
Abstract
Two mononuclear zinc(II) complexes of newly designed carboxamide derivatives, formulated as [Zn(L1)3](ClO4)2 (1) and [Zn(L2)3](ClO4)2 (2) [where L 1 = N-(furan-2-ylmethyl)-2-pyridinecarboxamide and L 2 = N-(thiophen-2-ylmethyl)-2-pyridine-carboxamide], have been isolated in pure form in the reaction of perchlorate salts of Zn(II) with ligands L 1 and L 2, respectively. The two complexes were characterized by physicochemical and spectroscopic tools, and by X-ray crystal structures of both ligands and the complex 1. In complex 1, zinc(II) is chelated by three ligands with a distorted octahedral geometry. The DNA-binding properties of zinc complexes 1 and 2 have been investigated by spectroscopic methods and viscosity measurements. The results suggest that both complexes 1 and 2 bind to DNA in an intercalation mode between the uncoordinated furan or thiophene chromophore and the base pairs of DNA.

Two distorted octahedral mononuclear zinc(II) complexes of newly designed carboxamide ligands bind to calf thymus DNA in an intercalation mode.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Carboxamide derivatives with nitrogen and oxygen as electron donor sites are effective chelating agents that can form complexes with a variety of metal ions. Recently, bi-, tri-, tetra- and hexapodal amide type ligands have been reported.[1–3] Such ligands, which are flexible in structure, can encapsulate the metal ion effectively shielding it from interactions with the surrounding. Nowadays there has been a growing interest in the transition metal complexes bearing amide type ligands due to their biochemical, photoluminescent, nonlinear optical and molecular recognition properties.[4–6] The interest in the coordination chemistry of transition metal complexes arises in part from the fact that they mimic the ligation of certain biomolecules in proteins. Zinc plays a significant role in medicinal, chemical, and biological events and it is the second most abundant transition metal next to iron. Zinc ions in the body (human beings contain an average of ∼2–3 g) play an essential role in many cellular processes,[7] including gene expression,[8] apoptosis,[9] enzyme regulation,[10] and neurotransmission.[11] Thus, there is no surprise that zinc complexes have attracted a considerable interest as synthetic structural imitators of the active site of a range of zinc(II) containing metalloenzymes, such as phosphatases[12,13] and aminopeptidases.[14]
In the last few years, a good amount of research works[15,16] have been focused on the interaction of small molecules with DNA to explore the role of the new complexes as anticancer drugs. So, the interaction between small molecules with DNA can cause damage in cells, blocking the division of cancer cells leading to apoptosis.[17] Small molecules can interact with DNA through two non-covalent modes: intercalation and groove binding. Among these interactions, the intercalation binding is one of the most relevant DNA-binding mode as this mode can be correlated with a possible antitumor activity of the compound.
Considering the above facts, herein we report the synthesis, structural characterization of two newly designed ligands (L 1 and L 2) and their corresponding zinc(II) complexes (1, 2) (scheme 1). The characterisation of the zinc(II) complexes and the carboxamide ligands have been carried out using physicochemical and spectroscopic tools along with the detailed structural analyses of both ligands and complex 1 by single crystal X-ray crystallography. The interaction of the complexes with calf thymus-DNA (CT-DNA) in intercalative mode has also been studied using by spectroscopic methods and viscosity measurements.

Synthetic procedure for the ligands and of the corresponding complexes.
2 Experimental
2.1 Materials and physical measurements
Reagent grade pyridine-2-carboxylic acid, furfuryl amine, 2-thiophenemethyl amine and zinc salts Zn(ClO 4)2.6H 2O were purchased from Sigma-Aldrich and used as received. Calf thymus-DNA was obtained from Bangalore Genie, India and ethidium bromide (EB) from Sigma were used as received. All other chemicals and solvents were of analytical grade.
