed with water and airdried. At this point, the minor impurities (TEMPO, PPor+-OH) found in the product were separated by silica gel column chromatography using dichloromethane:ethyl acetate (95:5) as the eluent. Yield: 110 mg (66%). MS (FAB): m/z 749.4788 [M-PF6]+ (calcd. 749.4792 for C46H64N5O2P).
2.2 Physical methods
NMR spectroscopy and Mass spectroscopy. NMR spectra were recorded with Bruker Avance 400 MHz spectrometer using CD3CN as the solvent. ESI mass spectra were recorded on a Bruker MicroTOF-III mass spectrometer.
UV-visible absorption and fluorescence spectroscopy. Steady-state UV-visible absorption spectra of PPor-TEMPO.PF6 and its reference compounds in acetonitrile were recorded with a Cary 100 UV-VIS spectrometer. The concentrations of the samples used for these measurements ranged from 10−6 M (porphyrin Soret band) to 10−5 M (Q-bands bands) solutions. Steady-state fluorescence spectra were recorded using a Photon Technologies International Quanta Master 8075-11 spectrofluorometer, equipped with a 75 W xenon lamp, running FelixGX software. An excitation wavelength of 550 nm, exclusively porphyrin, was used and the concentrations were held constant for all the compounds.
Electrochemistry. Cyclic and differential pulse voltammetric experiments (CH3CN, 0.1 M tetrabutylammonium hexafluorophosphate, (TBA.PF6)) were performed on a BAS Epsilon electrochemical analyzer (working electrode: glassy carbon, auxiliary electrodes: Pt wire, reference electrode: Ag wire). The ferrocene couple (E1/2 (Fc+/Fc) = 0.40 V in CH3CN, 0.1M TBA.PF6 under our experimental conditions) was used to calibrate the redox potential values.
Femtosecond transient absorption spectroscopy. Experiments were performed using an Ultrafast Femtosecond Laser Source (Libra) by Coherent incorporating diode-pumped, mode-locked Ti:Sapphire laser (Vitesse) and diode-pumped intracavity doubled Nd:YLF laser (Evolution) to generate a compressed laser output of 1.45 W. For optical detection, a Helios transient absorption spectrometer coupled with a femtosecond harmonics generator both provided by Ultrafast Systems LLC was used. The source for the pump and probe pulses were derived from the fundamental output of Libra (Compressed output 1.45 W, pulse width 100 fs) at a repetition rate of 1 kHz. 95% of the fundamental output of the laser was introduced into a harmonic generator which produces second and third harmonics of 400 and 267 nm besides the fundamental 800 nm for excitation, while the rest of the output was used for the generation of a white light continuum. In the present study, the second harmonic 400 nm excitation pump was used in all the experiments. Kinetic traces at appropriate wavelengths were assembled from the time-resolved spectral data. Data analysis was performed using Surface Xplorer software supplied by Ultrafast Systems. All measurements were conducted in degassed acetonitrile solutions at 298 K.
Transient EPR Spectroscopy. Transient EPR measurements were carried out at room temperature and 80 K using a modified Bruker EPR 200D-SRC X-band spectrometer (Bruker Canada, Milton ON, Canada). The samples were excited at 532 nm using 10 ns pulses from a Nd:YAG laser at a repetition rate of 10 Hz. For the room temperature experiments the samples were measured in the liquid crystal 4-pentyl-4′-cyanobiphenyl (5CB). The partial ordering of the molecules in the liquid crystal prevents complete averaging of the zero-field splitting allowing the contributions from the different spin states to be more easily distinguished from one another. For the low-temperature experiments ethanol was chosen as the solvent to ensure the formation of a glass and to prevent any aggregation during the freezing process.
Computations. Density functional theory computations were carried out using Orca 4.2.1.33,34 The geometry was optimized using the unrestricted Kohn-Sham method with B3LYP functional and the 6-31G(d,p) basis set. The resolution of identity and chain of spheres approximations (RIJCOSX)35 were used during the optimization. The influence of the acetonitrile solvent has been taken into account using the conductor-like polarizable continuum model.36
3 Results and discussion
3.1 Synthesis and characterization
The investigated compound was synthesized by exploiting the reactivity of the axial P–Cl bond of PPor+-Cl. The reaction of PPor-Cl.Cl with OH-TEMPO leads to a pairing of the TEMPO and porphyrin units. After purification and anion exchange, the desired product, PPor-TEMPO.PF6 was obtained in moderate yields. Preliminary characterization of PPor-TEMPO.PF6 was carried out by ESI High Resolution mass spectrometry. The mass spectrum of the dyad showed peak which correspond to the mass (m/z) [M – PF6]+, for details see the experimental section, and Scheme 1.
