
Abstract
Regardless of the progress made in the pathogenesis of ischemic stroke, it remains a leading cause of adult disability and death. To date, the most effective treatment for ischemic stroke is the timely recanalization of the occluded artery. However, the short time window and reperfusion injury have greatly limited its application and efficacy. Mitochondrial dysfunction and ATP depletion have become regarded as being hallmarks of neuropathophysiology following ischemic stroke. Mitochondrial transplantation is a novel potential therapeutic intervention for ischemic stroke that has sparked widespread concern during the past few years. This review summarizes and discusses the effects of mitochondrial transplantation in in vitro and in vivo ischemic stroke models. In addition, pharmacological interventions promoting mitochondrial transplantation are reviewed and discussed. We also discuss the potential challenges to the clinical application of mitochondrial transplantation in the treatment of ischemic stroke.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Stroke is a devastating disease caused by occlusion or rupture of blood vessels supplying the brain. It accounts for approximately 5.5 million deaths and 55% of neurological disabilities each year [1]. Stroke has imposed a significant burden on our society and every affected family due to its high mortality and disability rates [2]. There are two major types of strokes, ischemic stroke and hemorrhagic stroke. Ischemic stroke, also known as cerebral ischemia, represents approximately 80% of all stroke types [3]. Ischemic stroke mostly affected aging people [4], and in some cases ischemic stroke can be found in young people, which is caused by severe mitochondrial cytochrome c oxidase (COX) deficiency, along with a high proportion of mitochondrial DNA mutation in cortical blood vessels, leading to muscle weakness, fatigue, and pain [5]. Regardless of the significant progress made in knowledge surrounding the pathology and molecular mechanisms of ischemic stroke, the effective treatment for the condition is limited. To date, recombinant tissue plasminogen (rtPA) is the only effective drug for ischemic stroke that has been approved by the Food and Drug Administration of the United States [6, 7]. However, the short time window (about 4.5 h) and the possible side effects caused by the treatment (e.g., intracerebral bleed transformation and reperfusion injury) have largely limited its application [8,9,10]. These constraints necessitate the development of a new therapeutic approach.
The human brain is a high energy-consuming organ that uses nearly 20% of the energy produced by the human body, however, constitutes only 2% of the body mass [11, 12]. The high dependence of the brain on energy makes it vulnerable to energy crises in ischemic stroke. Mitochondria are the “powerhouse” of cells in most of the cell types in our human body. The energy produced by mitochondria supports important cellular functions such as protein transportation, neuronal cell membrane potential formation, and signal transduction [13, 14]. Mitochondrial dysfunction is thus inevitably related to neuronal cell death in ischemic stroke. Reducing or permanent loss of cerebral blood flow caused by cerebral vessel occlusion results in mitochondrial dysfunction and adenosine triphosphate (ATP) depletion, which trigger a series of cascade reactions including excessive reactive oxygen species (ROS) generation, excitotoxicity, and calcium overload [15, 16]. Mitochondria are also implicated in apoptotic neuronal death because of a long list of apoptosis-related proteins in the composition of mitochondria [17]. Taken together, mitochondria may be a good therapeutic target for ischemic stroke.
To date, there has been a notable advancement in preclinical research for therapeutic agents that target mitochondrial dysfunctions in mitochondrial diseases [18,19,20]. Nevertheless, just very few early-stage therapies succeed in the translation of these agents into clinical use, and their efficacy is still limited [19]. It was shown that under oxygen–glucose deprivation conditions, healthy adjacent astrocytes can transfer mitochondria to damaged neuronal cells to help them recover from ischemia damage [21]. However, this form of natural mitochondrial transfer under pathological conditions does not prove restoration of damaged cellular function. Mitochondrial transplantation is a novel therapeutic intervention for diseases with mitochondrial dysfunction aiming at transferring healthy mitochondria to cells or tissues with dysfunctional mitochondria to restore their energy supply [22, 23]. During the past several years, mitochondrial transplantation by using intercellular mitochondrial transfer or free mitochondrial transfer has shown attractive therapeutic efficacy in different kinds of diseases with mitochondrial dysfunction including myocardial infarction, Alzheimer’s disease, acute spinal cord injury, acute lung disease, and even cancers [24, 25]. The promising therapeutic efficacy of mitochondrial transplantation also was shown in ischemic stroke [26, 27]. In this review, evidence from articles regarding the effect of mitochondrial transplantation on ischemic stroke and pharmacological interventions targeting and facilitating this process from both in vitro and in vivo studies have been included and discussed. Challenges in promoting the clinical application of mitochondrial transplantation have also been discussed.
Mitochondria as a Potent Modulator in the Pathophysiology of Cerebral Ischemia and Cerebral Ischemia/Reperfusion Injury
Except for the well-known function as the major source of energy for most eucaryotic cell types, mitochondria also play a crucial role in a wide range of cellular functions. Mitochondrial dysfunction and ATP depletion caused by cerebral vessel occlusion have been previously related to oxidative stress, excitotoxicity, mitochondrial-based neuronal cells apoptosis, inflammation, and changes in mitochondrial quality and quantity control system, which are contributors to neuronal death in the infarct area. [15,16,17]. Even though blood flow is restored, it triggers more serious damage to the infarct area, which is known as reperfusion injury [8, 28]. All evidence above links mitochondria as a potent modulator in the pathophysiology of cerebral ischemia and cerebral ischemia/ reperfusion injury.
Oxidative stress is an important contributor to cerebral ischemia and cerebral ischemia/ reperfusion (I/R) injury [15, 29]. The role of mitochondrial dysfunction and oxidative stress has been widely reported in association with cerebral ischemia and cerebral I/R during the past few decades [15, 30]. Oxidative stress reflects the imbalance between the generation of ROS and the ability to remove them via the cellular antioxidant system [30]. Mitochondria are the major source of ROS generation [30]. Mitochondrial dysfunction induces excessive ROS production, and this situation is exacerbated during cerebral artery reperfusion [29, 31]. Excessive ROS generation in neuronal cells disrupts the balance of the antioxidant system and causes direct oxidative damage to important cellular components such as lipids, proteins, mitochondria, and DNA [29, 32]. ROS has also been associated with the opening of mitochondrial permeability transition pores (MPTP), resulting in the release of the mitochondrial intermembrane protein cytochrome C and the apoptosis-inducing factor (AIF) [13].
