
Abstract
The conserved band-interband pattern is thought to reflect the looped-domain organization of insect polytene chromosomes. Previously, we have shown that the chromodomain protein Chriz and the zinc-finger protein Z4 are essentially required for the maintenance of polytene chromosome structure. Here we show that both proteins form a complex that recruits the JIL-1 kinase to polytene chromosomes, enabling local H3S10 phosphorylation of interband nucleosomal histones. Interband targeting domains were identified at the N-terminal regions of Chriz and Z4, and our data suggest partial cooperation of the complex with the BEAF boundary element protein in polytene and diploid cells. Reducing the core component Chriz by RNAi results in destabilization of the complex and a strong reduction of interband-specific histone H3S10 phosphorylation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Interphase chromosomes are organized by a hierarchy of structural elements of increasing complexity that are stable enough to provide for the required nuclear order and sufficient flexible to allow the dynamic nuclear processes to take place. Thirty-five years ago, in a seminal paper, the Worcel group proposed a model for the organization of interphase chromatin into topologically closed domains (Benyajati and Worcel 1976) that predicted the folding of chromatin into a series of loops that are fixed at their basis and thus isolated from adjacent loops. This model provided a foundation for understanding the regulatory architecture of the genome. There is now substantial evidence for a domain model of genomic organization whereby long-range regulatory elements like enhancers and silencers are restricted to the regulation of transcription units within the same looped domain (Labrador and Corces 2002; Wallace and Felsenfeld 2007; De Wit and van Stensel 2009). Insulators placed at the domain boundaries form the basis of looped domains (Blanton et al. 2003; Murrell et al. 2004) and shield genes within the domains from the impact of outside regulatory elements (Bell et al. 1999; West et al. 2004).
Polytene chromosomes of genetically tractable insects like Drosophila provide an attractive system to study interphase chromatin by combining cytogenetics with molecular biology. The genetically conserved band-interband pattern is thought to reflect the looped-domain organization of chromosomes. A particular example is the Drosophila 87A7 heat-shock locus, a chromosomal band that contains the two divergently transcribed hsp70 genes flanked on either side by a pair of strong DNase I hypersensitive sites within the scs and scs′ boundary elements (Udvardy et al. 1985). Upon heat induction, the band decondenses and scs and scs′ locate to the edges of the puff formed, suggesting that these elements function to delimit a domain of gene expression. Both elements possess reported insulator activity (Kellum and Schedl 1992; Vazquez and Schedl 1994). The insulating activity of scs′ is mediated by a series of CGATA repeats that interact with the BEAF-32 proteins (Zhao et al. 1995; Cuvier et al. 1998). BEAF-32 immunolocalizes to many interband regions of polytene chromosomes (Zhao et al. 1995). The insulator activity of the scs element is mediated by the Zw5 protein (Gaszner et al. 1999) that localizes to more than 100 sites on polytene chromosomes and binds to the scs-containing edge of the hsp70 puff. Recently, it has been shown that Zw5 binds to BEAF-32 in vivo and this interaction is accompanied by a physical association of the genomic scs and scs′ elements, which might lead to the formation of a 15 kb higher-order looped domain (Blanton et al. 2003).
Besides BEAF and Zw5, genome-wide-acting boundary elements such as CP190, Su(Hw), Mod(mdg4), the GAGA factor or dCTCF (Mohan et al. 2007; Bartkuhn et al. 2009; Bushey et al. 2009; see recent reviews by Bartkuhn and Renkawitz 2008; Bushey et al. 2008) are present within interbands or at the boundaries between interbands and the more condensed bands. Genetically, this diverse family acts as insulators, providing position-independent expression of transgenes and/or enhancer-blocking function when placed between an enhancer and the promoter of a reporter gene (Bartkuhn and Renkawitz 2008; Bushey et al. 2008). The histone-H3S10-specific tandem kinase JIL-1 also located in interbands (Wang et al. 2001) may be required for the formation or maintenance of open chromatin. It results in local decondensation of the interphase chromosome when ectopically targeted to condensed sites in a process requiring its kinase activity (Deng et al. 2008). Previously, we and others identified Chriz and Z4 as two essential genes encoding chromatin proteins that are ubiquitously expressed in many tissues throughout Drosophila development. Z4 possesses seven zinc-fingers in its central domain and Chriz is a chromodomain protein interacting with Z4 (Eggert et al. 2004; Gortchakov et al. 2005; Rath et al. 2006). On polytene chromosomes, both proteins are found in many interbands and are essentially colocalized. Hypomorphic mutations in Z4 result in a progressive loss of band/interband structures of polytene chromosomes. In this article we present data to explain the targeting of Chriz and Z4 to interbands. We provide evidence that Chriz and Z4 form the core of a complex required for recruitment of the JIL-1 kinase to chromosomes as a prerequisite for local H3S10 phosphorylation during interphase.
2 Materials and methods
2.1 Drosophila stocks and breeding conditions
Fly strains were reared at a regular day-and-night cycle at room temperature on standard Drosophila cornmeal medium with the addition of dry yeast, soy bean meal and molasses. The w 1118 mutation was used as the genetic background for obtaining transgenic lines. Ectopic expression or knock-down by RNAi was achieved using the GAL4/UAS system of Brand and Perrimon (1993). The GAL4 drivers, GAL4-G231.1 (salivary gland specific from stage 13 embryos until early pupae; H Saumweber, unpublished) and GAL4-Sgs58 (Sgs4 enhancer, late 3rd instar larvae; G Korge, unpublished) were used for induction of ectopic expression or RNA interference (RNAi). UAS-lines used are listed in section 2.2. GAL4/UAS crosses were grown at RT (22–24°C) and were performed at least twice. The different transgenic lines were obtained following injection of the respective pUAST constructs and helper plasmid into w 1118 embryos as described (Eggert et al. 1998).