DFT calculations. Figure 1 shows the frontier orbitals and charge transfer difference density of the complex as predicted by DFT computations. The highest doubly occupied orbital (HDOMO) (Figure 1(a)) and lowest unoccupied orbital (LUMO) (Figure 1(b)) of the complex are localized on the PPor+ while the singly occupied orbital (SOMO) (Figure 1(c)) is localized on the nitroxide. The computations show that there is no significant mixing of the PPor+ and nitroxide frontier orbitals and that the two parts of the molecule can be treated as essentially separate entities. The difference density of the lowest excited state calculated by time-dependent DFT is shown in Figure 1(d). The red regions localized on the nitroxide indicate the loss of electron density while the green regions localized on the porphyrin are an increase in electron density. Thus, the lowest excited state is a charge transfer state in which the nitroxide has been oxidized by the porphyrin. The calculated ground-state energies of the HDOMO, HDOMO–1, LUMO and LUMO+1 in acetonitrile are given in Table 1 along with the energies of the excited doublet and quartet states.

Frontier molecular orbitals and charge transfer state difference density in PPor+-TEMPO. (a) Highest doubly occupied orbital (b) Lowest unoccupied orbital (c) singly occupied orbital (d) CT state density difference. The green surfaces are positive and red surfaces are negative. The orbitals and difference density were calculated using the program Orca33,34 and the surfaces were rendered using Avogadro.38
The calculations also predict that the HDOMO and HDOMO–1 are nearly degenerate with the HOMO–1 lying only 670 cm−1 below the HDOMO. Similarly, the LUMO+1 is only 104 cm−1 above the LUMO. Together the HDOMO–1. HDOMO, LUMO and LUMO+1 form the basis for Gouterman’s four orbital model of porphyrins37 that predicts two degenerate allowed transitions referred to as the Soret or B-bands in the blue of the visible spectrum and two degenerate Q-bands in the red. Energetically, the SOMO is also well-separated from the HDOMO and LUMO lying 0.415 eV above the HDOMO and 2.49 eV below the LUMO.
The state energies obtained from time-dependent DFT calculations are also shown in Table 1. As expected, the lowest excited doublet state is predicted to be the CT state, which is nearly doubly degenerate due to the approximate axial symmetry of the porphyrin and degeneracy of the corresponding orbitals. The CT state is predicted to lie 1.18 eV above the ground state. The excited sing-doublet state involving excitation of the porphyrin is also doubly degenerate and lies 2.3 eV above the ground state, while the lowest quartet state is at 1.85 eV.
UV−visible absorption spectroscopy. The UV−visible spectra of PPor-TEMPO.PF6 and its reference compounds (PPor-OMe.PF6 and OH-TEMPO) were measured in acetonitrile and the spectra are shown in Figure 2(a). The band positions (Q-band and B- or Soret band) and their molar extinction coefficients are summarized in Table 2. As shown in Figure 2(a), the absorption spectrum of the dyad is essentially a linear combination of its reference compounds and the positions and molar extinction coefficients (ε) of the porphyrin bands do not change significantly in the dyad compared to the corresponding monomer porphyrins. Overall, the absorption studies suggest that any ground state interaction between the porphyrin (PPor+) and the linked TEMPO is sufficiently weak that it causes no discernable change in their electronic structures. The absorption bands of PPor+ at 550 and 410 nm have been chosen to excite the PPor+ for steady-state fluorescence and transient absorption studies.

(a) Absorption spectra of PPor-OMe.PF6 (green), OH-TEMPO (red), and PPor-TEMPO.PF6 (purple) in acetonitrile. (b) Fluorescence spectra of PPor-OMe.PF6 (green), physical mixture of OH-TEMPO + PPor-OMe.PF6 (red), and PPor-TEMPO.PF6 (purple) in acetonitrile. Excitation wavelength was used 550 nm. All the sample concentrations maintained at 10 μM.
Electrochemistry and energetics. Cyclic voltammetry measurements of the newly synthesized compounds were performed in CH3CN with 0.1 M tetrabutylammonium hexafluorophosphate (TBA.PF6) as the supporting electrolyte and ferrocene as an internal standard. The cyclic voltammogram of PPor-TEMPO.PF6 and its reference compounds PPor-OMe.PF6 and TEMPO derivative (OH-TEMPO) are shown in Figure 3, and the oxidation and reduction midpoint potentials are summarized in Table 2. The redox processes of all of the compounds are found to be one-electron reversible processes based on the peak-to-peak separation values, and the cathodic-to-anodic peak current ratio. The observed voltammogram of PPor-TEMPO.PF6 is assigned to the combination of PPor-OMe.PF6 and OH-TEMPO derivative. During the cathodic scan, the dyad PPor-TEMPO.PF6 showed two reduction processes, which we assign to the first and second reduction of the PPor+ moiety. The anodic scan of the dyad reveals two oxidation processes, which are assigned to the first oxidation of TEMPO and first oxidation of PPor+ unit. The oxidation potential of the TEMPO is shifted slightly more positive due to the electron withdrawing ability of the PPor+ unit. Comparison of the dyads and their reference compounds shows that the redox processes are the sum of those of the reference compounds PPor-OMe.PF6 and OH-TEMPO, which again indicates that the porphyrin electronic structure is not perturbed by presence of TEMPO.