Excitotoxicity is the molecular mechanism first identified as being associated with cerebral ischemia and cerebral I/R injury [33, 34]. Mitochondrial dysfunction and ATP depletion are important contributors to excitotoxicity [16]. Glutamate is an important excitatory neurotransmitter in the human brain that serves as a crucial regulator in learning, synaptic plasticity, and memory [16, 35]. Uncontrolled aggregation of glutamate can lead to neuronal death because of accumulating toxic effects caused by triggering downstream receptors [35]. This phenomenon is now known as excitotoxicity [16, 35]. ATP depletion following cerebral ischemia results in electrolyte imbalance and subsequently neuronal cell membrane depolarization [35, 36]. This electrolyte imbalance prevents the uptake of glutamate from extracellular space, leading to the accumulation of glutamate [37]. Excessive accumulation of glutamate in the extracellular space induces excitotoxicity by activating N-methyl-d-aspartate receptors (NMDAR) on neuronal cells, resulting in an influx of calcium and subsequent calcium overload [16]. A high concentration of intracellular calcium in neuronal cells cascades the activation of calcium-dependent enzymes and protein kinases, causing important cellular component damage and ultimately cell death [38, 39]. Moreover, calcium overload can induce the opening of the MPTP, leading to mitochondrial dysfunction, ROS production, mitochondrial swelling, and cell death through apoptosis or necrosis pathways [40].
In addition, mitochondria are involved in neuronal cell death due to the presence of many apoptosis regulator proteins in mitochondria composition [13, 17]. Among these apoptosis regulator proteins, the B cell lymphoma 2 (Bcl-2) family are excellent regulators for the mitochondrial pathway of neuronal death [13]. The Bcl-2 family consists of two groups of proteins, specifically anti-apoptotic proteins (e.g., Bcl-xL, Bcl-w, and Bcl-2) and pro-apoptotic proteins (e.g., Bcl‐2 antagonist of cell death, BH3‐interacting domain death agonist, and Bcl‐2 antagonist) [13]. With the exception of the Bcl-2 family, several other mitochondrial proteins also play a crucial role in apoptotic cell death, for example, cytochrome C, endonuclease G, and AIF [13, 17]. Moreover, some of the mitochondrial proteins such as cytochrome C can trigger the activation of caspase and then initiate apoptotic cell death [13].
There is accumulating evidence to suggest that dysfunctional and damaged mitochondria play a crucial role in triggering inflammation and immune response during cerebral ischemia and cerebral I/R injury [17, 41, 42]. Following cerebral ischemia and cerebral I/R injury, dysfunctional and damaged mitochondria release excessive ROS and mitochondrial components including fragmented mitochondrial DNA (mtDNA). Previous studies have demonstrated that excessive ROS and fragmented mtDNA can serve as activators of the NLR family pyrin domain-containing 3 (NLRP3) inflammasomes, which finally induce the release of proinflammatory cytokines interleukin-1 (IL-1) and IL-8 [43,44,45]. The released fragmented mtDNA can also serve as a dangerous signal which can activate toll-like receptor 9 (TLR9) followed by activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway, resulting in upregulation of inflammatory cytokines (e.g., IL-6 and tumor necrosis factor-alpha) [46, 47].
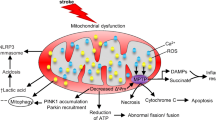
Mitochondria, themselves, have a quality and quantity control system allowing mitochondria to act against adverse situations in cerebral ischemia and cerebral I/R injury [41, 48, 49]. This system consists of mitochondrial fission, mitochondrial fusion, and mitophagy [48, 50, 51]. The process of mitochondrial fission enables mitochondria to divide into two separate mitochondrial organelles [49]. Mitochondrial fission is important in dealing with stressful situations because of its ability to divide damaged mitochondria and can protect against further damage to the healthy part [41, 49]. On the contrary, mitochondrial fusion allows the fusion of two individual mitochondria, which is important as it enables two slightly damaged mitochondria to share mitochondrial components (e.g., mitochondrial DNA and mitochondrial proteins) in a complementary fashion [41, 49]. Mitophagy is a process for the selective degradation of dysfunctional, damaged, or aging mitochondria by autophagy [41, 49]. All the mitochondrial quality and quantity control processes form a dynamic and coordinated system, which is crucial for the maintenance of mitochondrial quality, quantity, mitochondrial function, resistance to detrimental effects, and cell survival. The possible mechanisms that link mitochondria to the pathophysiology of cerebral ischemia and cerebral I/R injury are depicted in Fig. 1.

Possible mechanisms that link mitochondria and the pathophysiology of cerebral ischemia and cerebral I/R injury. Structurally, the infarct region can be divided into two areas, including the area where cell damage cannot be reversed (the infarct core) and the area in which cell damage can be reversed (the penumbra). The blockage of the cerebral artery results in ATP depletion and later electrolyte imbalance, which can restrain the uptake of glutamate from the extracellular space leading to glutamate aggregation. A high concentration of glutamate in the extracellular space over-activates NMDA receptors in the recipient neurons, leading to an influx of calcium, causing calcium overload. The overload of calcium and oxygen–glucose deprivation induce mitochondrial dysfunction, compromises ATP production, and causes excessive ROS generation. High concentrations of calcium and ROS in the mitochondria trigger the opening of MPTP, leading to the release of proapoptotic proteins (e.g., AIF, Cyt C, and Bad) and mitochondrial swelling. Proapoptotic proteins released from damaged mitochondria trigger cell death pathways and lead to apoptotic or necrotic cell death. The excessive production of ROS leads to oxidative stress and causes oxidative damage to important cellular components such as DNA, lipids, and mitochondria. ROS and mitochondrial DNA released from damaged neuronal cells trigger the activation of TLR9 and NLRP3 in immune cells, leading to the release of inflammatory cytokines such as IL-1, IL-6, IL-8, and TNF α causing inflammatory responses. Mitochondria have a quality and quantity control system allowing mitochondria to fight against cerebral ischemia and cerebral I/R injury, including mitochondrial fission, mitochondrial fusion, and mitophagy
Mitochondrial Transfer Under Pathological and Physiological Conditions and the Possible Mechanisms Involved in Mitochondrial Transfer
For a very long time, mitochondria were thought to be constrained within cells and transference between cells did not occur. The earliest report on mitochondrial transfer was in 1982 when Clark and colleagues reported that antibiotic ability in mitochondrial DNA can be transferred by simply cocultivation of the isolated mitochondria with recipient cells [52]. The first evidence for intercellular mitochondrial transfer was reported in 2006, when the authors found that mesenchymal stem cells (MSCs) restored the mitochondrial function of mtDNA mutated and depleted cells (A549 ρ° cells) by cocultivation of the two-cell lines [53]. The first in vivo study regarding mitochondrial transplantation was reported in 2009 by McCully and colleagues [54]. During the past two decades, many studies have been performed to investigate the therapeutic potential of mitochondrial transplantation on mitochondrial diseases. Mitochondrial transplantation is now considered a promising therapeutic intervention for different types of diseases, such as central nervous system diseases (e.g., spinal cord injury, Parkinson’s disease, and ischemic stroke), myocardial infarction, acute kidney injury, acute lung injury, and even various cancers [55,56,57,58,59].