2.2 Plasmid construction, transgene expression and yeast two hybrid assay
For plasmid construction, standard restriction enzymes, PCR methods and cloning protocols were used. Details are available upon request. For the yeast two hybrid assays, Chriz fragments were cloned into pGAD424 in-frame with the GAL4 activation domain in order to obtain the following constructs (in general, all constructs were verified by sequencing): pGAD424Chriz FL (aa 2–926), pGAD424ChrizN (aa 2–211), pGAD424ChrizNCD (aa 2–285), pGAD424ChrizCD (aa 193–285), pGAD424ChrizCDC (aa 193–926), pGAD424ChrizC (aa 273–926), pGAD424ChrizC1 (aa 279–509), pGAD424ChrizC2 (aa 500–768), pGAD424ChrizC3 (aa 700–926), pGAD424ChrizCF1 (aa 279–768) and pGAD424ChrizCF2 (aa 500–926). Similarly, the following Z4 fragments were cloned into pGBT9 in-frame with the GAL4 DNA-binding domain: pGBT9Z4 (aa 1–996), pGBT9Z4N (aa 1–237), pGBT9Z4M (aa 231–522) and pGBT9Z4C (aa 516–996). The Myc-tagged Chriz constructs used for pull-down experiments were as described by Gortchakov et al. (2004); the Z4 bait used was pGEX-6p-1Z4N with Z4 (aa 1–237) cloned in frame with GST; pGEX-6p-1 was used for expression of the GST negative control. For chromosomal-binding site analysis, transgenic strains were obtained by the injection of pUAST-MH plasmids (Gortchakov et al. 2004) carrying Z4 or Chriz fragments in-frame with an N-terminal NLS-Myc-his tag of a modified pUAST vector (Brand and Perrimon 1998). In this study, the following Z4 fragments were used Z4: pUAST-MH-GFP-Z4Zif1-3 (aa 232–328), pUAST-MH-GFP-Z4Zif4-6 (aa 356–446), pUAST-MH-GFP-Z4Zif4-7 (aa 356–522), pUAST-MH-GFP-Z4Zif1-7 (aa 232–522), pUAST-MH-Z4NZif1-7 (aa 1–516), pUAST-MH-Z4C (aa 517–996) and pUAST-MH-Z4N (aa 1–237). The following Chriz fragments were used: pUAST-MH-ChrizN (aa 2–211), pUAST-MH-ChrizCD (aa 160–327) and pUAST-MH-ChrizC (aa 275–926). Chriz-RNAi and Z4-RNAi constructs used were already published by Eggert et al. (2004) and Gortchakov et al. (2005).
For bacterial expression of the GST- and Myc-His-fusion proteins plasmid constructs were transformed into E. coli BL-21. The expression was at 37°C and induction at 0.5–0.8 OD600 by the addition of 0.5 mM IPTG for 2–3 h. The bacterial cell pellets were re-suspended in lysis buffer (Quiagen bacterial protein prep kit) containing protease inhibitors (1× PBS, 1 mM EDTA, 1 mM PMSF, 1 μg/ml Leupeptin, 1 μg/ml Aprotinin, 1 μg/ml Pepstatin) and lysed by sonication according to standard protocols. Expressed proteins were analysed on SDS-PAGE.
The yeast two hybrid interaction was performed using the yeast strain SFY526. Yeast transformation, reporter gene expression assay and quantification of protein–protein interaction were performed according to Clontech yeast protocols (http://www.clontech.com/). Interaction strength was determined by the activity of the LacZ reporter present in the system. β-Galactosidase activity was determined from freeze-thaw lysates of transformed yeast cells grown in liquid culture according to Clontech protocols.
2.3 In vitro protein interaction by GST-pull-down assay and Western blot
GST and GST-tagged proteins were purified using Glutathion S-Sepharose beads according to the GST purification protocol given by Amersham Biosciences. 2 μg purified GST-tagged fusion proteins or GST protein only as negative control was bound to 10 μl GST beads. Prebound beads were suspended in 200 μl 20 mM Tris pH7.5, 0.1 mM EDTA, 150 mM NaCl, and incubated with 4 μl cell extract containing the putative Myc-his-tagged interaction partner for 2 h at 4°C on a rotor. Beads were spun down by centrifugation and washed 5 times for each 10 min at 4°C in the same buffer with the addition of 0.3% (v/v) Triton X-100. The beads were transferred to a fresh tube, and the final pellet was eluted by addition of 20 μl SDS mix. After brief heating to 100°C, the beads were spun down, and the supernatant was removed and analysed by Western blot.
For immunoblotting, equal amounts of each probe were resolved on SDS-PAGE by electrophoresis. Blotting and blocking of the membrane was done according to standard procedures. The membrane was either incubated with mouse anti-GST antibody (Sigma) or with mouse anti-Myc antibody (Developmental Studies Hybridoma Bank, University of Iowa) overnight at 4°C. After washing 3× for each 20 min in 130 mM NaCl, 7 mM Na2HPO4, 3 mM NaH2PO4, pH 7.4, 0.3 % v/v Triton X-100 the membrane was incubated with alkaline phosphatase conjugated goat anti-mouse antibody (Dianova ) for 2 hrs. After 3× washing the membrane as before the blot was developed using NBT/BCIP solution.
2.4 Immunoprecipitation
Isolation of Kc-cell nuclei, preparation of nuclear extracts and immunoprecipitations were performed as described (Reim et al. 1999) with the following modifications: Nuclear extracts from Kc-cell nuclei were prepared in TEN300 (10 mM Tris pH 8.0; 300 mM NaCl) with the addition of each 1 μg/ml pepstatin and leupeptin and 1 mM PMSF. 200 μl of this extract was incubated with 25 μg purified antibody (for Z4 and anti-Myc monoclonal antibodies) or 3–5 μl of antiserum (for Chriz). Following 1 h rotation at 4°C, 50 μl Protein A Sepharose suspension (as a 50% slurry in TEN300) was added and the mixture was rotated for 2 h at 4°C. The beads were washed 3 times in 200 μl ice-cold TEN300 and transferred to a fresh tube, and after pelleting the beads the supernatant was removed carefully. Beads were eluted 3 times by the addition of one-pellet-volume SDS sample buffer and brief heating to 90°C. This resulted in 60 μl of combined elution fractions. Routinely, 20 μl (30%) of the combined elution fractions was analysed on Western blots in comparison with 20 μl of nuclear extract (10%) of the input fraction. Immunoblotting was performed as described in section 2.3. The first antibodies used were rabbit anti-BEAF (a gift by UK Laemmli) or rabbit anti JIL-1 (a gift by K Johansen) each at a 1:1000 dilution in PBS, 10% normal goat serum. The percentage of proteins immunoprecipitated was estimated by comparing signal intensities in Western blots of the input and precipitated fractions normalized by considering the volume of each fraction loaded.
2.5 Immunohistochemistry
Antibody staining of formaldehyde-fixed salivary gland squash preparations was performed as described previously (Saumweber et al. 1980). The Chriz and Z4 antibodies were as described previously (Eggert et al. 2004; Gortchakov et al. 2005) and mouse anti-c-Myc (9E10) was obtained from the Developmental Studies Hybridoma Bank, University of Iowa. Rabbit anti-phosphorylated H3S10 antibody was obtained from Abcam and rhodamine- or fluorescein-labelled secondary antibodies were purchased from Dianova. Preparations were inspected and documented by Olympus microscope based Deltavision Image Restoration microscopy (Applied Precision, USA) that allows quantitative fluorescence imaging. When indicated, filter settings and exposure time were essentially the same between two image stacks. Serial optical sections were deconvolved using the Resolve3D software provided with the instrument. Images were further processed and mounted using Corel Photopaint taking care to not distort brightness and contrast in comparison with the original data.