Left: Cyclic voltamogramas of OH-TEMPO (red), PPor-OMe.PF6 (green), and PPor-TEMPO.PF6 (purple) in acetonitrile with 0.1 M TBA.PF6. Right: Energy level diagram of PPor-TEMPO.PF6 in acetonitrile. The possible electron transfer (ET), internal conversion (IC) and intersystem crossing (ISC) processes are shown. The energies of the sing-doublet, trip-quartet and trip-doublet states (2.09 eV and 1.65 eV) are the literature values29 of the PPor+ excited singlet and triplet states and do not include exchange interaction between the nitroxide and the porphyrin.
The redox potentials can be used in combination with optical data to estimate the energetics of the electron transfer reactions in the PPor-TEMPO.PF6. The energy of the charge-separated state ECS (relative to the ground state) and the free-energy changes for charge-separation (ΔGCS) can be calculated using the Rehm and Weller equations. Because the magnitude of the exchange interaction between the nitroxide and the porphyrin is difficult to determine experimentally, the energies of the sing-doublet, trip-doublet and trip-quartet states have been taken as the literature values of first excited singlet-state and triplet-state energies (E0-0 = 2.09 eV and Etriplet =1.65 eV)29 of PPor+. The ECS is the energy of the charge-separated state and is the difference between the first oxidation potential of the donor TEMPO and first reduction potential of the acceptor PPor+ units.
where RD-A is the center-to-center distance between the donor (TEMPO) and the acceptor (PPor+), and has been taken as the distance between the phosphorus atom and the center of the nitroxide N–O bond (6.35 Å) using the optimized geometry from the DFT calculations. εS is the dielectric constant of the solvent used for the photophysical and redox potential measurements, in this case acetonitrile. The Gs value in acetonitrile was found to be −0.0605 eV. Using these equations, the energy of the charge-separated state PPor·-TEMPO+ is found to be 1.53 eV, which lies well below the excited sing-doublet, trip-doublet and trip-quartet state energies showing that PPor+ is energetically well suited to act as a photosensitizer for electron transfer from TEMPO to the 1(PPor+)* unit. With the above information, the energy level diagram was constructed as shown in Figure 3.
Qualitatively, the state energies predicted by the DFT calculations (Table 1) agree fairly well with the experimental values, although the energy difference between the trip-quartet state and CT state is significantly overestimated in the calculations.
Fluorescence Spectroscopy. The steady-state fluorescence spectra of PPor-TEMPO.PF6 and its reference compound PPor-OMe.PF6 in acetonitrile are shown in Figure 2(b). The spectra have been measured with excitation at 550 nm, where the absorption is exclusively due to PPor+. The spectral shapes and the emission maxima of the dyad are essentially the same as its reference compound, see Table 1. However, the fluorescence of the dyads is strongly quenched (87%) compared to that of the reference compound. The observed fluorescence quenching could be assigned to two possible intramolecular quenching mechanisms. One of them would be reductive electron transfer from TEMPO to the singlet excited state of the PPor+ (1(PPor+)*) as the anticipated charge-separated state is well placed with sufficient driving force (ΔGCS = −0.56 eV). The second possibility would be fast internal conversion (IC) to the trip-doublet state in the presence of the free-radical TEMPO. Additionally, intermolecular quenching was also tested by measuring the fluorescence spectra of PPor-OMe.PF6 and the 1: 1 physical mixture PPor-OMe.PF6 and OH-TEMPO, see Figure 2(b). As shown the fluorescence intensity is identical to the reference compound, which rules out the intermolecular mechanism. The exchange coupling between the unpaired electron on TEMPO and the electrons in the HDOMO and LUMO of the porphyrin splits the 3(PPor+)* state into trip-doublet and trip-quartet states as shown in Figure 3. The relative energies of the trip-doublet and trip-quartet is determined by the exchange coupling between the triplet and doublet spins and the trip-quartet is expected to be lower in energy than the trip-doublet. If the coupling between the nitroxide spin and the two unpaired electrons on the porphyrin is different the sing-doublet state becomes mixed with the trip-doublet state and fast relaxation occurs.39 The trip-doublet can then relax by spin-orbit coupling mediated intersystem crossing (ISC) to the trip quartet or the ground state. The trip-doublet state may also decay further to the charge separated state, but this process is spin forbidden from the trip-quartet state. Thus, the relative populations of the trip-quartet and CT states depend on the relative rates of the relaxation processes shown in Figure 3.
