
Abstract
We have synthesized five zinc complexes of molecular formulae [ZnCl2(2,6-dimethylphenyl-BIAO)]2 (1a), [ZnBr2(2,6-dimethylphenyl-BIAO)]2 (1b), [ZnI2(2,6-dimethylphenyl-BIAO)]2(1c), [ZnBr2(mes-BIAO)]2(2b) and [ZnBr2(dipp-BIAO)] (3b) with rigid unsymmetrical iminoacenaphthenone ligands, (2,6-dimethylphenyl-BIAO) (1), (mesityl-BIAO) (2) and (2,6-diisopropylphenyl-BIAO) (3). The zinc complex 1a was prepared by the reaction of ZnCl2 and neutral (mesityl-BIAO) (1). However, complexes 1b, 2b and 3b were obtained by the treatment of ZnBr2 and neutral ligands 1–3 respectively in 1:1 molar ratio in dichloromethane at ambient temperature. In a similar reaction of ZnI2 with (2,6-dimethylphenyl-BIAO) (1) in dichloromethane the corresponding iodo-complex 1c was obtained in good yield. All the zinc (II) complexes are characterized by FT-IR, 1H and 13C{1H} NMR spectroscopic techniques. The solid state structures of the complexes 1a, 1b, 1c, 2b and 3b are confirmed by single crystal X-ray diffraction analysis. The molecular structures of complexes 1a, 1b, 1c and 2b reveal the dimeric nature of the complexes and subsequently the centre atom zinc is penta-coordinated to adopt distorted trigonal bipyramidal geometry around it. In contrast, the complex 3b is in monomeric in nature due to bulkier size of the ligand and zinc ion is tetra coordinated to adopt distorted tetrahedral geometry.

We report five zinc complexes of molecular formulae [ZnCl2(2,6-dimethylphenyl-BIAO)]2 (1a), [ZnBr2(2,6-dimethylphenyl-BIAO)]2 (1b), [ZnI2(2,6-dimethylphenyl-BIAO)]2 (1c), [ZnBr2(mes-BIAO)]2 (2b) and [ZnBr2(dipp-BIAO)] (3b) with rigid unsymmetrical iminoacenaphthenone ligands, (2,6-dimethylphenyl-BIAO) (1), (mesityl-BIAO) (2) and (2,6-diisopropylphenyl-BIAO) (3). The solid state structures of all the zinc complexes were established by single crystal x-ray diffraction analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Zinc (II) complexes have been widely studied by several research groups for the past decades. The growing interest in the development of zinc catalysts for lactide polymerization[1–9] is one of the driving force. Recently N Zhao et al. have successfully reported some tri-dentate [NO] based ligands and their Zn(II), Ta(IV), Ti(IV), Zr(IV) complexes along with lanthanides, which are implemented as useful catalysts for a range of organic transformations.[10–16] As an alternative method, by switching the ligand systems from tridentate to bidentate nature, tuning of catalytic activity of the metal complexes can be anticipated. Clearly, most of the applied metals (e.g., Pd, Rh, Ru, Ir) displayed difficulties by their low abundance, high price or toxicity. But zinc complexes are less toxic, has high abundance and low price. On the basis of that, research groups are now focusing on different zinc complexes for catalytic activity in organic transformations. V Bette et al. have already reported the reduction of alkyl and aryl carbonyl by using zinc complex as a catalyst using Polymethylhydrosiloxane (PMHS).[17] Besides the reduction of carbonyl, zinc complexes can also catalyze hydrosilylation and exhibit polymerization of olefins. In recent years, we have developed a series of rigid bi-dentate [NO]-based N-(aryl) imino-acenapthenone (Ar-BIAO) ligands.[18] This class of ligands can be obtained from the single condensation of acenaphthequinone and aryl amine moiety in 1:1 ratio in acidic medium (scheme 1) as a major product. In comparison with doubly condensed product (Ar-BIAN) first introduced by van Asselt and Elsevier,[19] the Ar-BIAO ligand contains conjugated exocyclic carbonyl and imine groups. Secondly, the rigidity of the acenaphthene backbone forces the imine N-atoms to remain in a fixed cis orientation with the exocyclic carbonyl group which favours the chelating coordination to a metal centre. Similar to Ar-BIAN, 2,2 ′-bipyridine and 1,10-phenanthroline ligands,[20–22] the σ-donating and π-accepting properties of the Ar-BIAO ligands are observed due to the presence of exocyclic carbonyl and exocyclic imine groups attached with rigid acenaphthene backbone. Herein, we present the full accounts of the synthesis and structural studies of five zinc complexes with Ar-BIAO ligands, [ZnCl2(2,6-dimethylphenyl-BIAO)]2 (1a), [ZnBr2(2,6-dimethylphenyl-BIAO)]2 (1b), [ZnI2(2,6-dimethylphenyl-BIAO)]2 (1c), [ZnBr2(mes-BIAO)]2 (2b) and [ZnBr2(dipp-BIAO)] (3b) and their UV-vis spectroscopic properties in solution and in solid state.

Synthesis of (Ar-BIAO).
2 Experimental
2.1 General Information
All manipulations involving air- and moisture-sensitive compounds were carried out under argon using the standard Schlenk technique or argon-filled glove box. Dichloromethane and pentane were dried by standard methods and kept under molecular sieves prior to use. 1H NMR (400 MHz) and 13C { 1H} NMR (100 MHz) spectra were recorded on a BRUKER AVANCE III-400 spectrometer. BRUKER ALPHA FT-IR was used for FT-IR measurement. Elemental analyses were performed on a BRUKER EURO EA at the Indian Institute of Technology Hyderabad. Ar-BIAO ligands 1-3 were prepared according to the literature method[18,23] and anhydrous ZnCl2, ZnBr2 and ZnI2 were purchased from Sigma Aldrich and used without further purification.