2.6 Other methods
Analysis of colocalization was carried out on the digital fluorescent images manually. The image stacks of two colours were aligned on the z-axis and an optical section of each colour in the same z-plane was selected. A reference line was traced along the middle of the chosen chromosome section stained with antibody 1, and locations of fluorescent interband signals were recorded at the point of intersection with the reference line and marked with dots. The reference line including the dots of chromosome stained with antibody 1 was transferred by copy-and-paste to the chromosome section stained with antibody 2, and dots overlapping with signals on this chromosome were recorded as colocalized. Dots that did not match to a signal were assigned to be specific signals of antibody 1. In addition, chromosome 2 was inspected for signals that did not match one of the dots of antibody 1 at the midline and therefore are specific signals for antibody 2. In this way 50–100 sites chosen at random were evaluated and from the numbers obtained the percentage of costaining was estimated.
Information on the genomic distribution of BEAF, Chriz and JIL-1 in S2-cells was retrieved from Flybase in the Modencode section using the software available online (http://www.modencode.org/).
3 Results
3.1 The zinc-finger protein Z4 and the chromodomain protein Chriz physically interact
Previously we have shown that the chromatin proteins Chriz and Z4 are colocalized in interbands of polytene chromosomes and are coimunoprecipitated using nuclear extracts from Drosophila Kc cells. Yet, initial experiments to show a physical interaction failed (Gortchakov et al. 2005). However, data obtained by improved sensitivity of the yeast two hybrid assay demonstrate a physical interaction between both proteins (figure 1). Full-length protein and several N- and C-terminal truncations of the Chriz protein in-frame with the GAL4 activation domain (AD; figure 1a) were coexpressed in yeast with the Z4 full-length protein fused in-frame with the GAL4-DNA-binding domain (DBD; figure 1b). Lysates of transformed yeast cells were tested for β-galactosidase reporter activity. The units of β-galactosidase activity measured are given to the right of each construct to indicate strength of interaction with Z4 full-length protein (n.d.: not detectable). The same Chriz constructs when coexpressed with the DBD-only control were negative in this assay. As shown in figure 1a, the full-length Chriz protein (Chr-FL) and Chriz fragments containing at least aa 279–768 (Chr-C; Chr-CDC; Chr-C4) did show significant interaction. Notably, the chromodomain is not required for Z4 binding. To the contrary, different N- and C-terminal fragments of the Z4-protein fused in-frame to the DNA-binding domain were tested for interaction with full-length Chriz protein fused to the activation domain (figure 1b). In this assay the Z4 N-terminus (N-Z4; aa 1–237) was required and sufficient for interaction.

Chriz and Z4 interact in vitro as shown by yeast two hybrid assay. (a) Aminoterminal fusion proteins of the GAL4 activation domain with Chriz full-length protein and Chriz fragments shown as grey bars numbered to the left according to the position of their first and last amino acids were coexpressed in SFY526 yeast cells with aminoterminal fusions of the GAL4-DNA-binding domain and full-length Z4 (DBD-Z4-FL; see (b)). Lysates of cotransfected cells grown in liquid culture were assayed for β-galactosidase activity to indicate interaction. Measured units of β-galactosidase activity are listed to the right of each construct. n.d. indicates that no signal was detectable in the assay as in the cotransfection of pGBT9 expressing the DNA-binding domain only, which was used as a negative control. Chromodomain region (CD; aa 208–279) is labelled in light blue. (b) Aminoterminal fusion proteins of the GAL4-DNA-binding domain with full-length Z4 or Z4 fragments shown as grey bars numbered to the left according to the position of their first and last amino acids were coexpressed in SFY526 yeast cells with a fusion protein of the GAL4 activation domain and the Chriz full-length protein (AD-Chr-FL; see (a)). Lysates of transformed yeast cells were assayed for β-galactosidase as in (a). Interaction is indicated as units of β-galactosidase activity on the right of each construct with pGAD424 vector as a negative control. The pink section in Z4 labels the zinc-finger region (Zf1–7; aa 239–515).
As an independent proof for direct interaction, we tested the N-terminal Z4 fragment (N-Z4; aa1–237) as in-frame fusion with GST for its interaction with a series of N-terminal Myc-tagged Chriz fusion proteins by pull-down assay (figure 2a). The Chriz- and Z4-fusion proteins were readily expressed in E. coli as seen on the Western blot shown in figure 2b and d. Bound fragments were eluted as described in section 2.3 and tested by Western blot using α-Myc monoclonal antibody (figure 2c). When using N-Z4-GST as a bait, Myc-tagged Chriz full-length protein (aa 29–926) and C-terminal truncations aa 29–710 and aa 29–590 were bound. In contrast, Chriz fragments aa 29–456 and aa 29–346 did not bind (figure 2c). None of the fragments was bound when the same extracts were incubated with Glutathion S-Sepharose preloaded with GST (data not shown). This demonstrates that the interaction between both proteins is direct and that the Chriz site of interaction is N-terminal of aa 590. Combining this result with the data from yeast two hybrid assays, this indicates that the Z4 interaction site on the Chriz protein maps between aa 279–590.

Chriz and Z4 interact in vitro as shown by GST-pull-down assay. Similar amounts of purified GST or N-terminal GST-NZ4 fusion protein (aa 1–237) bound to Glutathion S-Sepharose beads were incubated with cell extracts containing ~2 μg Myc-tagged Chriz protein fragments with increasing C-terminal deletions. Proteins bound to the Glutathion S-Sepharose matrix were eluted and analysed on Western blots. (a) The grey bars show Myc-tagged Chriz proteins numbered according to the position of their first and last amino acids of the Chriz fragment contained; the blue section indicates position of chromodomain (CD); +/− indicates the resulting interaction according to the Western blot shown in (c). (b) Western blot of cell extracts expressing Myc-tagged Chriz fragments; the amount loaded on the gel was the same as used for the individual binding reactions shown in (c) (100% input); protein fragments indicated at the bottom of the lanes according to the first and last amino acid of the Chriz part contained were detected with anti-Myc antibodies. (c) Western blot of Myc-tagged Chriz protein fragments bound to Glutathion S-Sepharose that was preloaded with GST-NZ4 fusion protein; eluted proteins were detected with anti-Myc antibodies; protein fragments tested for interaction are indicated at the bottom of the lanes by the numbering of the Chriz fragment as in (a); note that the two largest C-terminal truncations (29–346 and 29–456) were not bound; when Glutathion S-Sepharose preloaded with GST was used as a control in the binding reaction, we did not observe binding of any of the Myc-tagged Chriz fragments (data not shown). (d) GST and GST-NZ4 fusion protein bound and eluted from Glutathione S-Sepharose beads as in binding reactions were detected by anti-GST antibodies; M in (b–d) indicates marker proteins with MWapp in kDa as indicated.