4 Time-resolved optical spectroscopy
Figure 4 shows pump-probe measurements of PPor+-OMe in two different time regions. Figures 4(a) and 4(b) show the absorbance changes in the picosecond to nanosecond time range, Figures 4(c) and 4(d) show corresponding data in the microsecond range. In both time regimes the negative peaks at 549 and 590 nm are due to the bleaching of the Q-bands when the molecule is excited. The weaker negative peak at ~640 nm arises from the porphyrin fluorescence. The strong absorbance increase band at ~440 nm and the weak broad band between 1100 and 1300 nm are due to excited state absorbance. The upper time trace is the absorbance change at 1185 nm and represents the excited singlet state absorbance that decays with a lifetime of 5.25 ns31 as expected for the excited singlet state of a porphyrin.

Transient absorption spectra of PPor-OMe.PF6 in acetonitrile. (a) Femtosecond absorbance changes at the indicated times. (b) Time dependent absorbance change at 1185 nm. (c) Nanosecond absorbance changes at the indicated times (d) Time dependent absorbance change at 465 nm.
In Figure 4(c) the absorption difference spectrum in the microsecond time region is shown. In this time range, the bleaching of the Q-bands is still observed but the negative fluorescence band at ~640 nm seen at an early time is no longer present. Instead, a broad absorption increase band between ~640 nm and 800 nm from the excited triplet state absorbance is seen. The time trace in the bottom right of Figure 4 shows the decay of the absorbance difference band at 465 nm. The red curve is a fit of the data with three exponentials with lifetimes of 8, 20 and 37 µs. Thus, as expected the data shows that in PPor+-OMe excitation leads to decay of the excited singlet state via fluorescence and intersystem crossing (ISC). The ISC lifetime is on the order of a few ns and the triplet state has a lifetime of a few tens of microseconds.
Figure 5 shows corresponding absorbance changes for PPor+-TEMPO in the picosecond time regime. These spectra reveal the significant impact that the presence of nitroxide has on the excited state dynamics. First, all of the absorption changes including the Q-band bleaching peaks decay nearly to zero within the 3 ns time window of the experiment. Thus, the overall excited state lifetime is much shorter. Second, the emissive band at ~640 nm due to the porphyrin fluorescence is barely visible underneath the excited state absorbance, again showing that the excited singlet state lifetime is much shorter. The decrease in the excited state lifetime could be due to the presence of the unpaired electron spin on the nitroxide and/or electron transfer from the nitroxide to the excited porphyrin. To better understand what lead to the absorbance changes that can be expected from the electron transfer, spectroelectrochemistry measurements of the reduction of PPor+-OMe are useful. Although the charge-transfer state of PPor+-TEMPO differs from the reduced form of the porphyrin, the spectra of the two species are expected to be similar. The UV/Vis spectra of PPor+-OMe as it undergoes its first reduction has been reported previously31 and show that the reduction leads to the loss of the Soret band at 416 nm and the two Q-bands at 549 and 590 nm, while new bands at 447 nm and 750 nm appear. Thus, we can expect to observe an increase in the absorbance at these two wavelengths in pump-probe measurements of PPor+-TEMPO if the CT is being formed. In the spectra of the reference compound in Figure 3, there are also absorbance increase bands at both of these wavelengths, thus it is difficult to distinguish between the porphyrin excited states and the reduced porphyrin. Nonetheless, the rapid decay of the PPor+-TEMPO absorbance changes and the observed absorbance increases around 450 nm and 750 nm suggest that the charge transfer state may be involved.

Femtosecond transient absorbance spectra of PPor-TEMPO.PF6 in acetonitrile.
5 Transient electron paramagnetic resonance spectroscopy (TREPR)
Figure 6 shows a comparison of the room temperature transient EPR spectra of PPor+-OMe (blue spectra), an equimolar mixture of PPor+-OMe and OH-TEMPO (red spectra) and PPor+-TEMPO (black spectra) in the liquid crystal 5CB. In Figure 6(a), the top spectra were extracted from the time/field dataset in a time window centred at 200 ns after the laser flash, while the bottom spectra are at 750 ns. The early spectra of PPor+-OMe and the physical mixture of PPor+-OMe and OH-TEMPO are identical and show an emission (E)/absorption (A) polarization pattern and arises from the triplet state of the porphyrin. In the nematic phase of the liquid crystal, the molecules undergo fast anisotropic tumbling and hence, the zero-field splitting of the triplet state is not averaged to zero but a non-zero value that is determined by the order parameters of the porphyrin. As is apparent in Figure 6(a) (top), the emissive peak at ~337 mT is more intense than the absorptive peak at ~357 mT. This net polarization of the triplet state probably arises from its dynamics. The porphyrin has two nearly degenerate excited triplet states associated with the Qx and Qy absorption bands. The calculated orbital and state energies of PPor+ (not shown), suggest that these two states are separated by an energy difference of a few hundred wavenumbers, hence some equilibration between them can be expected at room temperature. We have shown recently that such transitions lead to net polarization of the triplet state.40 However, the effect of the equilibration of the two triplet states is also similar to that caused by anisotropic motion, so that it is possible that the molecular motion may contribute to the net polarization.