The elemental (C, H and N) analyses were performed on a Perkin Elmer model 2400 elemental analyzer. IR spectra of 1 and 2 were recorded on a Perkin-Elmer FTIR model RX1 spectrometer (KBr disc, 4000-300 cm −1). Solution conductivities were measured using Systronics Conductivity Meter 304 model. 1H NMR spectra were recorded on a Bruker Avance DPX 500 MHz spectrometer using DMSO-d6 solution. The electronic absorption spectra were recorded on a JASCO UV-Vis/NIR spectrophotometer model V-570. The fluorescence spectra were obtained in the Hitachi-4500 fluorimeter at an excitation wavelength of 522 nm.
2.2 Preparation of ligands L 1 and L 2
The ligands L 1 and L 2 have been prepared following a common synthetic procedure. To a pyridine solution of pyridine-2-carboxylic acid (10 mmol, 1.231 g), furfurylamine (0.9712 g, 10 mmol) for L 1 or 2-thiophenemethyl amine (1.1312 g, 10 mmol) for L 2 was added at stirring condition. Then tri-phenylphosphite (3.101 g, 10 mmol) was added to this mixture and allowed to heat in oil bath at 80 ∘C for about 6 h (scheme 1). The resulting orange coloured solution was kept to evaporate for a few days to collect a white crystalline solid, after washing by methanol and water thoroughly. The crystalline material was dried in vacuo over silica gel was used for characterization by using physico-chemical and spectroscopic tools. Single crystals of L 1 and L 2 suitable for X-ray diffraction study were obtained by slow evaporation from an ethanolic solution.
L 1: Yield: 80-82%. C11H10N2O2: M.p. (∘C): 99 ± 2. Anal. Found: C, 65.21; H, 5.07; N, 13.98%; Calc.: C, 65.34; H, 4.98; N, 13.85%. IR (cm −1): ν C=N 1463, ν C=O1660, ν N−H 3344. 1H NMR (δ, ppm in dmso-d 6): 8.95 (d, 1H); 8.40-8.19 (m, 2H); 8.05-7.82 (m, 2H); 7.39 (m, 1H); 6.06-6.19 (m, 2H); 4.15 (s, 2H of CH 2).
L 2: Yield: 82-85%. C11H10N2OS: M.p. (∘C): 90 ± 2. Anal. Found: C, 60.39; H, 4.69; N, 12.93%; Calc.: C, 60.53; H, 4.62; N, 12.83%. IR (cm −1): ν C=N, 1462, ν C=O,1654, ν N−H 3317. 1H NMR (δ, ppm in dmso-d 6): 8.91 (d, 1H); 8.40-8.20 (m, 2H); 8.03-7.81 (m, 2H); 6.61-6.93 (m, 3H); 4.17 (s, 2H of CH 2).
2.3 Synthesis of zinc(II) complexes 1 and 2
Complexes 1 and 2 were prepared following a general procedure. The ligand (2.0 mmol, 608 mg of L 1 or 654 mg of L 2) was dissolved in dry ethanol by stirring the mixture for 15 minutes. To each of these solutions an ethanolic solution of zinc perchlorate hexahydrate (Zn(ClO4)2.6H2O, 373 mg, 1.0 mmol) was added while stirring and then refluxed for 4 h (scheme 1). The resulting mixture was filtered off and the clear filtrate was collected and kept aside at room temperature. The volume of the solutions was reduced by slow evaporation. The product was collected by washing with cold ethanol and water; and dried in vacuo. Pure crystalline product was obtained from ethanol. The crystallised material so obtained was used for elemental analysis, and for characterization using physico-chemical and spectroscopic tools. Single crystals of 1 suitable for X-ray diffraction study were obtained by slow evaporation of the ethanolic solution.
[Zn(L 1 ) 3 ](ClO 4 ) 2 : Yield: 60–65% (0.520 g); C 33 H 30Cl 2 N 6 O 14Zn: Anal. Found: C, 45.42; H, 3.20; N, 9.86%; Calc.: C, 45.63; H, 3.11; N, 9.63%. Conductivity (Λo, Ω−1.cm 2.mol −1) in MeOH: 202. IR (cm −1): ν C=N, 1476, \(\nu _{\mathrm {ClO4}}^{\mathrm {-}}\)1095, 626, ν C=O1639.