It was reported that damaged neurons caused by the hypoxia-ischemic condition can send “help-me” signals to adjacent cells to trigger a series of rescue procedures [60]. These “help-me” signals were later recognized as damage-associated molecular patterns (DAMPs), which include molecules varying from chemokines, cytokines to mitochondrial debris released from damaged neuronal cells [60]. Among these “help-me” signals, the mitochondria debris released from damaged neurons can trigger intercellular mitochondrial transfer from healthy adjacent cells to damage one [24, 60]. A recent report demonstrated that mitochondria can be released from the adjacent healthy astrocytes and transported to damaged neurons to support the viability and recovery of neuronal cells after an ischemic stroke attack [21].
Mitochondrial transfer also happens under physiological conditions and plays an important role in cell differentiation, proliferation, and tissue homeostasis [61, 62]. Acquistapace and colleagues found that human multipotent adipose-derived stem cells (hMADs) were able to reprogram mouse fully differentiated cardiomyocytes toward a progenitor-like state by partial cell fusion and mitochondrial transfer [61]. The promotion ability of the hMADs was significantly decreased after mutations and depletion of mitochondrial DNA in hMADs pretreated by ethidium bromide [61]. Furthermore, according to a previous report, intercellular transfer of mitochondria from vascular smooth muscle cells was found to initiate the proliferation of MSCs [62]. Intercellular mitochondrial transplantation was also regarded as a new form of mitochondrial dynamic, important in maintaining cellular homeostasis [24].
To date, the mechanism involving mitochondrial transfer has not been fully elucidated. Several possibilities have been reported regarding the mechanisms of intercellular mitochondrial transfer, including tunneling nanotubes (TNTs), extracellular vesicles (EVs), gap junctions, and cell fusion [24, 25]. Moreover, the transplantation of mitochondria can also be performed using isolated mitochondria. As to the transfer mechanism of free mitochondria, evidence from early research suggested that endocytosis may be the major mechanism [52]. Further studies confirmed that endocytosis can be modulated by different signaling pathways in different tissue types. For example, actin-based endocytosis was suggested as being the major mechanism for free mitochondria internalization in cardiomyocytes [22]. However, mitochondrial internalization in glioma cells was shown to be induced by the nicotinamide adenine dinucleotide (NAD+)- the cluster of differentiation 38 (CD38)- cyclic adenosine diphosphate ribose (cADPR)- calcium ion (Ca2+) pathway [63]. An increased understanding of the mechanisms involved in mitochondrial transplantation will help to improve the application of this intervention in mitochondrial diseases.
The Source of Mitochondria for Mitochondrial Transplantation
A good source of mitochondria is critical for successful mitochondrial transplantation, especially for future application in clinical therapy. The mitochondria used in mitochondrial transplantation have to be viable with high respiratory competence and the supply needs to be abundant. With a focus on the possible problems associated with the immune response and histocompatibility, early research regarding mitochondrial transplantation used autologous tissue as the source of mitochondria for mitochondrial isolation [22]. Various sources of tissues were assessed as a potential source of mitochondria for cardioprotection in a previous study [22]. They found that the liver has a higher mitochondrial number than skeletal muscles and atrial tissues if the starting weight of tissue was constant, and there was no significant difference in mitochondrial uptake or the efficacy of cardioprotection when using a different source of mitochondria [22]. Immune response in mitochondrial transplantation is a topic that remains not fully understood. Until now, just very few studies focus on this area [64]. Contradictory results were obtained, in which, some of the studies showed that there were immune responses when using allogeneic mitochondria, while the other studies showed no significant immune response [64]. Despite all this, many studies regarding mitochondrial transplantation using allogeneic or even xenogeneic mitochondrial sources have been reported and promising results were obtained [24, 65]. Allogeneic or xenogeneic mitochondria are also considered because, under some circumstances such as in patients with congenital mitochondrial dysfunction, mitochondria from this source of autologous tissue cannot be used for mitochondrial transplantation. However, the obtaining of some tissues from the body of the animal is invasive and hard to perform, which to some extent will increase pain for animals and violate the principles of animal care. Thus, researchers have tried to use different cell lines as a source of mitochondria for both mitochondrial isolation and intercellular mitochondrial transfer because a cell line is easy to access by simply cell culture. The most popular candidates are stem cells owing to them having the advantage of an abundance of mitochondria, exceptional ability for self-renewal, and low oxidative damage levels [25, 66]. However, due to the aggressive isolation procedures and detrimental extracellular environment (e.g., high calcium concentration), several studies used mitochondria-containing EVs as mitochondrial carriers instead of using free mitochondria in mitochondrial transplantation [55, 67]. EVs can be further divided into three groups according to their size, including microvesicles (MVs), exosomes, and apoptotic bodies [68]. Among these, MVs are larger vesicles that contain entire mitochondrial particles [67]. MVs have been regarded as good mitochondrial carriers because they offer a degree of protection to mitochondria from high extracellular calcium concentrations [55]. Moreover, the lipid bilayer of MVs can facilitate mitochondrial transfer through the membrane of the target cells [55]. However, recent evidence suggests that platelets may serve as a possible source of mitochondria which has a lot of benefits [69, 70]. On the one hand, platelets are an abundant component in peripheral blood and are easy to acquire by simply venipuncture from peripheral blood. On the other hand, mitochondria isolated from platelets from the target autologous source can avoid the possible problems regarding the immune response and histocompatibility. Also, mitochondria isolated from platelets can fulfill the requirements for minimally invasive and ethical issues.