3.2 Neither the Chriz chromodomain nor the Z4 zinc-finger region is required for interband targeting
Since the Z4 zinc-finger motif was not required for Chriz interaction, we speculated that it was required for chromosomal binding, possibly by specific DNA–protein interaction. Therefore, the full-length Z4 protein and several N- and C-terminal truncations were cloned in-frame with a N-terminal Myc-tag and a nuclear location sequence was added at the N-terminus. Constructs were inserted into pUAST P-element transformation vectors and several independent transgenic lines were established for each construct. Salivary-gland-specific overexpression was driven by crossing with G231.6-GAL4 line that is expressed in salivary glands from late embryo until the pupal stage (H Saumweber, unpublished). Significant salivary-gland-specific expression for each of the lines was established by Western blot analysis (data not shown) and chromosomal distribution was analysed by staining with Myc-specific antibodies (figure 3). The full-length protein, either in the wild-type or overexpressed, as Myc-tagged version is exclusively located in interbands of polytene chromosomes (shown for endogenous Z4 in figure 3a, bottom panel; Eggert et al. 2004). Surprisingly, the fragment Z4 Zif1–7 (aa 232–522), containing the region spanning the seven zinc-fingers, is located in bands and not in interbands (seen by the pink staining of the merged image in figure 3b, top right). Similar localization in bands was obtained for different subfragments of the zinc-finger domain, containing groups of zinc-fingers or single zinc-fingers, respectively: Z4 Zif1–3 (aa 232–328), Z4 Zif4–6 (aa 356–446), Z4 Zif4–7 (aa 356–522) and for the C-terminal fragment (aa 517–996) downstream of the zinc-finger region (data not shown). Only the N-terminal fragment N-Z4 (aa 1–237) showed an interband binding like the full-length protein (figure 3b bottom panel). Interestingly, the fragment Z4NZif1–7 (aa 1–516) including the zinc-finger region localized to interbands, too, again indicating that the N-terminal fragment specifies the chromosomal location. However, overexpression of this fragment showed a strong chromosomal phenotype, indicating a dominant negative effect of this fragment (data not shown).

N-terminal fragments of Chriz and Z4 are required for chromosomal targeting. Staining of polytene chromosomes with Hoechst (blue). Myc monoclonal antibody was used for detection of the fusion proteins and Chriz-antisera or Z4-monoclonal antibody for detection of the endogenous Z4 and Chriz proteins. Binding was visualized using the appropriate TRITC secondary antibodies (red). (a) Top: Chriz control; staining of the distal part of the X chromosome for Chriz and DNA or DNA alone; bottom: Z4 control; staining of the distal part of the X chromosome for Z4 and DNA or DNA alone; (b) top: staining of DNA, the Myc-tagged Z4-zinc-finger fragment 1–7 (Z4-ZF aa232–522) or both; the pink colour in the merged picture indicates that this fragment is located to condensed bands; bottom: staining of DNA, the Myc-tagged Z4 N-terminal fragment (N-Z4 aa1–237) or both at the distal part of X chromosome; this fragment is located in interbands; (c) Myc-tagged N-Chriz located in interbands. Top: DNA; middle: N-Chriz (aa2–211) stained with Myc-antibodies; bottom: merged. All images are single mid-chromosomal optical z-sections. Bar in (a), 2 μm.
In the same way we also mapped the region of the Chriz protein required for interband targeting by Myc-tagged transgene expression of Chriz fragments. The binding of the endogenous Chriz protein is shown in figure 3a, top panel. The N-terminal Chriz fragment ChrizN (aa 2–211) is located to interbands as the wild-type Chriz protein (figure 3 panel c). In contrast, the adjacent somewhat overlapping fragment ChrizCD (aa 160–327) containing the chromodomain is localized to bands, indicating that the chromodomain is not required for specific chromosomal targeting (data not shown). The C-terminal part of Chriz, ChrizC (aa 275–926), contains a self-interaction domain and therefore shows a slight preference for interband binding due to a self-interaction with the endogenous Chriz protein (data not shown). To conclude, for both Chriz and Z4, the N-terminal region is required for interband localization. Neither the zinc-finger region of Z4 nor the chromodomain of Chriz is needed for specific chromosomal targeting. The N-Z4 fragment responsible for Chriz interaction is also required for specific chromosomal targeting of Z4.
3.3 The presence of Chriz is required for Z4 interband-specific binding
We also wanted to know whether the two proteins are dependent on each other for specific chromatin binding. Therefore, we performed RNAi knock-down in larval imaginal discs and salivary glands by GAL4-induced hairpin RNA expression from pUAST transgenes. Significant reduction in nuclear Chriz staining was observed in the posterior compartment of wing imaginal discs when the UAS-Chriz-RNAi knock-down was induced by engrailed-GAL4, demonstrating the effectiveness of the knock-down (figure 4b). In the same cells, Chriz knock-down resulted in a significant reduction of nuclear staining by Z4, whereas cells in the anterior compartment serving as an internal control were not affected (figure 4c). In contrast, UAS-Z4-RNAi knock-down driven by engrailed-GAL4 significantly reduced nuclear Z4 staining in the posterior compartment (figure 4f) but did not affect amount and location of Chriz in these cells (figure 4g), indicating a dependence of Z4 localization on Chriz but not vice versa. A similar effect was observed for the mutual dependence of chromosomal binding of Chriz and Z4 as demonstrated by double staining of polytene chromosomes. Knock-down of UAS-Chriz RNAi induced by salivary-gland-specific G231.6-GAL4 (figure 5b; for Western results, refer to figure 10e) resulted in significant reduction of chromosomal binding of Z4 (figure 5b′). In contrast, salivary-gland-specific knock-down of Z4 by RNAi resulted in strongly reduced chromosomal binding of Z4 (figure 5d′) but only slightly affected Chriz binding (figure 5d), supporting the notion that Z4 chromosomal binding is dependent on the presence of the Chriz protein.