2.2 Synthesis of [ZnCl2(2,6-dimethylphenyl-BIAO)]2 (1a)
In a dry degassed Schlenk tube, ligand 1(200 mg, 0.70 mmol) was placed and about 10 mL of CH2Cl2 was added on to it. The solution was charged with anhydrous ZnCl2 (96 mg, 0.70 mmol) at ambient temperature under inert atmosphere. The reaction mixture was stirred for another 12 h and then the reaction mixture was filtered using cannula filtration. Deep reddish-orange coloured filtrate was evaporated to dryness under vacuo. The residue was re-dissolved in 2 mL chloroform and pentane (2 mL) was layered on to it and kept for crystallization at −40 ∘C. Single crystals of light orange colour were obtained after 2–3 days.Yield: 239 mg (81%). FT-IR (selected frequency): ν= 3057 (Ar- H), 2918 (C- H), 2854 (C- H), 1725 (C =O), 1645 (C =N), 1584, 1420, 1280, 1221, 840, 785 cm −1. 1H NMR (400 MHz, CDCl3, 25 ∘C): δ= 8.38 (t, 2H, An- H), 8.21 (t, 1H, An- H), 7.96 (t, 1H, An- H), 7.56 (t, 1H, An- H), 7.19 (m, 3H, Ar- H), 6.78 (d, 1H, An- H), 2.21 (s, 6H, C H3) ppm; 13C{1H} NMR (100 MHz, CDCl3, 25∘C): δ 189.3 (C=O), 161.2 (C=N), 147.4 (Ar-C), 140.9 (Ar-C), 135.0 (Ar-C), 132.1 (Ar-C), 130.0 (Ar-C), 128.8 (Ar-C), 128.4 (Ar-C), 127.4 (Ar-C), 127.3 (Ar-C), 127.0 (Ar-C), 126.8 (Ar-C), 126.5 (Ar-C), 125.4 (Ar-C), 124.6 (Ar-C), 122.6 (Ar-C), 19.7 (CH3) ppm. Elemental analysis: (C42H32Cl10 N 2O2Zn2) (1a.CHCl3) Calc. C 46.62, H 2.98, N 2.59; found C 46.22, H 2.61, N 2.33.
2.3 Synthesis of [ZnBr2(2,6-dimethylphenyl-BIAO)]2 (1b)
In a dry degassed Schlenk tube, ligand 1(200 mg, 0.70 mmol) was placed and about 10 mL of CH2Cl2 was added on to it. The solution was charged with anhydrous ZnBr2 (158 mg, 0.70 mmol) at ambient temperature under inert atmosphere. The reaction mixture was stirred for another 12 h and then the reaction mixture was filtered using cannula filtration. Deep reddish-orange coloured filtrate was concentrated to 2 mL and pentane (2 mL) was layered on to it and kept for crystallization at −40∘C. Single crystals of light orange colour were obtained after 2–3 days. Yield: 271 mg (76%). FT-IR (selected frequency): ν= 3065, 2919, 2851, 1726 (C =O), 1650 (C =N), 1587, 1434, 1280, 1221, 777, 730 cm −1. 1H NMR (400 MHz, CDCl3, 25 ∘C): δ= 8.47 (t, 2H, An- H), 8.27 (d, 1H, An- H), 8.05 (dd, 1H, An- H), 7.65 (dd, 1H, An- H), 7.30 (m, 3H, Ar-H), 6.86 (d, 1H, An-H), 2.29 (s, 6H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3, 25∘C): δ = 190.7 (C=O), 162.7 (C=N), 147.8 (Ar-C), 142.2 (Ar-C), 135.8 (Ar-C), 133.0 (Ar-C), 132.6 (Ar-C), 130.9 (Ar-C), 129.7 (Ar-C), 129.5 (Ar-C), 129.3 (Ar-C), 128.4 (Ar-C), 128.1 (Ar-C), 128.0 (Ar-C), 127.8 (Ar-C), 127.5 (Ar-C), 127.0 (Ar-C), 126.6 (Ar-C), 125.6 (Ar-C), 123.7 (Ar-C), 18.9 (CH3) ppm. Elemental analysis: (C44H40Br4Cl8 N 2O2Zn2) (1b.2CH2Cl2) Calc. C 38.78, H 2.96, N 2.06; found C 38.53 H 2.59, N 1.89.
2.4 Synthesis of [ZnI2(2,6-dimethylphenyl-BIAO)]2 (1c)
In a dry degassed Schlenk tube, ligand 1 (200 mg, 0.70 mmol) was placed and about 10 mL of CH2Cl2 was added on to it. The solution was charged with anhydrous ZnI2 (224 mg, 0.70 mmol) at ambient temperature under inert atmosphere. The reaction mixture was stirred for another 12 h and then the reaction mixture was filtered using cannula filtration. Deep reddish-orange coloured filtrate was concentrated to 2 mL and pentane (2 mL) was layered on to it and kept for crystallization at −40∘C. Single crystals of light orange colour were obtained after 2–3 days. Yield: 355 mg, 84%. FT-IR (selected frequency): ν= 3053, 2918, 2850, 1705 (C =O), 1645 (C =N), 1584, 1420, 1288, 1221, 833, 775 cm −1. 1H NMR (400 MHz, CDCl3, 25 ∘C): δ= 8.48 (q, 2H, An- H), 8.28 (d, 1H, An- H), 8.05 (t, 1H, An- H), 7.65 (t, 1H, An- H), 7.29 (m, 3H, Ar- H), 6.87 (d, 1H, An- H), 2.36 (s, 6H, C H3) ppm; 13C{1H} NMR (100 MHz, CDCl3, 25 ∘C): δ= 189.7 (C=O), 161.7 (C=N), 147.1 (Ar-C), 140.9 (Ar-C), 135.0 (Ar-C), 132.1 (Ar-C), 130.0 (Ar-C), 128.8 (Ar-C), 128.4 (Ar-C), 127.4 (Ar-C), 127.3 (Ar-C), 127.0 (Ar-C), 126.8 (Ar-C), 126.5 (Ar-C), 126.3 (Ar-C), 125.4 (Ar-C), 124.6 (Ar-C), 122.6 (Ar-C), 19.7 (CH3) ppm. Elemental analysis: (C42H34Cl4I4N2O2Zn2) (1c.CH2Cl2) Calc. C 36.58, H 2.49, N 2.03; found C 35.98 H 2.13, N 1.85.