Therapeutic Effects of Mitochondrial Transplantation on Neuronal Cells with Induced Oxygen–Glucose Deprivation and Oxygen–Glucose Deprivation/Reoxygenation Injury
Although it is impossible to absolutely mimic the human cerebral ischemia and cerebral I/R model using a single cell line in an in vitro system with the absence of cerebral components (glial cells, blood–brain barrier, blood flow, and the infiltration of peripheral leukocytes population), it provides a good opportunity to investigate specific molecular and biochemical mechanisms that are close to the disease in the human body [71]. There are two major ways to induce in vitro cerebral ischemia, one of these is oxygen–glucose deprivation (OGD) the other being enzymatic and chemical induction [71, 72]. According to a previous report, it takes a longer time to induce neuronal death in in vitro situations compared with an in vivo cerebral ischemia model [71]. Normally, the exposure time for OGD ranges from 1 to 24 h [71]. The rate of neuronal death in hypoxic conditions is dependent upon the concentration of glucose in the culture media [72, 73]. Usually, it takes 4–8 h to cause a half-maximal neuronal loss in a concentration of 2 mM of glucose, while it takes more than 24 h to induce a half-maximal neuronal loss in a concentration of 20 mM of glucose [72, 73]. Some of the in vitro studies used oxygen deprivation alone to model cerebral ischemia, this kind of in vitro model being known as the hypoxia model [74]. Neuronal cells can live a longer time under this circumstance as they can get access to glucose under hypoxic conditions.
To date, five in vitro studies have been carried out with a focus on the effect of mitochondrial transplantation on cerebral ischemia and cerebral I/R injury. We found that four in vitro models have been used in these five studies, including the OGD model, hypoxia model (just oxygen deprivation), oxygen–glucose deprivation/reoxygenation (OGD/R) model, and the hypoxia/ reoxygenation (H/R) model. Mitochondria from autologous or xenogeneic sources were used in these studies, including free mitochondria isolated from kidney fibroblast cells, N2a cells, and peripheral blood platelets, and microvesicles from the culture medium of astrocytes. Most of these studies showed promising therapeutic efficacy. Huang et al. reported that mitochondrial transplantation under OGD conditions increased cell viability in a primary cortical cell line treated with 4 h of OGD, whereas mitochondrial transplantation did not improve cell viability in their 6- and 12-h OGD models [75]. However, mitochondrial transplantation increased both intracellular LDH and neuronal proliferation in all OGD time point studies [75]. The results from this study indicated that mitochondrial transplantation under OGD conditions increased cell survival and reduced cell injury in neurons with induced OGD injury [75]. A study conducted by Xie and colleagues observed that mitochondrial transplantation under hypoxic conditions reduced the cell viability of N2a cells that had been exposed to 48 h of hypoxia injury, suggesting that mitochondrial transplantation under hypoxic conditions may not able to reverse hypoxic cell damage [74]. They presumed that exogenous mitochondria are a load to recipient cells. They postulated that when oxygen is provided, cells can deal with this load appropriately; however, in the absence of oxygen, the host cells need to provide additional energy to deal with this stress, and this will accelerate neuronal death [74]. We notice that in the OGD model, mitochondrial transplantation under OGD conditions improved cell viability while in the hypoxia model, mitochondrial transplantation under hypoxic conditions did not have this benefit [74, 75]. Our hypothesis is that this may be in part because of the difference in models and the hypoxic duration used in their studies. As we mentioned before, it takes a longer time for the hypoxia model to induce neuronal damage in comparison with the OGD model because neuronal cells in the hypoxia model can still freely access glucose in conditions of low oxygen [72, 73]. Therefore, the results of the two studies cannot be directly compared because of the difference in models and oxygen deprivation duration. However, it is known to all that mitochondria need continual access to oxygen and glucose for the process of ATP production. Thus, we can see that most of the in vitro and in vivo stroke models performed mitochondrial transplantation treatment under reperfusion conditions. We also found that in the study by Huang et al., cell viability was not increased when the OGD duration was longer than 4 h (6 and 12 h) [75]. This trend is similar to the results observed in the studies conducted by Xie et al. [74]. As to the reason why the mitochondrial treatment under OGD conditions attenuated cell viability after 4 h of the OGD procedure, we speculate that early treatment will get a greater efficacy under OGD conditions, probably by promoting mitochondrial fusion and inhibiting the mitochondrial-dependent apoptotic pathway. Moreover, in the study by Xie and colleagues, the hypoxia/reoxygenation model was used and they found that after 48 h of the hypoxia procedure, mitochondrial treatment under reperfusion conditions increased cell viability and mitochondrial fusion, and reduced ROS production and apoptosis in neuronal cells [74]. In an in vitro OGD/R study performed by Li and colleagues, mitochondrial transplantation using mitochondria containing microvesicles isolated from culture media of astrocytes increased neuronal viability and decreased neuronal death [76]. A study by Hayakawa and colleagues demonstrated that mitochondrial transplantation using mitochondrial particles isolated from culture media of astrocytes increased ATP production and cell viability in neuronal cells which had suffered from OGD/R injury [21]. Shi et al. reported that mitochondrial transplantation increased mitochondrial function and cell viability while reducing ROS levels and neuronal death in SH-SY5Y cells subjected to OGD/R injury [70]. Collectively, most of these findings suggest that mitochondrial transplantation attenuates OGD, H/R, and OGD/R-induced neuronal damage and neuronal death, in part by improving ATP production, mitochondrial fusion, and cell proliferation, and reducing apoptosis. However, we identified some important gaps in the knowledge in the papers studied. The dose of mitochondria is missing or not clearly elucidated in these papers [21, 74,75,76]. This quantity is essential as it is a crucial reference for future studies. Additionally, the hypoxia/OGD duration differed significantly, varying from 1 to 48 h; however, very few studies explained the reason why they used this duration in their text. The time used in hypoxia/OGD is of critical importance because 1 h of OGD induction has already been shown to cause widespread neuronal death [77]. Just five in vitro studies have been published with regard to the effect of mitochondrial transplantation on the treatment of oxygen–glucose deprivation and oxygen–glucose deprivation/reoxygenation injury. More studies with an optimized study design will be of great help in understanding the underlying mechanisms involved and also inform the development of mitochondrial therapy in oxygen–glucose deprivation and oxygen–glucose deprivation/reoxygenation injury. All of these results are shown in Table 1.