[Zn(L 2 ) 3 ](ClO 4 ) 2 : Yield: 60–65% (0.520 g); C 33 H 30Cl 2 N 6 O 11 S 3Zn: Anal. Found: C, 43.01; H, 3.38; N, 9.27%; Calc.: C, 43.12; H, 3.29; N, 9.1%. Conductivity (Ω−1.cm 2.mol −1) in MeOH: 195. IR (cm −1): ν C=N, 1476, \(\nu _{\mathrm {ClO4}}^{\mathrm {-}}\)1087, 628, ν C=O1639.
2.4 X-ray Crystallography
X-ray single crystal data of L 1, L 2 and complex 1 were collected at room temperature using Mo-Kα (λ = 0.71073 Å) radiation on a Bruker Smart Apex II diffractometer equipped with CCD area detector. Intensity data collection and reduction were carried out using the Bruker Smart Apex and Bruker Saint packages.[18] The structures were solved by direct methods using SHELXS-97 (Sheldrick, 2008) and refined by full-matrix least squares refinement method based on F 2, using SHELXL-97.[19] Crystallographic data and selected bond lengths and angles are tabulated in tables 1 and 2. In both the crystallographic independent molecules of L 2 the thiophene ring was found disordered over two coplanar orientations (with refined occupancies of 0.56/0.44 and 0.69/0.31) rotated by 180 ∘ about the C7-C8 and C18-C19 bond. All non-hydrogen atoms were refined anisotropically except for the disordered C atoms at lower occupancy in L 2. All calculations were performed using Wingx package.[20]
2.5 DNA binding experiments
Tris-HCl buffer solution was used in all the experiments involving CT-DNA. This tris–HCl buffer (pH 7.2) was prepared using deionised and sonicated HPLC grade water (Merck). The CT-DNA used in the experiments was sufficiently free from protein as the ratio of UV absorbance of the solutions of DNA in Tris-HCl at 260 and 280 nm (A 260/A 280) was almost ≈1.9.[21] The concentration of DNA was estimated with the help of the extinction coefficient (6600 M −1 cm −1) of DNA solution at 261 nm[22] and stock solution of DNA was always stored at 4 ∘C. Stock solution of the complex was prepared by adding 2 mL DMSO for dissolving and it was diluted with Tris-HCl buffer to get the required concentration for all the experiments. Absorption spectral titration experiment was performed by keeping the concentration of the complex constant and varying the CT-DNA concentration. To eliminate the absorbance of DNA itself, equal solution of CT-DNA was added to the reference solution. In the ethidium bromide (EB) fluorescence displacement experiment, 5.0 μL of the EB in Tris-HCl solution (1.0 mmol L −1) were added to 1.0 mL of DNA solution (at saturated binding levels),[23] and stored in the dark for 2.0 h. Then the solution of the complexes 1 and 2 were titrated into the DNA/EB mixture and diluted with Tris-HCl buffer to 5.0 mL to get the appropriate complex/CT-DNA mole ratio. Before measurements, the mixture was shaken up and incubated at room temperature for 30 min. The fluorescence spectra of EB bound to DNA were obtained at λ ex wavelength of 522 nm.
To adjudge the binding mode (groove/intercalative) of 1 and 2 with DNA, the viscosity measurement method by Ostwald’s viscometer was performed. Titrations were carried out by introducing complexes (0.5–3.5 μM) into the viscometer having a CT-DNA solution (5.0 μM). The viscosity values of the solutions were calculated from the observed flow time of CT-DNA-containing solution corrected from the flow time of buffer alone (t0), η = t − t0. The obtained data were used to plot the (η/η 0)1/3 versus the ratio of the concentration of complex and CT-DNA, where η is the viscosity of CT-DNA in presence of complex and η 0 is the viscosity of only CT-DNA.