2.5 Synthesis of [ZnBr2(Mes-BIAO)]2 (2b)
In a dry degassed Schlenk tube, ligand 2 (200 mg, 0.67 mmol) was placed and about 10 mL of CH2Cl2 was added on to it. The solution was charged with anhydrous ZnBr2 (150 mg, 0.67 mmol) at ambient temperature under inert atmosphere. The reaction mixture was stirred for another 12 h and the reaction mixture was filtered using cannula filtration. Deep reddish-orange coloured filtrate was concentrated to 2 mL and pentane (2 mL) was layered on to it and kept for crystallization at −40 ∘C. Single crystals of light orange colour were obtained after 2–3 days. Yield: 280 mg 80%. FT-IR (selected frequency): ν = 2920, 2851, 1729 (C =O), 1648 (C =N), 1587, 1435, 1290, 1224, 779, 730 cm −1. 1H NMR (400 MHz, CDCl3 25 ∘C): δ= 8.46 (q, 2H, An- H), 8.28 (d, 1H, An- H), 8.03 (d, 1H, An- H), 7.60 (q, 1H, An- H), 6.96 (d, 1H, An- H), 6.91 (d, 2H, Ar- H), 2.40 (s,3H, p-C H3), 2.34 (s, 3H, O-C H3), 2.25 (s, 3H, O-C H3) ppm; 13C{1H} NMR (100 MHz, CDCl3, 25∘C): δ 190.1 (C=O), 164.32 (C=N), 140.2 (Ar-C), 138.1 (Ar-C), 137.3 (Ar-C), 136.0 (Ar-C), 133.1 (Ar-C), 132.4 (Ar-C), 130.9 (Ar-C), 130.0 (Ar-C), 129.7 (Ar-C), 129.6 (Ar-C), 129.3 (Ar-C), 128.3 (Ar-C), 127.1 (Ar-C), 126.4 (Ar-C), 125.8, (Ar-C) 125.7 (Ar-C), 125.6 (Ar-C), 21.0 (CH3), 18.9(CH3) ppm. Elemental analysis: (C46H42Br4Cl8 N 2O2Zn2) (2b.2CH2Cl2) Calc. C 39.78, H 3.05, N 2.02; found C 39.31 H 2.79, N 1.89.
2.6 Synthesis of [ZnBr2(Dipp-BIAO)] (3b)
In a dry degassed Schlenk tube, ligand 3 (200 mg, 0.58 mmol) was placed and about 10 mL of CH2Cl2 was added on to it. The solution was charged with anhydrous ZnBr2 (132 mg, 0.58 mmol) at ambient temperature under inert atmosphere. The reaction mixture was stirred for another 12 h and then the reaction mixture was filtered using cannula filtration. Deep reddish-orange coloured filtrate was concentrated to 2 mL and pentane (2 mL) was layered on to it and kept for crystallization at −40 ∘C. Single crystals of light orange colour were obtained after 2–3 days. Yield: 259 mg 78%. FT-IR (selected frequency): ν= 3063, 2963, 2927, 1726 (C =O), 1649 (C =N), 1586, 1434, 1274, 778, 725 cm −1. 1H NMR (400 MHz, CDCl3, 25∘C): δ 8.40 (t, 2H, An-H), 8.21 (d, 1H, An-H), 7.98 (t, 1H, An-H), 7.55 (q, 1H, An-H), 7.41 (t, 1H, Ar-H), 7.32 (d, 2H, Ar-H), 6.68 (d, 1H, An-H), 3.01 (sept, 2H, CH(CH3)), 1.22 (d, 6H, CH(CH3)), 0.75 (d, 6H, CH(C H3)) ppm; 13C{1H} NMR (100 MHz, CDCl3, 25∘C): δ 189.8 (C=O), 162.3 (C=N), 146.9 (Ar-C), 138.4 (Ar-C), 138.2 (Ar-C), 135.1 (Ar-C), 132.1 (Ar-C), 129.9 (Ar-C), 128.8 (Ar-C), 128.0 (Ar-C), 127.8 (Ar-C), 126.2 (Ar-C), 125.7 (Ar-C), 125.4 (Ar-C), 124.1 (Ar-C), 122.6 (Ar-C), 27.9 (C-H), 23.9 (CH3), 23.7(CH3) ppm. Elemental analysis: (C25H25Br2Cl2NO Zn) (3b.2CH2Cl2) Calc. C 46.08, H 3.87, N 2.15; found C 45.71, H 3.43, N 1.99.