Therapeutic Effects of Mitochondrial Transplantation in Rodents with Induced Cerebral Ischemia/Reperfusion Injury
The experimental stroke model provides valuable opportunities to explore the pathophysiology and the pathogenesis of the disease. In recent years, the most popular in vivo stroke models in rodents are rats and mice in contrast to early experimental stroke studies which opted to use higher animal species [71]. Rats and mice have many advantages for mimicking the human stroke model, as well as a lower cost in keeping and acquisition, easier monitoring and tissue processing, and more acceptable with regard to ethical issues [71]. There are many ways to induce an in vivo stroke model, including the intraluminal suture MCAO model, craniectomy model, photothrombosis model, endothelin-1 model, and embolic stroke model [71]. Middle cerebral artery occlusion (MCAO) is the most frequently used method for the induction of cerebral ischemia [71]. The MCAO model can be achieved by introducing a monofilament into the internal carotid artery until reaching the proximal segment of the anterior cerebral artery followed by intraluminal blocking at the origin of the middle cerebral artery and the blood flow of the middle cerebral artery can be achieved by removing this intraluminal suture [78].
Currently, there are eight in vivo studies regarding the therapeutic effect of mitochondrial transplantation on cerebral I/R injury. All these studies used the MCAO model. Rats or mice were used as the animal models. Free mitochondria isolated from skeletal muscles, the livers, or cell lines were used as sources of mitochondria. Two of the studies performed intercellular mitochondrial transfer using stem cells as sources of mitochondria. Both free mitochondrial transplantation and intercellular mitochondrial transplantation showed promising efficacy. The results showed that mitochondrial transplantation improved mitochondrial function in animals with cerebral I/R injury. In a study by Zhang et al., mitochondrial transplantation immediately after reperfusion increased ATP levels in rats with induced middle cerebral artery occlusion/ reperfusion (MCAO/R) injury [79]. Mitochondrial transplantation also restored mitochondrial respiration as indicated by increased oxygen consumption rate and extracellular acidification rate in the MCAO/R rat model study conducted by Liu et al. [80]. In addition, in a study by Yip and colleagues, mitochondrial transplantation reduced mitochondrial fission and mitochondrial damage as well as increased mitochondrial biogenesis in an MCAO/R rat model [81]. Mitochondrial transplantation protected against mitochondrial dysfunction in the early stages of stroke onset and the protective effect can be observed even 28 days after reperfusion [79,80,81]. Evidence showed that mitochondrial transplantation plays an important role in regulating oxidative stress [79, 81]. In a study by Zhang et al., mitochondrial transplantation reduced oxidative stress parameters and increased antioxidative enzymes in rats with induced MCAO/R injury [79]. Similarly, Yip and colleagues reported that reduced nicotinamide adenine dinucleotide phosphate oxidase 1 (NOX-1), NOX-2, and p22phox, known as enzymes that can promote ROS generation, were downregulated when treated with isolated mitochondria in a MCAO/R rat model [81]. In addition to the role mentioned above, mitochondrial transplantation alleviated cerebral inflammation caused by excessive glial cell activation as evidenced by decreasing glial fibrillary acidic protein (GFAP) and ionized calcium binding adaptor molecule (Iba-1) expression [79, 81, 82]. However, we noticed that in a study by Huang and colleagues, it was shown that mitochondrial transplantation increased levels of both GFAP-positive cells and Iba-1-positive cells, which are opposite to the results published by other studies [75]. Unfortunately, these results were not extensively discussed in the paper. Astrocytes and microglia are crucial components of the central nervous system. DAMPs such as fragmented mitochondria, intracellular proteins, and ROS released from damaged or necrotic cells can trigger the recruitment and activation of astrocytes and microglia after the onset of cerebral ischemia and cerebral I/R injury [83, 84]. The activation status of astrocytes and microglia can continue for several weeks after stroke onset [83, 84]. Inflammatory cytokines released by activated astrocytes and microglia are vital components of neuronal inflammation during cerebral ischemia and cerebral I/R injury [85]. There is an accumulating body of evidence to demonstrate that mitochondrial transplantation could alleviate cerebral I/R injury in part by inhibiting the activation of glial cells and glial scar formation [26, 27]. There is the possibility that glial activation might be alleviated after mitochondrial transplantation owing to its anti-inflammatory role. These results need to be extensively investigated and confirmed in future research. Mitochondrial dysfunction, oxidative stress, and inflammation caused by glial cell activation ultimately contribute to neuronal death and brain infarction after the onset of cerebral ischemia and cerebral I/R injury. Recent evidence showed that mitochondrial transplantation also plays an important role in the inhibition of neuronal death pathways. Mitochondrial transplantation reduced neuronal death in MCAO/R rat models as evidenced by a reduction in terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells, pro-apoptotic proteins (e.g., cleaved caspase 3, BCL‐2‐associated X protein, and cleaved poly polymerase), and DNA damage markers (e.g., cyclophilin D and H2A histone family member X) [75, 79, 81, 82]. All these beneficial effects following mitochondrial transplantation contribute to the reduction of infarct areas in the brain. As is shown in Table 2, mitochondrial transplantation significantly reduced the infarct area in rats with induced MCAO/R injury [74, 75, 79,80,81,82, 86, 87]. This protective effect can be observed early on day 1 after arterial reperfusion, and also be found at different time points after 28 days of reperfusion. Neurological function was assessed by different systems after mitochondrial transplantation. All the results showed that mitochondrial transplantation attenuated neurological deficits in animals with induced cerebral I/R injury [74, 75, 79,80,81,82, 87]. These outcomes may be in part attributed to the beneficial effects of mitochondrial transplantation. In the articles listed, we found that both local and systemic delivery of mitochondria can salvage cerebral I/R injury in an in vivo stroke model. Valuable data provided by Huang and colleagues showed that injection of free mitochondria into both the intra-femoral artery and intracerebral can alleviate cerebral I/R injury in rats [75]. Interestingly, they found that local administration of mitochondria seems to have a better efficacy [75]. They showed that local administration of mitochondria can more effectively reduce infarct area and cell apoptosis and induce glial activity when compared with systemic administration [75]. Moreover, local administration of mitochondria in rats attenuated a neurological deficit 7 days after stroke onset, while systemic administration showed no significant effects on neurological function at the same time in their study [75]. These results suggested that local administration of mitochondria might have greater efficacy in cerebral I/R injury. The possible explanation may be that the intracerebral injection can directly deliver mitochondria into the specific infarct area, while mitochondria that were delivered by systemic administration had to travel a long way before they arrived at the infarct site. Importantly, mitochondria delivered via systemic administration need to pass through the blood–brain barrier before they arrive in the parenchyma of the brain, and this barrier will largely confine the efficacy of the delivery of mitochondria. However, systemic administration has other advantages regarding mitochondrial delivery. Firstly, systemic administration can be achieved by simply venous injection and is less invasive therefore secondary injury caused by intracerebral injection can be avoided. Secondly, systemic administration is more acceptable to patients and is, therefore, more prone to be widely applied in clinical application. In our review, most of the mitochondrial transplantation studies were performed before or at the onset of reperfusion; therefore, mitochondrial transplantation would provide the best efficacy when it was given together with rtPA or given to the subjects after 4.5 h since at the time rtPA is not able to protect the brain against ischemic stroke. All these findings are described in Table 2.