Nuclear location of Z4 requires the presence of Chriz. Chriz knock-down decreases Z4 nuclear location, but Z4 knock-down does not affect Chriz nuclear location. (a–d) Wing disc in which Chriz was knocked down by en-GAL4 in the posterior compartment; (e–h) wing disc in which Z4 was knocked down by en-GAL4 in the posterior compartment; (a, e) DNA stained by Hoechst; (b, g) staining by Chriz antiserum and TRITC-labelled secondary antibody; (c, f) staining by Z4 antibody and FITC-conjugated secondary antibody; (d, h) merged images of (b, c) and (f, g), respectively; the thin stippled line marks the boundary between the anterior and posterior compartment; anterior compartment on the left of this line; insets in (a–d) show enlarged views of the image on cells at the compartment boundary from the regions indicated by the squares in (a) or (e) respectively. Western blot to demonstrate the extent of Chriz knock-down is shown in figure 9. Bar in (a), 30 μm.

Chromosomal binding of Z4 requires the presence of Chriz. Chriz knock-down affects Z4 chromosomal binding but not vice versa. (a–d) Staining by Chriz antiserum and FITC-labelled secondary antibody (red); (a′–d′) staining by Z4 antibody and TRITC-conjugated secondary antibody (red); (a, a′, c, c′) wild-type controls; (b, b′) Chriz-RNAi knock-down; note that Z4 staining is also reduced; Western blot to demonstrate the extent of Chriz knock-down is shown in figure 9; (d, d′) Z4-RNAi knock-down; note that Chriz binding is not affected under these conditions. All images shown are mid-chromosomal optical sections taken and printed at the identical exposure for the green (a–d) and red (a′–d′) filter sets, respectively. Bar in (a), 10 μm.
To further demonstrate that the Z4 chromosomal binding depends on interaction with Chriz, we overexpessed Myc-tagged N-Z4 in a wild-type background and studied the binding of the endogenous Z4 protein. We reasoned that if Z4 chromosomal binding was mainly dependent on Chriz interaction, this binding should be competed by the overexpression of the N-Z4 fragment containing the Chriz interaction site. This was indeed the case (figure 6). Myc-N-Z4 overexpression in salivary glands diminishes chromosomal binding of endogenous Z4 significantly (figure 6b′). In contrast, Chriz chromosomal binding is only slightly affected by overexpressing Myc-N-Z4 (figure 6b′′). At the same time, Myc-N-Z4 replaces Z4 on interbands as demonstrated by α-Myc staining of the twin gland from the same animal (figure 6c′). As shown by the merged image (figure 6c′′′) Myc-N-Z4 protein is located in interbands under these conditions similar to the endogenous Z4 protein (figure 6a′′′). We conclude that Z4 is mainly targeted to interbands by its interaction with Chriz and that this targeting depends on its N-terminal region.

Interband binding of endogenous Z4 can be blocked by N-Z4 overexpression. (a–a′′′) wild-type control; (b–b′′′ and c–c′′′) polytene chromosomes of larvae overexpressing N-Z4 (aa1–237); note that chromosomes shown in (b–b′′′) and (c–c′′′) are from the same animal; (a, b, c) DNA staining by Hoechst; (a′, b′) staining of Z4 antibody detected by FITC-labelled secondary antiserum; note that N-Z4 competes Z4 for chromosomal binding; (c′) staining of Myc antibody detected by FITC-labelled secondary antiserum to visualize N-Z4 interband binding; (a′′–c′′) Chriz antiserum detected by TRITC-labelled secondary antibodies; (a′′′–c′′′) merged images of the rows (a, b, c), respectively. All images shown are mid-chromosomal optical z-sections taken at the same exposure for the green and red filter sets. Bar in (a), 2 μm.
3.4 Chriz and Z4 colocalize and coimmunoprecipitate with BEAF and JIL-1
Experiments on how the Chriz N-terminus provides a mechanism for chromosomal targeting are still in progress. As an alternative route to elucidate interband targeting of the Chriz–Z4 complex, we investigated the interaction with known chromosomal proteins that were reported as interband binding. The BEAF protein shows a considerable overlap in chromosomal staining with Z4 (figure 7a–d). We estimate an overlap in binding sites of roughly 60% (for the calculation, see section 2.6). However, a number of interbands is occupied by Z4 (figure 7b; red) or BEAF only (figure 7c; green). At higher resolution, significant colocalization is evident from ChIP on chip data extracted from Flybase/ModEncode. Figure 7f shows for comparison the distribution of BEAF and Chriz on a 300 kb region of distal chromosome 3 L obtained by chromatin immunoprecipitation from Drosophila S2 cells (http://www.modencode.org/). Note that although the shapes of the peaks differ in detail, the peak position is almost identical between both profiles. The colocalization is corroborated by our results from coimmunoprecipitation of BEAF with Chriz antiserum from nuclear extracts of Drosophila Kc-cells. A Western blot of proteins immunopreciptated from nuclear extracts with Z4 monoclonal antibodies and probed by BEAF antibodies is shown in figure 7e. Comparing the signal intensity of the eluate and input fraction used for immunoprecipitation (see section 2.4), we estimate that ~50% of the BEAF protein is coimmunoprecipitated with Z4 protein. Note that under the conditions used, not more than ~60% of Z4 protein is recovered by immunoprecipitation using the Z4 antibody in this assay. Taken together, our data suggest that the BEAF protein might be a component in the Chriz–Z4 complex.

BEAF colocalizes and coimmunoprecipitates with Z4. Mid-chromosomal optical z-section of a triple-stained wild-type chromosome. DNA (Hoechst, blue), Z4 (mouse monoclonal antibody; red) and BEAF (rabbit polyclonal antiserum; green) are as indicated. (a) DNA and Z4 staining; (b) Z4 staining; (c) BEAF staining and (d) the merged image of Z4 and BEAF staining with costaining shown in yellow. The extent of costaining was quantified as described in section 2.6. Bar in (a), 2 μm. (e) Western blot of BEAF coimmunoprecipitated with Z4 monoclonal antibody; lane IP: 18 μl (30%) of the eluate containing BEAF (red arrows) coimmunoprecipitated from 200 μl nuclear extract; lane input: 20 μl (10%) of nuclear extract used for the immunoprecipitation, HC, LC heavy and light chains of Z4 antibody used for IP; (f) ChIP on chip profile of a 300 kb region of distal chromosome 3 L using chromatin from S2 cells and Chriz or BEAF antisera as indicated. Data in (f) were obtained from Flybase (http://www.modencode.org/).