2.7 Single-crystal x-ray structure determinations
Single crystals of compound 1a, 1b, 1c, 2b and 3b were grown from a solution of CH2Cl2 (CHCl3 for 1a) and pentane under argon atmosphere at a temperature of −40 ∘C. In each case a crystal of suitable dimensions was mounted on a CryoLoop (Hampton Research Corp.) with a layer of light mineral oil and placed in a nitrogen stream at 150(2) K. All measurements were made on a Agilent Supernova X-calibur Eos CCD detector with graphite-monochromatic CuK α (1.54184 Å) and MoK α (0.71073 Å, for 2b) radiation. Crystal data and structure refinement parameters are summarized in the table 1. The structures were solved by direct methods (SIR92)[24] and refined on F 2 by full-matrix least-squares methods; using SHELXL-97.[25] Non-hydrogen atoms were anisotropically refined. H-atoms were included in the refinement on calculated positions riding on their carrier atoms. The function minimized was [\(\sum w(F\textit {o}^{\mathrm {2}}-F\textit {c}^{\mathrm {2}})^{\mathrm {2}}\)] (w= 1 / [ σ 2 (FO2)+ (aP) 2+bP]), where P = (Max(FO2,0) + 2 FC2) / 3 with σ 2(FO2) from counting statistics. The function R1 and wR2 were (Σ∥Fo|−|Fc∥) / Σ|Fo| and [ Σw(FO2−FC2)2 / Σ(wFo 4)], 1/2 respectively. The ORTEP-3 program was used to draw the molecule.
3 Results and Discussion
The 2, 6-dimethylphenyl-BIAO ligand (1) was treated with anhydrous zinc dichloride in dichloromethane in a 1:1 molar ratio to afford [ZnCl2(2,6-dimethylphenyl-BIAO)]2 (1a) as orange solid in good yield (scheme 2). Other zinc complexes 1b, 1c, 2b and 3b were also isolated in similar reactions with respective Ar-BIAO ligands and zinc halides (scheme 2). The zinc complexes are soluble in THF, toluene, benzene and dichloromethane at room temperature. All the complexes were characterized by spectroscopic and analytical techniques. The solid state structures of all the complexes 1a-c, 2b and 3b were characterized by single crystal X-ray diffraction analysis.

Synthesis of zinc complexes of 1a-c, 2b and 3b.
In FT-IR spectra, all the compounds 1a–c, 2b and 3b show strong absorption bands at 1725, 1726, 1705, 1729 and 1726 cm −1 respectively for respective C =O bond stretching, which is slightly deviated from the C =O bond stretching frequencies of the corresponding ligands.[18,23] The strong absorption bands at 1645 (1a), 1650 (1b), 1645 (1c), 1648 (2b) and 1649 (3b) cm−1 can be assigned to C =N bond stretching of the iminoacenapthenone moiety. In the 1H NMR spectrum measured in CDCl3, compound 1a shows a sharp singlet signal at 2.21 ppm which can be assigned to six protons from two methyl group at O-position of the phenyl group. For zinc dibromo and diiodo complexes 1b and 1c, the respective methyl protons appeared as sharp singlet at 2.29 and 2.36 ppm which is slightly low field shifted compared to that of 1a. However, the chemical shift values are in similar ranges and the slight differences can be accounted for by the gradual increase in sizes from chlorine to iodine atom attached to the zinc atom.
However, these chemical shift values are slightly high field shifted with respect to the corresponding values of ligand 1 (δ 2.04 ppm). The multiplets in the range of 6.68–8.48 ppm for the complexes 1a-c are due to the aromatic protons present in the phenyl group as well in the acenapthene moiety. Compound 2b which has mesityl-BIAO ligand, shows two sharp singlets at 2.34 and 2.25 ppm in a 1:1 ratio and can be assigned to each of three methyl protons at O-positions of the phenyl group indicating the free rotation around the N(imine)-C(mesityl) is restricted in NMR time scale. The singlet resonance signal at 2.40 ppm for compound 2b is assigned to the three methyl protons of the mesityl group at p-position. These chemical shifts are in the similar range with that of free ligand 2 (δ 2.04, (six o-protons) and 2.35 (three p-protons)[23]. The resonances of the aryl protons present in acenaphthene back bone and aryl ring are in the expected region in between 6.91–8.46 ppm similar with the corresponding values of complexes 1a–c and in well agreement with the free ligand 2. For compound 3b, where more bulky 2,6-disopropyl-BIAO ligand is present, two doublet signals at 0.74 and 1.22 ppm in 1:1 ratio and the coupling constants of (12 Hz) can be assigned to the methyl protons attached to the isopropyl groups and this is in similar range with that of free ligand 3 (δ 0.8 and 1.1 ppm).[18] The characteristic septet at 3.01 ppm corresponds to the resonance of the CH proton present in the isopropyl group is also observed slightly down field shifted than the corresponding value (2.80 ppm) of ligand 3. The resonances of the protons of acenaphthene back bone and aryl ring for compound 3b are also in the expected region between 6.68-8.40 ppm and in good agreement with the free ligand 3. In 13C {1H} NMR spectra of the compounds 1a–c, 2b and 3b, carbonyl carbon is mostly de-shielded and appear at δ= 189.3, 190.7, 189.7, 189.7 and 189.8 ppm respectively. The imine carbon is observed at 161.2, 162.7, 161.7, 164.3 and 162.3 ppm respectively and the ipso carbon attached to nitrogen atom is observed at 147.4, 147.8, 147.1, 147.0 and 146.9 ppm respectively for complex 1a–c, 2b and 3b. These values are slightly shifted to the high field region compared to the respective Ar-BIAO ligands[18,23] due to the attachment of zinc ion.