3 Result and Discussion
3.1 Synthesis and characterization
The ligands L 1 and L 2 were isolated in solid state from the reaction of pyridine-2-carboxylic acid with the corresponding amine (furfurylamine for L 1 or 2-thiophenemethyl amine for L 2) in pyridine solvent (scheme 1). The structural characterizations of both L 1 and L 2 have been confirmed by spectroscopic and physico-chemical tools along with the single crystals X-ray diffraction by collecting the crystals from the ethanolic solution of the carboxamide derivatives on slow evaporation. Two hexacoordinated zinc(II) complexes formulated as [Zn(L1)3](ClO4)2 (1) and [Zn(L2)3](ClO4)2 (2) were obtained by reacting each ligand with zinc(II) perchlorate in ethanol. The complexes are soluble in common organic solvents. The spectroscopic and elemental analyses confirm the formulations of the complexes. The molar conductivities of freshly prepared solutions (∼ 1×10−3 M concentration) of 1 (ΛM= 202 Ω−1.cm 2.mol −1) and 2 (ΛM= 195 Ω−1.cm 2.mol −1) in methanol are in good agreement with the 1:2 electrolytic nature of the complexes.
3.2 X-ray crystal structure analysis
The molecular structures of ligands L 1 and L 2 are depicted in figures 1 and 2 and selected bond lengths and angles are listed in table 2. The ligand L 2 crystallizes with two independent molecules in unit cell (figure S1, in Supplementary Information). These two molecules present a close comparable conformation with similar geometrical parameters and differ for the disorder in the thiophene rings over two coplanar orientations (see Experimental section). A rotation about the C5-C6 bond is required in order to allow the coordination of the ligand to the metal through the carboxamide oxygen and the pyridine nitrogen.

ORTEP view (ellipsoid at 40% probability) of ligand L 1.

ORTEP view (ellipsoid at 30% probability) of one of the two crystallographic independent ligand L 2 (thiophene ring disordered, only the conformational isomer at higher occupancy shown).
The ORTEP drawing of complex 1 is depicted in figure 3 and a selection of bond lengths and angles is listed in table 2. The zinc ion is chelated by three asymmetric bidentate chelating ligands through the carboxamide oxygen and the pyridine nitrogen, in a distorted octahedral geometry. The geometrical isomer separated in solid state is such that in the N3O3 chromophore the three N and thus O donors occupy mer positions about the metal. The Zn-N and Zn-O bond distances are within the range expected for such species varying in the range 2.1149(19)–2.1453(19) and 2.1065–2.1450(16) Å, respectively. It is evident that the formation of the five-membered rings upon coordination with a chelating N,O bond angles in between 75.87–77.53(6) ∘, leads to octahedral angle values (table 2) that significantly deviate from the ideal ones. Each NH group interacts with perchlorate oxygen atoms of the counter anion via weak H-bonds (N ⋯O distance ca. 3.0 Å) (figure S2).

ORTEP view (ellipsoid at 40% probability) of [Zn(L 1)3] 2+ cation of complex 1.
3.3 FTIR spectral analysis
The IR spectrum of ligand L 1 shows strong absorptions at 3344 and 1660 cm −1, which are assignable to the ν N−H and ν C=O stretching. Upon complexation the ν C=O band is shifted to a lower frequency (1639 cm−1), suggesting the coordination of the amide carbonyl oxygen to the Zn(II) centre. Similarly, in IR spectrum of ligand L 2, the characteristics absorption bands at 3317 cm −1 for the ν N−H and at 1654 cm −1 for ν C=O stretching were observed, whereas in complex 2, the characteristic band for the ν N−H stretching is almost undisturbed but the absorption band ν C=O observed in L 2 have been shifted to lower frequency at ca. 1639 cm −1. This shift demonstrates that the amide oxygen formed a coordinative bond with zinc(II) ion. In both complexes, the symmetric (ν sClO4−) bands at 1095 cm −1 and 1087 cm −1; and asymmetric (ν asClO4−) bands at 626 cm −1 and 628 cm −1, respectively, in the spectra 1 and 2 were recorded. The medium-to-low intensity bands at 525 and 460 cm −1 in complex 1 and at 528 and 450 cm −1 in complex 2 are attributed to the Zn–O and Zn–N coordination bonds, respectively, confirming the coordination mode of ligands to the metal.
3.4 Theoretical calculation
Since we did not get the crystal structure of complex 2 so, to clarify the ground state configurations of complex 2 DFT calculations were performed using Gaussian-09 software over a Red Hat Linux IBM cluster using density functional theory (DFT) with the B3LYP/6-31G (d, p) functional model and basis set.[24] The optimized structure shows similar arrangement as that of complex 1 (figure S3).