In a similar manner we also investigated the colocalization and coimmunoprecipitation of Z4 and JIL-1, a H3S10 specific tandem kinase (Wang et al. 2001). As evident from figure 8a–c both proteins significantly colocalize on polytene chromosomes. We estimate an extent of ~80% colocalization. However, as marked in the figure some interbands bind one or the other of the two proteins exclusively. By using nuclear extracts of Drosopila Kc-cells ~60% of JIL-1 protein was coimmunoprecipitated with Chriz (figure 8e bottom, for estimation see section 2.4). Using Z4 monoclonal antibody for JIL-1 coimmunoprecipitation a somewhat reduced yield was obtained (figure 8e top; we estimate ~30% since 30% of eluate give a similar signal than 10% of input fraction).

JIL-1 colocalizes and coimmunoprecipitates with Z4 and Chriz. (a–d) Mid-chromosomal optical z-section of a triple-stained wild-type chromosome; DNA (Hoechst, blue), Z4 (mouse monoclonal antibody; green) and JIL-1 (rabbit polyclonal antiserum; red) are as indicated. (a) Z4 (green); (b) JIL-1 (red); (c) merge of Z4 and JIL-1 (colocalization in yellow); (d) merge of JIL-1 and DNA (blue). Red dots in (a–d) mark loci labelled by JIL-1 only; green dots mark sites labelled by Z4 only. Bar in (a–c), 2 μm; in (d), 2.5 μm. (e) Western blot of JIL-1 coimmunoprecipitated with Chriz and Z4; top left: 18 μl (30%) of the eluate containing JIL-1 coimmunoprecipitated from 200 ml nuclear extract using Z4 monoclonal antibody; top right: 20 μl (10% input) of nuclear extract used for the immunoprecipitation; bottom left: 18 μl (30%) of the eluate containing JIL-1 coimmunoprecipitated from 200 ml nuclear extract using Chriz antiserum; bottom right: 20 μl (10% input) of nuclear extract used for immunoprecipitation.
We then compared the colocalization of Chriz, BEAF and JIL-1 at higher resolution using data from the Flybase obtained by chromatin immunoprecipitation from Drosophila S2 cells (http://www.modencode.org/), focussing on two well-studied interband regions that were mapped on polytene chromosomes at 3C6-7 (Rykowski et al. 1988) and 61C7-8 (Demakov et al. 2004). ChIP on chip data for the 3C6-7 interband upstream the Notch promoter (black stippled rectangles) are shown in figure 9a. Data were obtained for each two different BEAF and Chriz antisera and one JIL-1 antiserum by chromatin immunoprecipitation from Drosophila S2 cells. All antisera show at least one peak at approximately the same position, within the interband upstream of the Notch promoter. However, additional peaks nearby do not exactly match and the sequence enrichment for the two BEAF antisera and the JIL-1 antiserum is too low to be statistically significant. Similarly, data were inspected for binding to the 61C7-8 interband (red stippled rectangles shown in figure 6b) for the same antisera. BEAF and Chriz show a strong signal at the same molecular position that overlaps a broader JIL-1 binding region with signal intensities that are too low to be statistically significant. We conclude that in two regions of diploid cells that were mapped as interbands on polytene chromosomes the proteins Chriz, BEAF and JIL-1 are colocalized.

Chriz, JIL-1 and BEAF bind to sequences in S2 cells that were mapped as interbands on polytene chromosomes. (a) Top: kirre/Notch region framing the 3C6-7 interband (black stippled rectangle); line drawing below: ChIP on chip data from S2 cells for two independent BEAF and two independent Chriz antisera and for JIL-1antiserum; interband region marked by black stippled rectangle. (b) Bottom: region framing the 61C7-8 interband (red stippled rectangle), next to bantam micro-RNA gene; line drawing above: ChIP on chip data from S2 cells for two independent BEAF and two independent Chriz antisera and for JIL-1 antiserum; interband region marked by red stippled rectangle; data obtained from Flybase (http://www.modencode.org/).
3.5 Interband-specific histone H3S10 phosphorylation depends on the integrity of the Chriz–Z4 complex
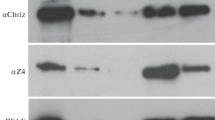
Reduction of Z4, Chriz and JIL-1 results in loss of band/interband boundaries and a distorted and partially condensed polytene chromosome phenotype (Wang et al. 2001; Eggert et al. 2004; Gortchakov et al. 2005; Rath et al. 2006; M Gan, unpublished). Targeted H3S10 phosphorylation by JIL-1 kinase, on the other hand, was demonstrated to contribute to local chromatin decondensation (Deng et al. 2008). We, therefore, wanted to know if the displacement of the Chriz–Z4 complex would affect H3S10 phosphorylation on interphase chromosomes, since the complex may serve as an endogenous targeting mechanism for the JIL-1 kinase. Therefore, we decreased Chriz and hence the chromosomally bound Z4 by UAS-Chriz RNAi induced by salivary-gland-specific G231.6-GAL4 expression and studied the consequences for H3S10 phosphorylation (figure 10). In wild-type chromosomes, H3S10 phosphorylation is present in many interbands (figure 10b). In contrast, this histone mark is strongly reduced following Chriz knock-down (figure 10d; note that in this case the exposure was slightly increased to show chromosomal background staining) along with the strong reduction of chromosomally bound Z4 protein (compare figure 5b′) and a diminished level of chromosomal JIL-1 protein (data not shown). On Western blots of salivary gland proteins from Chriz-RNAi-induced larvae, Chriz protein was strongly reduced and phosphorylated H3S10 was decreased to ~20% of its wild-type level (figure 10e). Together these findings indicate that Chriz localization to interbands is a requirement for H3S10 phosphorylation of these regions by JIL-1.

RNAi-induced Chriz knock-down strongly reduces histone H3S10 phosphorylation in interbands. (a, b) Polytene chromosomes from wild-type control. (c, d) Chriz-RNAi knock-down animals were stained for DNA by Hoechst (a, c: blue) or by phospho-H3S10 antisera (b, d: red). All images shown are mid-chromosomal optical z-sections; the image shown in (c) is somewhat overexposed to show chromosomal background and is thus lacking H3S10-phosphorylation. Bar in (a), 4 μm. (e) Western blot of proteins extracted from salivary glands of Chriz-RNAi knock-down animals (left column) or wild-type controls (right column) probed with Chriz (top) or phospho-H3S10 antisera (bottom); lamin antibodies were used as a loading control (middle).
4 Discussion
The chromatin proteins Chriz and Z4 both are ubiquitous and essential proteins that are required for the maintenance of interphase chromosome structure (Eggert et al. 2004; Gortchakov et al. 2005; Rath et al. 2006). Chriz and Z4 directly interact by their central and N-terminal domains, respectively. The Z4 N-terminal domain is required for Z4 interband targeting, mediated by Chriz interaction. When overexpressed, it competes the chromosomal binding of the endogenous Z4 protein. The Chriz N-terminal domain is required for interband targeting of the Chriz protein by an as-yet unknown mechanism. Destabilization of the complex results in strong down-regulation of H3S10 phosphorylation on polytene chromosomes.