Pharmacological Interventions that Promote Mitochondrial Transplantation
Despite promising results reported by a large number of mitochondrial transplantation studies involving different kinds of disease, the uptake ratio of transplanted free mitochondria is quite low. The uptake ratio of transplanted mitochondria in cardiomyocytes was less than 10%, while in brain mitochondrial transplantation, the uptake ratio of mitochondria was also only 7–14% [69, 70, 88]. Facilitating the improvement in mitochondrial uptake will be of great help in the outcome and development of mitochondrial therapy. For the past few years, several techniques have been developed to improve the uptake of mitochondria, including the use of centrifugation, the application of high pressure, and the help of magnetic beads and cell-penetrating peptides [25]. Emerging evidence shows that melatonin plays an important role in modulating mitochondrial function and promoting mitochondrial transfer under cerebral I/R conditions [81, 89]. Melatonin has been linked to mitochondrial protection owing to its crucial role in free radical scavenging, the promotion of mitochondrial biogenesis, and antiapoptotic ability [90]. Melatonin levels, particularly nocturnal melatonin levels, were significantly decreased in stroke patients, compared to the healthy subjects [91, 92]. A previous study suggested that a decrease of melatonin levels by 1.0 pg/ml might link to an increase in stroke risk of approximately 2% [92]. To date, two preclinical studies have carried out investigations into the possible role of melatonin in mitochondrial function and mitochondrial transplantation [81, 89]. In the in vitro study conducted by Nasoni and colleagues, treatment with melatonin immediately after reoxygenation significantly increased mitochondrial mass, mitochondrial biogenesis, and mitochondrial fusion, while reducing mitochondrial fission and oxidative stress in a murine model with induced OGD/R injury [89]. More importantly, they found that melatonin can promote mitochondrial transfer via promoting the formation of TNTs and the transfer of mitochondria through TNTs [89]. In the in vivo study by Yip et al., pretreatment with melatonin 3 h before induction of MCAO/R injury followed by treatment with free mitochondria 1 h after reperfusion significantly increased mitochondrial function and neurological function, while reducing cell death, oxidative stress, astrogliosis, and brain infarction in rats with induced cerebral I/R injury [81]. At the same time, Yip et al. also conducted an in vitro study to investigate the effect of melatonin on mitochondrial function and mitochondrial transfer [81]. Similar to the results of their in vivo study, they found that pretreatment with melatonin before induction of cerebral I/R-like injury significantly increased mitochondrial function, mitochondrial mass, and antioxidative ability while reducing neuronal death in N2a cells with H2O2-induced injury [81]. Importantly, they found that pretreatment with melatonin significantly improved mitochondrial transfer via the formation of TNTs [81]. Collectively, these results suggested that melatonin protects against mitochondrial dysfunction and promotes mitochondrial transfer under conditions of cerebral I/R injury. Melatonin may be a useful pharmacological intervention for improving the efficacy of mitochondrial transplantation. However, these promising results should be carefully confirmed in future research. Also, the pre-treatment of melatonin for ischemic stroke disease is not possible to translate the clinical application. So, future research should be conducted post-treatment of melatonin in ischemic stroke research. As melatonin is a hormone that is secreted by the human body and is vitally important in maintaining body homeostasis, the safety and possible side effects caused by the administration of melatonin cannot be excluded and should also be carefully investigated before it can be used in the treatment of the human body. All of these results are summarized in Table 3 and Table 4.