3.5 DNA binding experiments
The interaction of the zinc (II) complexes with calf thymus DNA (CT-DNA) has been investigated using absorption and emission spectra. To examine the binding mode of the metal complexes with DNA, electronic absorption spectroscopy is used as a distinctive characterization tool. In general, the interaction of the metal complex to the DNA helix is testified by an increase of the n →π* band of zinc (II) complex due to the involvement of strong intercalative interactions between the effective chromophore of the complexes and the base pairs of DNA.[25] The absorption spectra of the free metal complexes and their adducts with CT-DNA are shown in figures 4A and S4. The extent of the hyperchromism indicates that the binding mode of the complex to DNA is electrostatic or by intercalation, which can stabilize the DNA duplex[26,27] and the observed spectral changes indicate a strong interaction of the zinc(II) complexes with CT-DNA. To establish the binding affinity of the Zn(II) complexes with CT-DNA, the apparent association constant K b was determined from the spectral titration data using the following equation:[28]
where [DNA] is the concentration of DNA, ε f, and ε b correspond to the extinction coefficient, respectively, for the free complex and for the zinc (II) complex in the fully bound form. ε a is a variable quantity, corresponds to A obs/[DNA] and varies for each addition of DNA to the zinc (II) complex. Plots of [DNA]/(ε a−ε f)vs [DNA] (figures 4B and S5) gave the apparent association constant Kb as the ratio of slope to the intercept. The calculated Kb values show that Kb of complex 1 (Kb: 1.34 × 105M−1) is higher than that of 2 (K b:4.12×104 M −1). However, these values suggest that both complexes (1 and 2) likely bind via intercalative mode to DNA, involving a stacking interaction between the furan or thiophene chromophore and the base pairs of DNA.

(A) Electronic spectral titration of complex 1 with increasing CT-DNA concentration at 287 nm in tris-HCl buffer; [1] = 2.28 × 10 −5 mol L −1; [DNA]: (a) 0.0, (b) 1.52 × 10 −6, (c) 3.12 × 10 −6, (d) 4.27×106, (e) 6.17 × 10 −6, (f) 8.25×10−6 mol L −1; (B) Plot of [DNA]/(ε a- ε f) vs. [DNA] from the absorption titration of CT-DNA with complex 1 in Tris-HCl buffer. Association constant K\(_{\mathrm {b}} = 1.34 \times 10^{\mathrm {5}}\textit {M}^{\mathrm {-1}}\)(R = 0.93953, n = 5 points).
The DNA-binding modes of the two complexes were further monitored by a fluorescent ethidium bromide (EB) displacement assay.[29] EB is a conjugate planar molecule with a very weak fluorescence intensity that greatly increases when EB is specifically intercalated into the base pairs of double-stranded DNA. When EB is expelled by another intercalator, the fluorescence of the DNA–EB complex is quenched.[30] The binding propensity of the complex to CT-DNA has been investigated by the fluorescence spectral technique using the emission intensity of EB. Although EB does not exhibit any emission in the buffer medium due to fluorescence quenching by solvent molecules, it shows an emission band in the presence of CT-DNA due to intercalative binding to the helix. The decrease in fluorescence intensity of the DNA-bound EB (with excitation wavelength of 522 nm) decreases with the increasing concentration of the complexes (see figures 5A and S6) because the binding of the zinc(II) complexes to DNA promotes the release of EB molecules from the double helix and concomitant decrease in the fluorescence emission.
The quenching induced by the addition of the zinc(II) complexes follows the linear Stern-Volmer equation.[31]
where I 0 and I represent the fluorescence intensity in the absence and presence of the quencher, respectively. K sv is a linear Stern-Volmer quenching constant, and Q is the concentration of the quencher. In the quenching plot of I0/I vs. [complex] (see figures 5B and S7), Ksv value is given by the slope of the regression line. The Ksv values for complexes 1 and 2 are 3.52 × 104 M−1 (R = 0.92507, n = 5 points) and 2.103 × 104 M−1 (R2 = 0.96083, n = 5 points), respectively, suggesting a strong affinity of zinc(II) complexes to CT-DNA.