Room temperature TREPR spectra in the liquid crystal 5CB at different delay times. (a): PPor+-OMe (blue) and an equimolar mixture of PPor+-OMe and OH-TEMPO (red). The top and bottom spectra are at 200 ns and 750 ns after the laser flash. (b): Corresponding spectra of PPor+-TEMPO at 70 ns and 750 ns after the laser flash.
At 750 ns after the laser flash, the spectrum of PPor+-OMe (Figure 6(a), blue spectrum, bottom) retains the same polarization pattern but the intensity of the spectrum decays as a result of equalization of the populations of the spin sublevels due spin-lattice (T1) relaxation. In contrast, in the presence of OH-TEMPO, strong emissive features from the nitroxide, appear in the middle of the spectrum and the polarization of the triplet state changes. The emissive polarization of the nitroxide occurs as a result of collisions between the nitroxide and triplet state and can be explained with the radical-triplet pair mechanism (RTPM).26,41,42,43,44,45 The change in the polarization of the high-field part of the triplet spectrum is due to the depopulation of the triplet sublevels that interact most strongly with the radical. The hyperfine component of the nitroxide at the highest field is also much more intense than the other two peaks. This is partly due to the narrower linewidth of the peak as a result of incomplete anisotropic motional averaging. However, it also appears to have stronger spin polarization. This implies that the doublet-triplet mixing that leads to polarization of the radical may be dependent on the orientation of the nitroxide nitrogen nuclear spin.
Figure 6(b) shows corresponding spectra for PPor+-TEMPO at 70 ns (upper black spectrum) and 750 ns (lower black spectrum) after the laser flash. The early spectrum shows broad absorptive features about 20 mT wide centred around a narrow absorptive peak. The width of the broad features is too large to be associated with any of the possible doublet states of PPor+-TEMPO and we assign them to the ±3/2 ↔ ±1/2 transitions of the lowest trip-quartet state. The narrow peak in the centre of the spectrum is assigned to the +1/2 ↔ −1/2 transition of the quartet state. However, it could also potentially contain contributions from the trip-doublet and ground doublet and charge-transfer doublet states. At a late time, an emissive signal from the ground-state is observed. We postulate that the emissive polarization is generated by collisions between molecules in the excited quartet state and ground state in analogy to the polarization generated by collisions between the triplet and doublet states in the physical mixture. The linewidths of the peaks in the ground state spectrum are broader than in the physical mixture as a result of slower tumbling of the molecule due to its larger size. The relative heights of the three hyperfine components are also different than in the physical mixture. This is at least partly due to the fact that the ordering and rotational correlation time of PPor+-TEMPO in the liquid crystal environment is different than that of OH-TEMPO. It is also possible that the polarization of the nitroxide shows different selectivity for the hyperfine components than in the physical mixture. However, the most important observation is that the trip-quartet state of PPor+-TEMPO decays with a lifetime on the order of a few hundred ns and only the ground state is observed at a longer time.
Figure 7 shows a comparison of the low-temperature TREPR spectra of PPor+-TEMPO and PPor+-OMe in ethanol glass at 80 K. The spectrum of PPor+-OMe is typical of the triplet state of a porphyrin populated by spin-orbit coupling mediated intersystem crossing and is essentially identical to the spectrum reported recently in 2-methyltetrahydrofuran.46 The spectrum of PPor+-TEMPO is purely absorptive and is significantly narrower than that of the PPor+-OMe. We assign the spectrum to the trip-quartet state of the complex based on its width and general shape. Intersystem crossing to the trip-quartet state of coupled quartet-doublet systems generally leads to a polarization pattern in which the wings of the spectrum show predominant multiplet polarization (E/A or A/E) with some net polarization that results in an absorptive or emissive central peak.10,11,12,15,18 However, in cases in which there are nearby quartet and doublet states that are thermally accessible and/or the exchange interaction between the triplet and doublet states is comparable to the Zeeman energy net polarization can develop.16,25,40,47,48 Here, we postulate that equilibration between the low-lying trip-doublet and trip-quartet states leads to partial averaging of the zero-field splitting tensor and generates the observed predominant absorptive polarization.