3.1 Solid state structures
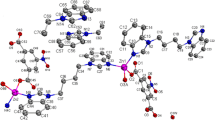
The compounds 1a–c, 2b and 3b were re-crystallized from dichloromethane (CHCl3 for 1a) and pentane (1:1) mixture and their molecular structures in the solid state were established by single crystal X-ray diffraction analysis. Compound 1a crystallizes in monoclinic space group C2/c with four molecules in unit cell along with one chloroform molecule as solvate. In contrast, complexes 1b, 1c and 2b crystallize in triclinic space group P−1 having only one molecule in their respective unit cell along with dichloromethane molecule (two for 1b and 2b and one for 1c) as solvents. The details of structural and refinement parameters of all the complexes 1a–c and 2b are given in table 1. The molecular structures of complexes 1a–c and 2bare shown in figure 1a–d respectively.


a-d Solid-state structures of Q2 1a-c and 2a showing the atom labelling scheme omitting hydrogen atoms for clarity. Selected bond lengths in [Å]: 1a. Zn(1)-N(1) 2.087(3), Zn(1)-Cl(2) 2.2103(15), Zn(1)-Cl(1) 2.3091(14), Zn(1)-Cl(1) i 2.4404(13), Zn(1)-O(1) 2.468(3), Cl(1)-Zn(1) i 2.4404(13), O(1)-C(1) 1.225(5), N(1)-C(2) 1.277(5), N(1)-C(13) 1.459(5), C(2)-C(3) 1.462(5), C(2)-C(1) 1.522(6), C(1)-C(11) 1.469(6); Selected bond angles in [ o]: N(1)-Zn(1)-Cl(2) 116.30(12), N(1)-Zn(1)-Cl(1) 115.46(11), Cl(2)-Zn(1)-Cl(1) 124.18(5), N(1)-Zn(1)-Cl(1) i 96.67(10,) Cl(2)-Zn(1)-Cl(1) i 104.78(5), Cl(1)-Zn(1)-Cl(1) i 88.50(4), N(1)-Zn(1)-O(1) 74.84(12), Cl(2)-Zn(1)-O(1) 89.41(9), Cl(1)-Zn(1)-O(1) 84.97(9), Cl(1) i-Zn(1)-O(1) 165.68(9,) Zn(1)-Cl(1)-Zn(1) i 91.50(4), C(1)-O(1)-Zn(1) 105.5(3), C(2)-N(1)-C(13) 119.9(3), C(2)-N(1)-Zn(1) 118.9(3), C(13)-N(1)-Zn(1) 121.2(2), N(1)-C(2)-C(3) 134.5(4), N(1)-C(2)-C(1) 117.8(4), C(3)-C(2)-C(1) 107.7(4), O(1)-C(1)-C(11) 130.7(5), O(1)-C(1)-C(2) 122.7(4), C(11)-C(1)-C(2) 106.5(4), C(10)-C(11)-C(1) 132.9(5), C(12)-C(11)-C(1) 105.8(4). 1b.Br(1)-Zn(1) 2.3266(6), Br(2)-Zn(1) i1 2.4243(6), Br(2)-Zn(1) 2.5980(7), Zn(1)-N(1) 2.073(3), Zn(1)-Br(2) i1 2.4243(6), Zn(1)-O(1) 2.483(3), O(1)-C(2) 1.223(5), N(1)-C(1) 1.274(5), N(1)-C(13) 1.455(5), C(1)-C(11) 1.464(5), C(1)-C(2) 1.536(5); Selected Bond Angles in [ o]: Zn(1) i1-Br(2)-Zn(1) 88.50(2), N(1)-Zn(1)-Br(1) 116.29(9), N(1)-Zn(1)-Br(2) i1 117.64(9), Br(1)-Zn(1)-Br(2) i1 121.54(2), N(1)-Zn(1)-O(1) 74.91(11), Br(1)-Zn(1)-O(1) 89.52(7), Br(2) i1-Zn(1)-O(1) 84.01(7), N(1)-Zn(1)-Br(2) 96.30(9), Br(1)-Zn(1)-Br(2) 103.50(2), Br(2) i1-Zn(1)-Br(2) 91.50(2), O(1)-Zn(1)-Br(2) 166.65(7), C(2)-O(1)-Zn(1) 105.8(3), C(1)-N(1)-C(13) 118.8(3), C(1)-N(1)-Zn(1) 118.6(3), C(13)-N(1)-Zn(1) 122.4(2) N(1)-C(1)-C(11) 134.2(4), N(1)-C(1)-C(2) 119.0(3), C(11)-C(1)-C(2) 106.9(3), O(1)-C(2)-C(3) 132.7(4), O(1)-C(2)-C(1) 121.4(4). 1c. I(1)-Zn(1) 2.6529(13), I(1)-Zn(1) i1 2.8263(15), I(2)-Zn(1) 2.5377(13), Zn(1)-N(1) 2.104(8), Zn(1)-O(1) 2.392(8), Zn(1)-I(1) i1 2.8263(15), O(1)-C(10) 1.222(14), N(1)-C(9) 1.259(13), N(1)-C(1) 1.452(11), C(2)-C(1) 1.409(14); Selected Bond Angles in [ o]: Zn(1)-I(1)-Zn(1) i1 87.89(4), N(1)-Zn(1)-O(1) 76.0(3), N(1)-Zn(1)-I(2) 118.0(2), O(1)-Zn(1)-I(2) 91.63(19), N(1)-Zn(1)-I(1) 111.4(2), O(1)-Zn(1)-I(1) 80.23(18), I(2)-Zn(1)-I(1) 126.07(5), N(1)-Zn(1)-I(1) i1 95.6(2), O(1)-Zn(1)-I(1) i1 165.42(19), I(2)-Zn(1)-I(1) i1 102.89(5), I(1)-Zn(1)-I(1) i1 92.11(4), C(10)-O(1)-Zn(1) 106.7(6), C(9)-N(1)-C(1) 119.0(8), C(9)-N(1)-Zn(1) 117.2(6), C(1)-N(1)-Zn(1) 123.8(6). 1d. Br(2)-Zn(1) 2.4512(7), Br(2)-Zn(1) i1 2.5639(7), Br(1)-Zn(1) 2.3435(7), Zn(1)-N(1) 2.104(4), Zn(1)-O(1) 2.414(3), Zn(1)-Br(2) i1 2.5639(7), O(1)-C(2) 1.212(6), N(1)-C(1) 1.286(6), N(1)-C(13) 1.442(5), C(1)-C(11) 1.466(6), C(1)-C(2) 1.535(6), C(2)-C(3) 1.466(6); Selected bond Angles in [ o]: Zn(1)-Br(2)-Zn(1) i1 89.08(2), N(1)-Zn(1)-Br(1) 114.62(10), N(1)-Zn(1)-O(1) 75.58(12), Br(1)-Zn(1)-O(1) 87.64(8), N(1)-Zn(1)-Br(2) 117.29(10), Br(1)-Zn(1)-Br(2) 122.18(3), O(1)-Zn(1)-Br(2) 82.14(8), N(1)-Zn(1)-Br(2) i1 96.74(10), Br(1)-Zn(1)-Br(2) i1 106.71(3), O(1)-Zn(1)-Br(2) i1 165.60(8), Br(2)-Zn(1)-Br(2) i1 90.92(2), C(2)-O(1)-Zn(1) 107.3(3), C(1)-N(1)-C(13) 118.9(4), C(1)-N(1)-Zn(1) 117.3(3), C(13)-N(1)-Zn(1) 123.7(3), N(1)-C(1)-C(2) 117.6(4), O(1)-C(2)-C(3) 131.4(4), O(1)-C(2)-C(1) 122.1(4), C(3)-C(2)-C(1) 106.5(4).