(A) Emission spectra of the CT-DNA-EB system in tris-HCl buffer upon titration with complex 1. λ ex=522 nm; [EB] =4.77×10−6 mol L −1, [DNA] =1.23×10−5 mol L −1; [Complex]: (a) 0.0, (b) 1.03×10−5, (c) 2.15×10−5, (d) 3.17×10−5, (e) 4.11×10−5, (f) 5.19×10−5 mol L −1. The arrow denotes the gradual increase of complex concentration. (B) Plot of I 0/I vs. [complex 1] ×105. K sv=3.52×104 M −1 (R = 0.92507, n = 5 points).
The apparent binding constant (K app) was calculated using the equation, K EB[EB] = K app[complex],[32] where the complex concentration was the value corresponding to a reduction of 50% of the fluorescence intensity of EB and KEB is the DNA-binding constant of EB, KEB = 1.0 × 107 M−1 for EB.[32] The value for K app for complexes 1 and 2 are 9.2×105 and 2.03×105 M −1, respectively, which are less than the binding constants of classical intercalators and metallointercalators (∼10 7 M −1),[33] suggesting that the complex binds to DNA via an intercalative mode with moderate strength.
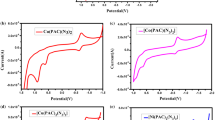
Furthermore, the interaction between the complexes and DNA was investigated by viscosity measurements, which are regarded as the most effective means to study intercalative binding mode of DNA in solution.[34,35] A classical intercalative mode causes significant increase in viscosity of the DNA solution due to an increase in separation of base pairs at the intercalation sites and hence an increase in overall DNA length. In contrast, a partial, non-classical intercalation of ligand could bend (or kink) the DNA helix, reduce its effective length and, concomitantly, its viscosity.[36] As seen in (figure 6), the viscosity of CT-DNA increases by increasing the ratio of complexes 1 and 2 to CT-DNA, resembling the binding mode of EB to the double helix. The results are in agreement with the above UV spectroscopic data where hypochromism and red-shift of complexes 1 and 2 are observed in the presence of DNA.

Change in the relative viscosity (η/η
o)1/3 of CT-DNA as function of r, the molar ratio of the compound to the DNA base pairs: complex 1 ( ), 2(
), 2( ), and EB (
), and EB ( ).
).
4 Conclusion
Two newly designed amide type ligands (L 1, L 2) and their correspondent zinc(II) complexes (1, 2) have been synthesized and characterized using physicochemical and spectroscopic tools. Detailed X-ray structural analysis of complex 1 shows that zinc(II) ion is in a distorted octahedral coordination geometry with N and O donors located in meridional positions. The binding of the two complexes with calf thymus DNA (CT-DNA) have been studied by UV-Vis spectra, fluorescence quenching and viscosity measurement techniques. The experimental results clearly indicate that the zinc(II) complexes bind to CT-DNA in an intercalative mode and the calculated intrinsic binding constants K b indicates a stronger interaction of complex 1 with DNA compared to complex 2.
References
Song X Q, Wen X G, Liu W S and Wang D Q 2010 J. Solid State Chem. 183 1
Song X Q, Yu Y, Liu W S, Dou W, Zheng J R and Yao J N 2007 J. Solid State Chem. 180 2616
Tang Y, Zhang J, Liu W S, Tan M Y and Yu K B 2005 Polyhedron 24 1160
Lee T G, Lee J H, Hyun M Y, Jang S P, Lee H G, Kim C, Kim S J and Kim Y 2010 Inorg. Chem. Commun. 13 1493
Kang D, Seo J, Lee S Y, Lee J Y, Choi K S and S S L. 2007 Inorg. Chem. Commun. 10 1425
Adarsh N N, Kumar D K and Dastidar P 2008 Inorg. Chem. Commun. 11 636
Vallee B L and Falchuk K H 1993 Physiol. Rev. 73 79
Falchuk K H 1998 Mol. Cell. Biochem. 188 41
Zalewski P D, Forbes I J and Betts W H 1993 J. Biochem. 296 403
Maret W, Jacob C, Vallee B L and Fischer E H 1999 Proc. Natl. Acad. Sci. USA 96 1936
Cuajungco M P and Lees G J 1997 Neurobiol. Dis. 4 137
Dealwis C G, Chen L, Brennan C, Mandecki W and Abadzaptero C 1995 Protein Eng. 8 865
Zhang Y, Liang H, Huang J Y, Ke H and Lipscomb W N 1993 Biochemistry 32 7844
Roderick S and Mathews B W 1993 Biochemistry 32 3907
Song Y M, Wu Q, Yang P J, Luan N N, Wang L F and Liu Y M 2006 J. Inorg. Biochem. 100 1685
Tan C, Liu J, Chen L, Shi S and Ji L 2008 J. Inorg. Biochem. 102 1644
Zuber G, Quada J C and Hecht S M 1998 J. Am. Chem. Soc. 120 9368
Bruker, 2000 SMART, SAINT. Software Reference Manual (Madison, Wisconsin Bruker AXS Inc.)