Low temperature TREPR spectra PPor+-TEMPO (black spectrum) and PPor+-OMe (red spectrum) in ethanol glass at 80 K. The spectra were extracted from the time/field datasets in a time window centred at 800 ns after the laser flash.
6 Conclusions
The fast decay observed in the femtosecond transient absorbance data suggests that in acetonitrile the sing-doublet state may decay by electron transfer from the nitroxide to PPor+. This process is not spin selective. Thus, the CT state should not contribute to the TREPR signals if it is populated via this pathway. However, the optical data do not provide unambiguous evidence for ET from the sing-doublet and further studies are in progress to investigate whether it occurs or not. The broad wings of the room temperature TREPR spectra show clear evidence for the formation of the quartet state. Taken together with the observed fluorescence quenching it suggests that IC to the trip-doublet state and ISC to the trip-quartet state compete favourably with ET. This is somewhat surprising since the mixing of the sing-doublet and trip-doublet states induced by the exchange coupling between the triplet and doublet spins should be relatively weak and therefore is not expected to have a strong influence on the depopulation of the excited sing-doublet state.
In contrast to ET from the sing-doublet, decay of the trip-doublet to the CT state could generate spin polarization and would retain any polarization of the trip-doublet state.32 Thus, it may contribute to the absorptive central peak in the TREPR spectrum of PPor+-TEMPO observed at an early time (Figure 6(b)). In addition, any polarization of the CT state should be transferred to the ground state during reverse ET. At room temperature, it is possible that some of the ground state polarization observed at late times is generated in this way. At low temperature (Figure 7) the spectrum does not show any features that can be assigned to the ground state. Since the trip-quartet state displays strong net polarization and the trip-doublet state can probably be thermally populated from the trip-quartet, decay to the ground state either directly from the trip-quartet or via the trip-doublet and/or CT state appears to be slow.
The very strong absorptive net polarization and the broadening of the wings of the low-temperature trip-quartet state spectrum suggest that the excited state undergoes rapid dynamics. The near degeneracy of the two lowest porphyrin triplet states means that transitions between them can occur and depending on how large the exchange coupling is between the triplet and doublet spins the trip-doublet may also be thermally accessible from the trip-quartet. This creates a rather complex situation in which four (partially) averaged spectra can contribute to the observed signal.
To better understand the excited state dynamics in this system we are extending these studies to investigate the dependence of the polarization on the length of the bridge between porphyrin and the nitroxide.
References
Maddah HA, Berry V and Behura SK 2020 Biomolecular photosensitizers for dye-sensitized solar cells: Recent developments and critical insights Renew. Sustain. Energy Rev. 121 109678
Fukuzumi S, Lee Y-M and Nam W 2018 Mimicry and functions of photosynthetic reaction centers Biochem. Soc. Trans. 46 1279
Mora SJ, Odella E, Moore GF, Gust D, Moore TA and Moore AL 2018 Proton-Coupled Electron Transfer in Artificial Photosynthetic Systems Acc. Chem. Res. 51 445
Sanvito S 2011 Molecular spintronics Chem. Soc. Rev. 40 3336
Bruss D and Leuchs G 2019 Quantum Information: From Foundations to Quantum Technology Applications (Weinheim, Germany: John Wiley)
Rugg BK, Krzyaniak MD, Phelan BT, Ratner MA, Young RM and Wasielewski MR 2019 Photodriven quantum teleportation of an electron spin state in a covalent donor–acceptor–radical system Nat. Chem. 11 981
Tripathi A and Rane V 2019 Toward Achieving the Theoretical Limit of Electron Spin Polarization in Covalently Linked Radical-Chromophore Dyads J. Phys. Chem. B 123 6830
Liu G, Liou S-H, Enkin N, Tkach I and Bennati M 2017 Photo-induced radical polarization and liquid-state dynamic nuclear polarization using fullerene nitroxide derivatives Phys. Chem. Chem. Phys. 19 31823