The complexes 1a–c and 2b are dimeric in nature and two zinc ions are bridged through two μ 2-halide ions in each case. In complexes 1a–c, the coordination polyhedron in each case is formed by the chelation of imine nitrogen, carbonyl oxygen atoms of the 2,6-dimethyl-BIAO ligand and the halide atoms (three chloride for 1a, three bromide for 1b and three iodide ions for 1c) attached to the zinc ion. In similar fashion, the coordination polyhedron of compound 2b is formed by the ligation of nitrogen and oxygen atoms present in mesityl-BIAO ligand and chloride ions. The complexes 1a–c and 2b are centrosymmetric due the presence of an inversion center in the middle of each of the molecule each of them form a four membered core Zn1-Cl1-Zn1 i-Cl1 i (1a), Zn1-Br1-Zn1 i-Br1 i (1b and 2b), and Zn1-I1-Zn1 i-I1 i (1c). In addition, two more metallacycles are formed in each case Zn1-N1-C2-C1-O1 and Zn1 i-N1 i-C2 i-C1 i-O1 i (1a), Zn1-N1-C1-C2-O1 and Zn1 i-N1 i-C1 i-C2 i-O1 i (1b), Zn1-N1-C9-C10-O1 and Zn1 i-N1 i-C9 i-C10 i-O1 i (1c) and Zn1-N1-C1-C2-O1 and Zn1 i-N1 i-C1 i-C2 i-O1 i (2b). In these complexes, the geometry around each zinc ion can be best described either a distorted square pyramidal having N1, O1, Cl1, Cl1 i atoms for 1a, N1, O1, Br2, Br2 i for 1b and 2b, N1, O1, I1, I1 i for 1c in the basal position and Cl2 (1a), Br1 (1b and 2b) and I1 (1c) atoms in the apical position or distorted trigonal bipyramidal having N1 Cl1 Cl2 atoms (for 1a), N1 Br1 Br2 (for 1b and 2b) and N1 I1 I2 (for 1c) are in the equatorial position and O1, Cl1 i (1a), O1, Br2 i (1b and 2b), O1, I1 i (1c), atoms are in the apical position. Thus, all three complexes 1a-c and 2b are examples of unusual penta- coordinated zinc atom. Such kind of unusual zinc complexes are rare in literature.[26,27] The Zn-O distances [2.469(2) Å (1a), 2.484(3) Å (1b), 2.392(8) Å (1c) and 2.414(3) Å (2b)] which are well in agreement with the zinc oxygen coordination bond rather covalent bond (Zn-O 1.976 (2) Å for covalent bond.[27] The Zn1-N1 distances 2.087(3) Å (for 1a), 2.073(3) Å (for 1b), 2.104(8) Å (for 1c and 2b) are in the range of the zinc nitrogen coordination bond as the similar Zn-N distance 2.023(3) -2.34(3) Å is observed for Zn(mmpcd)]ClO4 (mmpcd = Me2pzCH2)2 NC 2H3MeNHC5H6 CSSCH 3) (pz = pyrazole) reported by Chaudhury et al.[28] and 1.994(2) - 2.050(2) Å observed for [{CH(Ph2PNSiMe3)2} ZnN(SiMe3)2] reported by Roesky et al.[29] Three different Zn-halide distances in each complex [Zn-Cl 2.210(2), 2.309(2), 2.440(1), Å for 1a; Zn-Br 2.327(6), 2.424(6), 2.598(7) Å for 1b; Zn-I 2.536(1), 2.653(1), 2.826(2) Å for 1c and Zn-Br 2.343(7), 2.451(7), 2.564(7) Å for 2b] indicate that one halide ion is purely covalent bonded, whereas second halide ion is bonded covalently as well as in bridging fashion which elongate Zn-halide distance slightly, and the third halide ion is purely a coordination bond and has the longest Zn-halide distance. Thus, an asymmetric attachment of the halide ions to the zinc centres are observed in each complexes. The average distance of 2.217(7) Å was observed as Zn-Cl distance in BIANZnCl2[30] and 2.337–2.347Å was reported as Zn-Br distance (ZrBr2(TEEDA)) (tetraethylethylenediamine) in literature. 31 The Zn-I bond distance of 2.550(8)–2.553(7) Å is reported for BIANZnI2 by Schumann et al.[30]
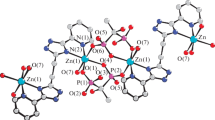