Challenges in Promoting the Clinical Application of Mitochondrial Transplantation in Cerebral Ischemia and Cerebral Ischemia/ Reperfusion Injury
Although several preclinical studies, both in vitro and in vivo, have published findings in support of the promising role of mitochondrial transplantation as a possible therapeutic intervention for cerebral ischemia and cerebral I/R injury, there is still a long way to go before it can be finally applied to the clinical situation. Nowadays, an optimal procedure clearly defining the source, the dose, the route, and timing of administration of the mitochondria has not been established in studies investigating mitochondrial transplantation and ischemic stroke, procedures evidently vital in ensuring the successful application of the therapy. Moreover, the mechanisms which support the role of mitochondrial transplantation in such a wide range of functions that can reverse not only energy deficit, but also functions such as oxidative stress, apoptosis, and modulating inflammation have not yet been fully understood. Furthermore, regardless of the fact that the uptake ratio of transplanted mitochondria is just around 7 ~ 14%, effective drugs that can promote mitochondrial uptake in ischemic stroke have not been successfully developed [70]. Even though few studies have shown that melatonin can promote intercellular mitochondrial transfer by promoting TNTs formation, further studies are needed to confirm this result [89]. Several open questions have been raised questioning the authenticity of the promising therapeutic efficacy of mitochondrial transplantation. One of these is “how can free mitochondria survive in the high calcium concentration in the extracellular space before they are taken up by recipient cells [88]?”. When mitochondria are placed in a high calcium concentration condition, calcium overload induces the opening of deadly permeability transition pore formation, which allows the leak out of mitochondrial components, resulting in mitochondrial swelling, mitochondrial fragmentation, and apoptosis [88, 93]. Mitochondria are unlikely to withstand such high calcium concentration (around 1.8 mM) in the blood or extracellular space [88]. Another is “how can extracellular mitochondria generate ATP without the help of enzymes present in the cytoplasm [88]?”. Normally, before mitochondria generate ATP, substrates such as glucose and fatty acids need to be converted into pyruvate and coenzyme A. Thus, it seems like impossible for extracellular mitochondria to generate ATP without the help of enzymes in the cytoplasm. Last but not least, “how can the few mitochondria that get into the recipient cells (mostly less than 10%) produce enough ATP to support such high energy-consuming cells or organs [88]?”. As we know, the human brain and heart are high energy-consuming organs. The brain consumes nearly 20% of the energy produced by the human body while constituting just around 2% of body mass [11]. The human heart also has a very high energy demand in order to sustain its contractile function. The heart will run out of ATP within around 2 to 10 s if there is no continued energy supply and mitochondrial phosphorylation contributes nearly 95% of the ATP requirement for the heart [94]. Thus, it is hard to understand how can such a few internalized mitochondria reverse the energy deficit of the recipient cells. These open questions are critically important and should be answered to ensure successful clinical use. A diagram illustrating the current challenges in the promotion of the clinical application of mitochondrial transplantation on cerebral ischemia and cerebral I/R injury is shown in Fig. 2.

Current challenges that prevent the clinical application of mitochondrial transplantation in cerebral ischemia and cerebral I/R injury. A To date, there are no optimal procedures with regard to the mitochondrial source, mitochondrial dose, time-point to give mitochondria, or administration route for mitochondrial transplantation in cerebral ischemia and ischemia I/R injury. B–D Several open questions that are of vital importance for mitochondrial transplantation were raised in previous reports. These questions are listed in pictures (B), (C), and (D) and the main text of our review article. The answer to these questions is important for the application of mitochondrial transplantation
Conclusion
Mitochondrial transplantation is a new and promising therapeutic intervention for cerebral ischemia and cerebral I/R injury. Evidence from both in vitro and in vivo studies suggest that mitochondrial transplantation can restore mitochondrial function, reduce oxidative stress, and inhibit apoptotic cell death. In in vivo studies, mitochondrial transplantation has also been shown to effectively reduce neuronal inflammation and the infarct area and attenuate neurological deficit. An optimal procedure for mitochondrial transplantation should be established to ensure the success of this procedure. Further clarification of the mechanism needed for successful mitochondrial transplantation and the answering of the major concerns that challenge this promising therapy will be of great help in promoting the development of this exciting treatment. We believe that mitochondrial transplantation will have broad application prospects in mitochondrial diseases, especially in the case of cerebral ischemia and cerebral I/R injury.
Data Availability
Not applicable.
References
Nian K, Harding IC, Herman IM, Ebong EE (2020) Blood-brain barrier damage in ischemic stroke and its regulation by endothelial mechanotransduction. Front Physiol 11:605398. https://doi.org/10.3389/fphys.2020.605398
Ding Q, Liu S, Yao Y, Liu H, Cai T, Han L (2022) Global, regional, and national burden of ischemic stroke, 1990–2019. Neurology 98(3):e279–e290. https://doi.org/10.1212/wnl.0000000000013115
Global Burden of Disease Study 2013 Collaborators (2015) Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386(9995):743–800. https://doi.org/10.1016/S0140-6736(15)60692-4
Soriano-Tárraga C, Giralt-Steinhauer E, Mola-Caminal M, Vivanco-Hidalgo RM, Ois A, Rodríguez-Campello A, Cuadrado-Godia E, Sayols-Baixeras S, Elosua R, Roquer J, Jiménez-Conde J (2016) Ischemic stroke patients are biologically older than their chronological age. Aging (Albany NY) 8(11):2655–2666. https://doi.org/10.18632/aging.101028
Terni E, Giannini N, Brondi M, Montano V, Bonuccelli U, Mancuso M (2015) Genetics of ischaemic stroke in young adults. BBA Clin 3:96–106. https://doi.org/10.1016/j.bbacli.2014.12.004
Feske SK (2021) Ischemic Stroke. Am J Med 134(12):1457–1464. https://doi.org/10.1016/j.amjmed.2021.07.027
Barthels D (1866) Das H (2020) Current advances in ischemic stroke research and therapies. Biochim Biophys Acta Mol Basis Dis 4:165260. https://doi.org/10.1016/j.bbadis.2018.09.012
Gauberti M, Lapergue B, Martinez de Lizarrondo S, Vivien D, Richard S, Bracard S, Piotin M, Gory B (2018) Ischemia-reperfusion injury after endovascular thrombectomy for ischemic stroke. Stroke 49(12):3071–3074. https://doi.org/10.1161/strokeaha.118.022015