Fujisawa J, Ishii K, Ohba Y, Iwaizumi M and Yamauchi S 1995 Electron spin polarization transfer from excited triplet porphyrins to a nitroxide radical via a spin exchange mechanism J. Phys. Chem. 99 17082
Ishii K, Hirose Y, Fujitsuka H, Ito O and Kobayashi N 2001 Time-resolved EPR, fluorescence, and transient absorption studies on phthalocyaninatosilicon covalently linked to one or two TEMPO radicals J. Am. Chem. Soc. 123 702
Ishii K, Ishizaki T and Kobayashi N 1999 Experimental evidence for a selection rule of intersystem crossing to the excited quartet states: Metallophthalocyanines coordinated by 4-amino-TEMPO J. Phys. Chem. A 103 6060
Tarasov VF, Islam SS, Ohba Y, Forbes MD and Yamauchi S 2011 Multifrequency TREPR Investigation of Excited-State ZnTPP/Nitroxide Radical Complexes Appl. Magn. Reson. 41 175
Chernick ET, Casillas R, Zirzlmeier J, Gardner DM, Gruber M, Kropp H, et al. 2015 Pentacene appended to a TEMPO stable free radical: the effect of magnetic exchange coupling on photoexcited pentacene J. Am. Chem. Soc. 137 857
Teki Y 2020 Excited-State Dynamics of Non-Luminescent and Luminescent pi-Radicals Chem.- Eur. J. 26 980
Kandrashkin Y and van der Est A 2003 Light-induced electron spin polarization in rigidly linked, strongly coupled triplet–doublet spin pairs Chem. Phys. Lett. 379 574
Poddutoori PK, Kandrashkin YE, Karr P and van der Est A 2019 Electron spin polarization in an Al (III) porphyrin complex with an axially bound nitroxide radical J. Chem. Phys. 151 204303
Moons H, Goovaerts E, Gubskaya VP, Nuretdinov IA, Corvaja C and Franco L 2011 W-band transient EPR and photoinduced absorption on spin-labeled fullerene derivatives Phys. Chem. Chem. Phys. 13 3942
Poddutoori PK, Pilkington M, Alberola A, Polo V, Warren JE and van der Est A 2010 Spin− Spin Interactions in Porphyrin-Based Monoverdazyl Radical Hybrid Spin Systems Inorg. Chem. 49 3516
Teki Y, Miyamoto S, Iimura K, Nakatsuji M and Miura Y 2000 Intramolecular spin alignment utilizing the excited molecular field between the triplet (S= 1) excited state and the dangling stable radicals (S= 1/2) as studied by time-resolved electron spin resonance: observation of the excited quartet (S= 3/2) and quintet (S= 2) states on the purely organic π-conjugated spin systems J. Am. Chem. Soc. 122 984
Teki Y, Kimura M, Narimatsu S, Ohara K and Mukai K 2004 Excited High-Spin Quartet (S= 3/2) State of a Novel π-Conjugated Organic Spin System, Pyrene-Verdazyl Radical Bull. Chem. Soc. Jpn. 77 95
Takemoto Y and Teki Y 2011 Unique Dynamic Electron-Spin Polarization and Spin Dynamics in the Photoexcited Quartet High-Spin State of an Acceptor-Donor-Radical Triad ChemPhysChem 12 104
Nolden O, Fleck N, Lorenzo ER, Wasielewski MR, Schiemann O, Gilch P and Richert S 2020 Excitation energy transfer and exchange-mediated quartet state formation in porphyrin-trityl systems Chem. Eur. J. 27 2683
Zhang X, Sukhanov AA, Yildiz EA, Kandrashkin YE, Zhao J, Yaglioglu HG and Voronkova VK 2021 Radical-Enhanced Intersystem Crossing in a Bay-Substituted Perylene Bisimide- TEMPO Dyad and the Electron Spin Polarization Dynamics upon Photoexcitation ChemPhysChem 22 55
Giacobbe EM, Mi Q, Colvin MT, Cohen B, Ramanan C, Scott AM, et al. 2009 Ultrafast intersystem crossing and spin dynamics of photoexcited perylene-3, 4: 9, 10-bis (dicarboximide) covalently linked to a nitroxide radical at fixed distances J. Am. Chem. Soc. 131 3700
Kandrashkin YE and van der Est A 2019 The triplet mechanism of electron spin polarization in moderately coupled triplet-doublet rigid complexes as a source of the enhanced +1/2 ↔ −1/2 transitions J. Chem. Phys. 151 184301
Fujisawa J, Ohba Y and Yamauchi S 1997 Electron-spin polarizations generated from interactions between excited triplet porphyrins and stable radicals studied by time-resolved electron paramagnetic resonance J. Phys. Chem. A 101 434
Furhop JH, Kadish KM and Davis DG 1973 Redox behavior of metallo oxtaethylporhyrins J. Am. Chem. Soc. 95 5140
Akiba K, Nadano R, Satoh W, Yamamoto Y, Nagase S, Ou Z, et al. 2001 Synthesis, structure, electrochemistry, and spectroelectrochemistry of hypervalent Phosphorus (V) octaethylporphyrins and theoretical analysis of the nature of the PO bond in P (OEP)(CH2CH3)(O) Inorg. Chem. 40 5553