Surprisingly, neither the Chriz chromodomain nor the Z4 zinc-finger region is needed for chromosomal targeting and no interaction partners for these signature protein domains are yet identified. However, both domains provide important functions. Point mutations in the Chriz chromodomain are unable to complement Chriz mutations (M Gan, unpublished). Point mutations within the Z4 zinc-finger region are not yet available, but overexpression of the N-terminal Z4 fragment aa 1–516 including the zinc-finger region results in a ‘shrunken’ interband phenotype (data not shown), although neither overexpression of the Z4 full-length protein nor of N-Z4 aa 1–240 affect chromosomal structure. This indicates a dominant negative effect for the zinc-finger region in the aa 1–516 construct.
Data from coimmunoprecipitation and colocalization in situ suggest that Chriz and Z4 interact with the boundary element binding protein BEAF and the H3S10-specific histone kinase JIL-1. Chriz and JIL-1 directly interact in vitro (Rath et al. 2006), but it remains to be shown if the JIL-1–Z4 interaction is also direct or mediated by Chriz protein. Also, direct interaction between Chriz–Z4 and BEAF still has to be demonstrated. Unlike the Chriz–Z4 interaction (Gortchakov et al. 2005), neither the BEAF–Z4 nor the JIL-1–Z4 colocalization is 100%. All four proteins are located in many interbands, but in each case there are sites that are occupied by one or the other protein exclusively. The mechanism explaining this promiscuity in interaction is not yet fully understood. However, it has been demonstrated that interband proteins can be shared between different chromatin protein complexes (Mohan et al. 2007; Kind et al. 2008; Bartkuhn et al. 2009; Bushey et al. 2009; Raja et al. 2010). For instance, CP190 is present in complexes with BEAF or Su(Hw) but not both. Furthermore, CP190 binds to CTCF with ~50% of CP190–CTCF complexes also bound by BEAF (Bushey et al. 2009). Mutual interactions could be mediated both by cooperation or competition between different factors for local binding. It is worth mentioning that Chriz also shows a significant overlap with CP190 in polytene chromosome binding in situ and in ChIP on chip experiments on Drosophila cell lines (H Saumweber, unpublished, and Modencode, data not shown). The overlap with both BEAF and CP190, both well known for their activity as boundary element factors (Bartkuhn et al. 2009; Bushey et al. 2009), suggests that Chriz and Z4 may be involved in the control of boundaries, a hypothesis that is currently being tested in our laboratory.
The qualitative impression of colocalization gained from polytene chromosome staining is confirmed at higher resolution by ChIP on chip data from diploid S2 cells. Although these data still await a more systematic analysis, it is evident that in some regions the match in BEAF/Chriz binding sites is rather convincing (compare figure 7) but elsewhere it is more moderate (figure 10). JIL-1 also colocalizes with Chriz but often shows a broader distribution overlapping several Chriz peaks or extending from a Chriz peak into an adjacent region (figure 10b). Strikingly, for the two cases of well-studied interband regions at 3C6-7 (Rykowski et al. 1988) and 61C7-8 (Demakov et al. 2004), binding of BEAF, Chriz and JIL-1 is also observed in S2 diploid cells, suggesting that this stretch of open chromatin is conserved in structure between cell lineages. In the case of 3C6-7, the binding may be correlated with Notch transcription in S2 cells. However, to our knowledge there is no Notch transcription in salivary glands. The transcriptional activity at interband 61C7-8 is not known.
The effect of reducing the function of Chriz, Z4 and JIL-1 on polytene chromosome structure was studied by the use of hypomorphic alleles and tissue-specific RNAi knock-down (Wang et al. 2001; Eggert et al. 2004, Rath et al. 2006; M Gan unpublished). In general, it results in a phenotype of progressive loss of distinct band interband structure, sometimes concomitant with partial condensation or torsional distortion of polytene chromosomes. Deng et al. (2008) reported that targeting of active JIL-1 kinase resulted in ectopic histone H3S10 phosphorylation and local chromosome decondensation that was dependent on JIL-1 kinase activity. We reasoned that a major role of the Chriz–Z4 complex might be the recruitment of JIL-1 to the chromosome that would result in local phosphorylation of nearby nucleosomes, allowing the ≥30 nm chromatin fibre to unfold and form a less condensed interband chromatin. In support of this hypothesis, gland-specific Chriz-RNAi resulted in decreased levels of chromosomally bound Z4 and JIL-1 and in a significant loss of interband specific H3S10 phosphorylation. However, not all chromosomes exhibited the loss of structure phenotype mentioned above. Conceivably, RNAi induction might have been too mild and there was still enough Chriz protein left to provide for the maintenance of chromosome structure. The experiments were done at 22–24°C, a temperature when the GAL4 inducer is still not at its maximal activity. Raising the temperature to 29°C early on made the inducer more effective but resulted in a tiny salivary gland phenotype that was not amenable to cytological analysis. Using the Sgs4 enhancer active in late 3rd instar at 29°C was not effective since the Chriz protein with its long half-life masked the RNAi effect. A different explanation for the lack of chromosomal phenotype may be that 10%–20% of the JIL-1 binding sites that do not depend on Chriz/Z4 are not affected by Chriz-RNAi and are sufficient to sustain the chromosome structure.
In conclusion, we provide evidence for a chromatin complex with the chromodomain protein Chriz at its core. The complex, which, besides the Z4 protein, may contain the sequence-specific DNA-binding protein BEAF, is required for H3S10 phosphorylation of interphase chromosomes, presumably by local recruitment of the tandem kinase JIL-1. Local binding and activity of the complex on polytene chromosomes may result in decondensation of interband regions and related sites on nonpolytene diploid chromosomes.