The monomeric zinc complex 3b crystallizes in monoclinic space group P21/c having two independent molecules in the unit cell along with two dichloromethane molecules. The details of structural and refinement parameters of the complex 3b are given in table 1. The molecular structure of complex 3bis shown in figure 2. The central ion zinc is tetra-coordinated through nitrogen and oxygen atoms present in the 2,6-diisopropylphenyl-BIAO ligand and two bromide ions attached to zinc. The geometry around the zinc can be best described as distorted tetrahedral. With respect to the dimeric complexes 1a–c and 2b, the monomeric form of 3b can be understood due to the presence of 2,6-isopropylphenyl group on the iminoacenapthenone moiety which prevents the formation of dimer. The Zn-O distances [2.162(2) and 2.154(2) Å for molecule 1 and 2 respectively] are slightly shorter than that of complexes 1a–c and 2b (see above). This can be explained due to expansion of coordination number from four to five for zinc ion as we move from tetrahedral geometry of 3b to trigonal bipyramidal geometry of 1a–c and 2b. Nevertheless, the Zn-N distances [2.086(2) and 2.105(2) Å for molecule 1 and 2 respectively] are in the similar range with that of 1a–c and 2b. The Zn-Br distances [2.318(6) and 2.328(6) Å for molecule 1 and 2.307(6) and 2.332 (6) Å for molecule 2) are also similar with that of zinc complexes 1b and 2b. In 3b, the four membered zinc metallacycles Zn1-N1-C1-C2-O1 (molecule 1) and Zn2-N2-C25-C26-O1 (molecule 2) are formed and almost co-planar with the acenapthene plane (dihedral angle 6.79 ∘).

Solid-state structure of 3b showing the atom labelling scheme omitting hydrogen atoms for clarity. Selected bond lengths in [Å]: Zn(1)-N(1) 2.086(2), Zn(1)-O(1) 2.162(2), Zn(1)-Br(1) 2.3181(6), Zn(1)-Br(2) 2.3280(6), O(1)-C(2) 1.227(4), N(1)-C(1) 1.278(4), N(1)-C(13) 1.460(4), C(1)-C(11) 1.460(4), C(1)-C(2) 1.539(4), C(2)-C(3) 1.463(4); Selected bond angles in [ o]: N(1)-Zn(1)-O(1) 81.03(9), N(1)-Zn(1)-Br(1) 114.77(7), O(1)-Zn(1)-Br(1) 107.52(6), N(1)-Zn(1)-Br(2) 113.13(7), O(1)-Zn(1)-Br(2) 104.90(6), Br(1)-Zn(1)-Br(2) 125.08(2), C(2)-O(1)-Zn(1) 109.00(19), C(1)-N(1)-C(13) 118.1(3), C(1)-N(1)-Zn(1) 111.5(2), C(13)-N(1)-Zn(1) 130.30(19), N(1)-C(1)-C(11) 135.3(3), N(1)-C(1)-C(2) 117.4(3), O(1)-C(2)-C(3) 132.4(3), O(1)-C(2)-C(1) 121.0(3), C(3)-C(2)-C(1) 106.5(3).
3.2 UV-visible spectra
UV-visible absorption spectra of Ar-BIAO ligands 1, 2 and 3 (figure 3a) were measured in dichloromethane at room temperature and displayed a nearly comparable absorption pattern at 230, 303 nm. The absorption spectra of the ligands 1, 2 and 3 can be attributed to the π→π* transitions and n → π* transitions respectively. The UV-visible absorption spectra of complexes 1a, 1b, 1c, 2b and 3b (figure 3b) were also measured in dichloromethane at room temperature and displayed a similar kind of absorption pattern at 230 nm for π→π* transition. Upon coordination with zinc (II) ion, the absorption peaks are slightly shifted compared to that of the respective ligands. The solid-state UV-visible absorption spectra of 1a, 1b, 1c, 2b, 3b were significantly different from that of solution (figure 4). In the solid-state UV-visible absorption spectra of 1a, 1b, 1c, 2b, 3b a broad absorption peak from 250 to 500 nm were attributed to the π to π* transition of ligand. In the solid-state UV-visible absorption spectra of all zinc (II) complexes, the π→π* transition intensity is in the same order for complexes 1a, 1b, 2b and 3b but for complex 1c, the intensity drastically decreases compared to that of other complexes.

a-b. The UV-vis absorption spectra of ligands 1–3 and zinc complexes 1a-c, 2b and 3b in CH2Cl2 at room temperature (3.207 × 10 −6 M) respectively.

The solid state UV-vis absorption spectra of complexes 1a-c,2b and 3b.