Poddutoori PK, Thomsen JM, Milot RL, Sheehan SW, Negre CF, Garapati VK, et al. 2015 Interfacial electron transfer in photoanodes based on phosphorus (v) porphyrin sensitizers co-deposited on SnO 2 with the Ir (III) Cp* water oxidation precatalyst J. Mater. Chem. A 3 3868
Poddutoori PK, Dion A, Yang S, Pilkington M, Wallis JD and van der Est A 2010 Light-induced hole transfer in a hypervalent phosphorus (V) octaethylporphyrin bearing an axially linked bis (ethylenedithio) tetrathiafulvalene J. Porphyr. Phthalocy. 14 178
Poddutoori PK, Lim GN, Pilkington M, D’Souza F and van der Est A 2016 Phosphorus (V) Porphyrin-Manganese (II) Terpyridine Conjugates: Synthesis, Spectroscopy, and Photo-Oxidation Studies on a SnO2 Surface Inorg. Chem. 55 11383
Rozenshtein V, Berg A, Stavitski E, Levanon H, Franco L and Corvaja C 2005 Electron spin polarization of functionalized fullerenes. Reversed quartet mechanism J. Phys. Chem. A 109 11144
Neese F 2012 The ORCA program system Wiley Interdiscip. Rev. Comput. Mol. Sci. 2 73
Neese F 2018 Software update: the ORCA program system, version 4.0 Wiley Interdiscip. Rev. Comput. Mol. Sci. 8 e1327
Izsák R and Neese F 2013 Speeding up spin-component-scaled third-order pertubation theory with the chain of spheres approximation: the COSX-SCS-MP3 method Mol. Phys. 111 1190
Barone V and Cossi M 1998 Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model J. Phys. Chem. A 102 1995
Gouterman M, Wagnière GH and Snyder LC 1963 Spectra of porphyrins: Part II. Four orbital model J. Mol. Spectrosc. 11 108
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E and Hutchison GR 2012 Avogadro: an advanced semantic chemical editor, visualization, and analysis platform J. Cheminformat. 4 17
Ake RL and Gouterman M 1969 Porphyrins XIV. Theory for the luminescent state in VO Co, Cu complexes Theor. Chim. Acta 15 20
Kandrashkin YE, Di Valentin M and van der Est A 2020 Reversible triplet energy hopping in photo-excited molecules: A two-site model for the spin polarization J. Chem. Phys. 153 094304
Blattler C, Jent F and Paul H 1990 A Novel Radical-Triplet Pair Mechanism for Chemically-Induced Electron Polarization (CIDEP) of Free-Radicals in Solution Chem. Phys. Lett. 166 375
Blättler C and Paul H 1991 CIDEP after laser flash irradiation of benzil in 2-propanol. Electron spin polarization by the radical-triplet pair mechanism Res. Chem. Intermed. 16 201
Kobori Y, Takeda K, Tsuji K, Kawai A and Obi K 1998 Exchange interaction in radical-triplet pairs: Evidences for CIDEP generation by level crossings in triplet-doublet interactions J. Phys. Chem. A 102 5160
Kawai A, Watanabe Y and Shibuya K 2002 Time resolved ESR study on energy difference of quartet and doublet states in radical-triplet encounter pairs Mol. Phys. 100 1225
Shushin AI 1993 The relaxational mechanism of net CIDEP generation in triplet—radical quenching Chem. Phys. Lett. 208 173
Zarrabi N, Bayard BJ, Seetharaman S, Holzer N, Karr P, Ciuti S, et al. 2021 A charge transfer state induced by strong exciton coupling in a cofacial μ-oxo-bridged porphyrin heterodimer Phys. Chem. Chem. Phys. 23 960
Kandrashkin YE, Asano MS and van der Est A 2006 Light-induced electron spin polarization in vanadyl octaethylporphyrin: I. Characterization of the excited quartet state J. Phys. Chem. A 110 9607
Kandrashkin YE, Asano MS and van der Est A 2006 Light-induced electron spin polarization in vanadyl octaethylporphyrin: II. Dynamics of the excited states J. Phys. Chem. A 110 9617
Acknowledgements
This work was supported by the Grant-in-Aid program from the University of Minnesota Duluth to PPK, the Natural Sciences and Engineering Research Council Canada (Discovery Grant 2015-04021 to AvdE) and the National Science Foundation (Grant No. 2000988 to FD).
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue on Beyond Classical Chemistry
Rights and permissions
About this article

Cite this article
PODDUTOORI, P.K., HOLZER, N., BAYARD, B.J. et al. Excited state dynamics and electron transfer in a phosphorus(V) porphyrin – TEMPO conjugate. J Chem Sci 133, 65 (2021). https://doi.org/10.1007/s12039-021-01925-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-021-01925-2