Sheldrick G M 2008 Acta. Crystallogr. Sect. A A64 112
Farrugia L J 1999 J. Appl. Crystallogr. 32 837
Marmur J 1961 J. Mol. Biol. 3 208
Reichmann M E, Rice S A, Thomas C A and Doty P 1954 J. Am. Chem. Soc. 76 3047
Barton J K, Goldberg J M, Kumar C V and Turro J N 1986 J. Am. Chem. Soc. 108 2081
Cossi M, Rega N, Scalmani G and Barone V 2003 J. Comput. Chem. 24 669
Dhara K, Ratha J, Manassero M, Wang X Y, Gao S and Banerjee P 2007 J. Inorg. Biochem. 101 95
Tysoe S A, Morgan R J, Baker A D and Strekas T C 1993 J. Phys. Chem. 97 1707
Vijayalakshmi R, Kanthimathi M, Subramanian V and Nair B U 2000 Biochem. Biophys. Acta 1475 157
Pyle A M, Rehmann J P, Meshoyrer R, Kumar C V, Turro N J and Barton J K 1989 J. Am. Chem. Soc. 111 3051
Boger D L, Fink B E, Brunette S R, Tse W C and Hedrick M P 2001 J. Am. Chem. Soc. 123 5878
Lepecq J B 1971 Methods. Biochem. Anal. 20 41
Stern O and Volmer M 1919 Z. Phys. 20 183
Lee M, Rhodes A L, Wyatt M D, Forrow S and Hartley J A 1993 Biochemistry 32 4237
Qian J, Gu W, Liu H, Gao F X, Feng L, Yan S P, Liao D Z and Cheng P 2007 Dalton. Trans. 10 1060
Kathiravan A and Renganathan R 2009 Polyhedron 28 1374
Baguley B C and Lebret M 1984 Biochemistry 23 937
Zhang S, Zhu Y, Tu C, Wei H, Yang Z, Lin L, Ding J, Zhang J and Guo Z 2004 J. Inorg. Biochem. 98 2099
Acknowledgement
Financial support from the Council of Scientific and Industrial Research (CSIR), New Delhi, India is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information
Crystallographic data for the structural analyses of ligands L 1, L 2 and of complex 1 have been deposited with the Cambridge Crystallographic Data Centre bearing the CCDC Nos. 1058930-1058932, respectively. Copies of this information are available on request at free of charge from CCDC, 12 Union Road, Cambridge, CB21EZ, UK (fax: + 44-1223-336-033; e-mail: deposit@ccdc.ac.uk or http://www.ccdc.cam.ac.uk). Two crystallographic figures, plots of DNA binding experiments for complex 2 from the absorbance and emission study and its comparison with complex 1 in Table S1 and CIF files of ligands L 1, L 2 and of complex 1 are available at www.ias.ac.in/chemsci.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
MONDAL, B., SEN, B., ZANGRANDO, E. et al. Zinc(II) complexes of carboxamide derivatives: Crystal structures and interaction with calf thymus DNA. J Chem Sci 127, 1747–1755 (2015). https://doi.org/10.1007/s12039-015-0954-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-015-0954-3