4 Conclusion
We have successfully synthesized and characterized four dimeric penta-coordinated zinc complexes [ZnCl2(2,6-dimethylphenyl-BIAO)]2 (1a), [ZnBr2(2,6- dimethylphenyl-BIAO)]2 (1b), [ZnI2(2,6-dimethylphenyl- BIAO)]2 (1c) and [ZnBr2(Mes-BIAO)]2 (2b) along with one tetra-coordinated monomeric zinc complex [ZnBr2(Dipp-BIAO)] (3b). The molecular structures of all the complexes were established and they revealed a bidentate ligation from the Ar-BIAO ligands in each case through lone pairs of nitrogen and oxygen atoms. Thus, it was observed that by changing the steric crowding on the Ar-BIAO ligand, the nuclearity of the zinc complexes can be changed.
References
Sun H, Ritch J S and Hayes P G 2011 Inorg. Chem. 50 8063
Pastor M F, Whitehorne T J J, Oguadinma P O and Schaper F 2011 Inorg. Chem. Commun. 14 1737
Drouin F, Oguadinma P O, Whitehorne T J J, Prud’homme R E and Schaper F 2010 Organometallics 29 2139
Boerner J, Floerke U, Doering A, Kuckling D, Jones M D, Steiner M, Breuning M and Herres-Pawlis S 2010 Inorg. Chem. Commun. 13 369
Darensbourg D J and Karroonnirun O 2010 Inorg. Chem. 49 2360
Howard R H, Alonso-Moreno C, Broomfield L M, Hughes D L, Wright J A and Bochmann M 2009 Dalton Trans. 8667
Labourdette G, Lee D J, Patrick B O, Ezhova M B and Mehrkhodavandi P 2009 Organometallics 28 1309
Chisholm M H, Eilerts N W, Huffman J C, Iyer S S, Pacold M and Phomphrai K 2000 J. Am. Chem. Soc. 122 11845.
Chakraborty D and Chen E Y -X 2003 Organometallics 22 769
Zhao N, Chen L, Ren W, Song H and Zi G 2012 J. Organomet. Chem 712 29
Zi G 2009 Dalton Trans. 9101
Zi G 2011 J. Organomet. Chem. 696 68
Xiang L, Wang Q, Song H and Zi G 2007 Organometallics 26 5323
Wang Q, Xiang L, Song H and Zi G 2008 Inorg. Chem. 47 4319
Zi G, Xiang L and Song H 2008 Organometallics 27 1242
Xiang L, Song H and Zi G 2008 Eur. J. Inorg. Chem. 1135
Bette V, Mortreux A, Savoia D and Carpentier J F 2004 Tetrahedron 60 2837
(a) Anga S, Paul M, Naktode K, Kottalanka R K and Panda T K 2012 Z. Anorg. Allg. Chem. 637 1311; (b) Anga S, Pal T, Kottalanka R K, Paul M and Panda T K 2013 Can. Chem. Trans. 1 105; (c) Anga S, Biswas S, Kottalanka R K, Mallik B S and Panda T K 2014 Can. Chem. Trans. 2 72
van Asselt R and Elsevier C J 1992 Organometallics 11 1999
Cavell K J, Stufkens D J and Vrieze K 1980 Inorg. Chim. Acta 47 672
Reinhold J, Benedix R, Birner P and Hennig H 1979 Inorg. Chim. Acta 33 209
(a) Jeon M, Han C J and Kim S Y 2006 Macromol. Res. 14 306; (b) Small B L, Rios R, Fernandez E R, Gerlach D L, Halfen J A and Carney M J 2010 Organometallics 29 6723; (c) Schmiege B M, Carney M J, Small B L, Gerlach D L and Halfen J A 2001 Dalton Trans. 2547
Kovach J, Peralta M, Brennessel W W and Jones W D 2011 J. Mol. Struct. 992 33
Sheldrick G M SHELXS-97 Program of Crystal Structure Solution University of Göttingen, Germany 1997
Sheldrick G M SHELXL-97 Program of Crystal Structure Refinement University of Göttingen, Germany 1997
Guru S and Ramana Rao D V 1968 Z. Anorg. Allg. Chem. 362 108
Bhattacharyya S, Kumar S B, Dutta S K, Tiekink E R T and Chaudhury M 1996 Inor. Chem. 35 1967
Seetawan U, Jugsujinda S, Seetawan T, Ratchasin A, Euvananont C, Junin C, Thanachayanot C and Chainaronk P 2011 Mater. Sci. Appl. 2 1302
Marks S, Köppe R, Panda T K and Roesky P W 2010 Chem. Eur. J. 16 7096
Fedushkin I L, Skatova A A, Eremenko O V, Hummert M and Schumann H 2007 Z. Anorg. Allg. Chem. 633 1739
Eckert P K, Vieira I S, Gessner V H, Börner J, Strohmann C and Herres-Pawlis S 2013 Polyhedron 49 151
Acknowledgements
This work was supported by the Council of Scientific and Industrial Research (CSIR) scheme ((No. 01(2530)/11/EMRII)) and start-up grant from IIT Hyderabad. S.A. thanks CSIR, India and K. N. thanks University Grant Commission (UGC), India for their PhD fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as a supplementary publication no. CCDC 1003687-1003691. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB21EZ, UK (fax: + (44)1223-336-033; email: deposit@ccdc.cam.ac.uk).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
ANGA, S., REJ, S., NAKTODE, K. et al. Syntheses and solid state structures of zinc (II) complexes with Bi-dentate N-(Aryl)imino-acenapthenone (Ar-BIAO) ligands. J Chem Sci 127, 103–113 (2015). https://doi.org/10.1007/s12039-014-0756-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-014-0756-z