References
Bartkuhn M and Renkawitz R 2008 Long range chromatin interactions involved in gene regulation. Biochim. Biophys. Acta 1783 2161–2166
Bartkuhn M, Straub T, Herold M, Herrmann M, Rathke C, Saumweber H, Gilfillan GD, Becker PB and Renkawitz R 2009 Active promoters and insulators are marked by the centrosomal protein 190. EMBO J. 28 877–888
Bell AC, West AG, and Felsenfeld G 1999 The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98 387–396
Benyajati C and Worcel A 1976 Isolation, characterization, and structure of the folded interphase genome of Drosophila melanogaster. Cell 9 393–407
Blanton J, Gaszner M and Schedl P 2003 Protein:protein interactions and the pairing of boundary elements in vivo. Genes Dev. 17 664–675
Brand AH and Perrimon N 1993 Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118 401–415
Bushey AM, Ramos E and Corces VG 2009 Three subclasses of a Drosophila insulator show distinct and cell type-specific genomic distributions. Genes Dev. 23 1338–1350
Bushey AM, Dorman E and Corces VG 2008 Chromatin insulators: regulatory mechanisms and epigenetic inheritance. Mol. Cell 32 1–9
Cuvier O, Hart CM and Laemmli UK 1998 Identification of a class of chromatin boundary elements. Mol. Cell Biol. 18 7478–7486
Demakov S, Gortchakov A, Schwartz Y and Zhimulev I 2004 Molecular and genetic organization of Drosophila melanogaster polytene chromosomes: evidence for two types of interband regions. Genetica 122 311–324
Deng H, Bao X, Cai W, Blacketer MJ, Belmont AS, Girton J, Johansen J and Johansen KM 2008 Ectopic histone H3S10 phosphorylation causes chromatin structure remodelling in Drosophila. Development 135 699–705
De Wit E and van Stensel B 2009 Chromatin domains in higher eukaryotes: insights from genome wide mapping studies. Chromosoma 118 25–36
Eggert H, Bergemann K and Saumweber H 1998 Molecular Screening for P Element Insertions in a Large Genomic Region of Drosophila melanogaster using polymerase chain reaction mediated by the vectorette. Genetics 149 1427–1431
Eggert H, Gortchakov A and Saumweber H 2004 Identification of the Drosophila interband-specific protein Z4 as a DNA-binding zinc-finger protein determining chromosomal structure. J. Cell Sci. 117 4253–4264
Gaszner M, Vazquez J and Schedl P 1999 The Zw5 protein, a component of the scs chromatin domain boundary, is able to block enhancer-promoter interaction. Genes Dev. 13 2098–2107
Gortchakov A, Kaltenhäuser J, Eggert H and Saumweber H 2004 Construction of pMH, a covenient Escherichia coli protein expression vector. Appl. Mol. Biol. 38 713–716
Gortchakov A, Eggert H, Gan M, Mattow J, Zhimulev IF and Saumweber H 2005 Chriz, a chromodomain protein specific for the interbands of Drosophila melanogaster. Chromosoma 114 54–66
Kellum R and Schedl P 1992 A group of scs elements function as domain boundaries in an enhancer-blocking assay. Mol. Cell Biol. 12 2424–2431
Kind J, Vaquerizas JM, Gebhardt P, Gentzel M, Luscombe NM, Bertone P and Akhtar A 2008 Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell 133 813–828
Labrador M and Corces VG 2002 Setting the boundaries of chromatin domains and nuclear organization. Cell 111 151–154
Mohan M, Bartkuhn M, Herold M, Philppen A, Heinl N, Bardenhagen I, Leers J, White RAH, et al. 2007 The Drosophila insulator proteins CTCF and CP190 link enhancer blocking to body patterning. EMBO J. 26 4203–4214
Murrell A, Heweson S and Reik W 2004 Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 36 889–893
Raja SJ, Charapitsa I, Conrad T, Vaquerizas JM, Gebhardt P, Holz H, Kadlec J, Fraterman S, Luscombe NM and Akhtar A 2010 The non-specific lethal complex is a transcriptional regulator in Drosophila. Mol. Cell 38 827–841
Rath U, Ding Y, Deng H, Qi H, Bao X, Zhang W, Girton J, Johansen J and Johansen K 2006 The chromodomain protein, chromator, interacts with JIL-1 kinase and regulate the structure of polytene chromosomes. J. Cell Sci. 119 2332–2341
Reim I, Mattow J and Saumweber H 1999 The RRM protein NonA from Drosophila forms a complex with the RRM proteins Hrb87F and S5 and the Zn finger protein PEP on hnRNA. Exp. Cell Res. 253 573–586
Rykowski MC, Parmelee SJ, Agard DA and Sedat JW 1988 Precise determination of the molecular limits of a polytene chromosome band: regulatory sequences for the Notch gene are in the interband. Cell 54 461–472
Saumweber H, Symmons P, Kabisch R, Will H and Bonhoeffer F 1980 Monoclonal antibodies against chromosomal proteins of Drosophila melanogaster: establishment of antibody producing cell lines and partial characterization of corresponding antigens. Chromosoma 80 253–275
Udvardy A, Maine E and Schedl P 1985 The 87A7 chromomere. Identification of novel chromatin structures flanking the heat shock locus that may define the boundaries of higher order domains. J. Mol. Biol. 20 341–358
Vazquez J and Schedl P 1994 Sequences required for enhancer blocking activity of scs are located within two nuclease-hypersensitive regions. EMBO J. 13 5984–5993
Wallace JA and Felsenfeld G 2007 We gather together: Insulators and genome organization. Curr. Opin. Genet. Dev. 17 400–407
Wang Y, Zhang W, Jin Y, Johansen J and Johansen KM 2001 The JIL-1 tandem kinase mediates histone H3 phosphorylation and is required for maintenance of chromatin structure in Drosophila. Cell 105 433–443
West AG, Huang S, Gaszner M, Litt MD and Felsenfeld G 2004 Recruitment of histone modifications by USF proteins at a vertebrate barrier element. Mol. Cell 16 453–463
Zhao K, Hart CM and Laemmli UK 1995 Visualization of chromosomal domains with boundary element-associated factor BEAF-32. Cell 81 879–889
Acknowledgements
We thank Kristen M Johansen for her generous gift of JIL-1 antibodies, Ulrich K Laemmli for providing us with BEAF antibodies, and Andrey Gortchakov and Michael Cieslak for providing us with constructs. We are grateful to all our colleagues for providing their data to the Modencode data source in Flybase. MG was funded by a fellowship of the International PhD Program in Molecular Cell Biology that was provided by the MDC Berlin.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Gan M, Moebus S, Eggert H and Saumweber H 2011 The Chriz-Z4 complex recruits JIL-1 to polytene chromosomes, a requirement for interband-specific phosphorylation of H3S10. J. Biosci. 36 425–438] DOI 10.1007/s12038-011-9089-y
Rights and permissions
About this article
Cite this article
Gan, M., Moebus, S., Eggert, H. et al. The Chriz–Z4 complex recruits JIL-1 to polytene chromosomes,a requirement for interband-specific phosphorylation of H3S10. J Biosci 36, 425–438 (2011). https://doi.org/10.1007/s12038-011-9089-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12038-011-9089-y