Kalogeris T, Baines CP, Krenz M, Korthuis RJ (2012) Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298:229–317. https://doi.org/10.1016/b978-0-12-394309-5.00006-7
Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D (2008) Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. The New England J Med 359(13):1317–1329. https://doi.org/10.1056/NEJMoa0804656
Bélanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14(6):724–738. https://doi.org/10.1016/j.cmet.2011.08.016
Alle H, Roth A, Geiger JR (2009) Energy-efficient action potentials in hippocampal mossy fibers. Science (New York, NY) 325(5946):1405–1408. https://doi.org/10.1126/science.1174331
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science (New York, NY) 305(5684):626–629. https://doi.org/10.1126/science.1099320
Jordan J, de Groot PW, Galindo MF (2011) Mitochondria: the headquarters in ischemia-induced neuronal death. Cent Nerv Syst Agents Med Chem 11(2):98–106. https://doi.org/10.2174/187152411796011358
Orellana-Urzúa S, Rojas I, Líbano L, Rodrigo R (2020) Pathophysiology of ischemic stroke: role of oxidative stress. Curr Pharm Des 26(34):4246–4260. https://doi.org/10.2174/1381612826666200708133912
Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188. https://doi.org/10.1016/j.pneurobio.2013.11.006
Yang JL, Mukda S, Chen SD (2018) Diverse roles of mitochondria in ischemic stroke. Redox Biol 16:263–275. https://doi.org/10.1016/j.redox.2018.03.002
Singh A, Faccenda D, Campanella M (2021) Pharmacological advances in mitochondrial therapy EBioMedicine 65:103244. https://doi.org/10.1016/j.ebiom.2021.103244
Garone C, Viscomi C (2018) Towards a therapy for mitochondrial disease: an update. Biochem Soc Trans 46(5):1247–1261. https://doi.org/10.1042/bst20180134
Russell OM, Gorman GS, Lightowlers RN, Turnbull DM (2020) Mitochondrial diseases: hope for the future. Cell 181(1):168–188. https://doi.org/10.1016/j.cell.2020.02.051
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535(7613):551–555. https://doi.org/10.1038/nature18928
McCully JD, Levitsky S, Del Nido PJ, Cowan DB (2016) Mitochondrial transplantation for therapeutic use. Clin Transl Med 5(1):16. https://doi.org/10.1186/s40169-016-0095-4
Park A, Oh M, Lee SJ, Oh K-J, Lee E-W, Lee SC, Bae K-H, Han BS, Kim WK (2021) Mitochondrial transplantation as a novel therapeutic strategy for mitochondrial diseases. Int J Mol Sci 22(9):4793. https://doi.org/10.3390/ijms22094793
Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y, Shi L, Meloni BP, Zhang C, Zheng M, Gao J (2021) Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther 6(1):65. https://doi.org/10.1038/s41392-020-00440-z
Huang T, Zhang T, Gao J (2022) Targeted mitochondrial delivery: a therapeutic new era for disease treatment. J Control Release 343:89–106. https://doi.org/10.1016/j.jconrel.2022.01.025
Chen W, Huang J, Hu Y, Khoshnam SE, Sarkaki A (2020) Mitochondrial transfer as a therapeutic strategy against ischemic stroke. Transl Stroke Res 11(6):1214–1228. https://doi.org/10.1007/s12975-020-00828-7
Liu F, Lu J, Manaenko A, Tang J, Hu Q (2018) Mitochondria in ischemic stroke: new insight and implications. Aging Dis 9(5):924–937. https://doi.org/10.14336/ad.2017.1126
Lin L, Wang X, Yu Z (2016) Ischemia-reperfusion injury in the brain: mechanisms and potential therapeutic strategies. Biochem Pharmacol (Los Angel) 5(4):213–228. https://doi.org/10.4172/2167-0501.1000213
Rodrigo R, Fernández-Gajardo R, Gutiérrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W (2013) Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord: Drug Targets 12(5):698–714. https://doi.org/10.2174/1871527311312050015
Allen CL, Bayraktutan U (2009) Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke 4(6):461–470. https://doi.org/10.1111/j.1747-4949.2009.00387.x
Jelinek M, Jurajda M, Duris K (2021) Oxidative stress in the brain: basic concepts and treatment strategies in stroke. Antioxidants (Basel) 10(12):1886. https://doi.org/10.3390/antiox10121886
Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C (2007) Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 27(27):7083–7093. https://doi.org/10.1523/jneurosci.1645-07.2007
Rothman SM, Olney JW (1986) Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol 19(2):105–111. https://doi.org/10.1002/ana.410190202
Shen Z, Xiang M, Chen C, Ding F, Wang Y, Shang C, Xin L, Zhang Y, Cui X (2022) Glutamate excitotoxicity: potential therapeutic target for ischemic stroke. Biomed Pharmacother 151:113125. https://doi.org/10.1016/j.biopha.2022.113125
Kaplan-Arabaci O, Acari A, Ciftci P, Gozuacik D (2022) Glutamate scavenging as a neuroreparative strategy in ischemic stroke. Front Pharmacol 13:866738. https://doi.org/10.3389/fphar.2022.866738
Luoma JI, Kelley BG, Mermelstein PG (2011) Progesterone inhibition of voltage-gated calcium channels is a potential neuroprotective mechanism against excitotoxicity. Steroids 76(9):845–855. https://doi.org/10.1016/j.steroids.2011.02.013
Rossi DJ, Oshima T, Attwell D (2000) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403(6767):316–321. https://doi.org/10.1038/35002090
Papazian I, Kyrargyri V, Evangelidou M, Voulgari-Kokota A, Probert L (2018) Mesenchymal stem cell protection of neurons against glutamate excitotoxicity involves reduction of NMDA-triggered calcium responses and surface GluR1, and is partly mediated by TNF. Int J Mol Sci 19(3):651. https://doi.org/10.3390/ijms19030651
Verma M, Wills Z, Chu CT (2018) Excitatory dendritic mitochondrial calcium toxicity: implications for parkinson’s and other neurodegenerative diseases. Front Neurosci 12:523. https://doi.org/10.3389/fnins.2018.00523
Bauer TM, Murphy E (2020) Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ Res 126(2):280–293. https://doi.org/10.1161/circresaha.119.316306
Carinci M, Vezzani B, Patergnani S, Ludewig P, Lessmann K, Magnus T, Casetta I, Pugliatti M, Pinton P, Giorgi C (2021) Different roles of mitochondria in cell death and inflammation: focusing on mitochondrial quality control in ischemic stroke and reperfusion. Biomedicines 9(2):169. https://doi.org/10.3390/biomedicines9020169
Alishahi M, Farzaneh M, Ghaedrahmati F, Nejabatdoust A, Sarkaki A, Khoshnam SE (2019) NLRP3 inflammasome in ischemic stroke: as possible therapeutic target. Int J Stroke 14(6):574–591. https://doi.org/10.1177/1747493019841242
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225. https://doi.org/10.1038/nature09663
Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD (2013) Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191(10):5230–5238. https://doi.org/10.4049/jimmunol.1301490
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36(3):401–414. https://doi.org/10.1016/j.immuni.2012.01.009
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464(7285):104–107. https://doi.org/10.1038/nature08780
Zhang JZ, Liu Z, Liu J, Ren JX, Sun TS (2014) Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int J Mol Med 33(4):817–824. https://doi.org/10.3892/ijmm.2014.1650
Tian H, Chen X, Liao J, Yang T, Cheng S, Mei Z, Ge J (2022) Mitochondrial quality control in stroke: From the mechanisms to therapeutic potentials. J Cell Mol Med 26(4):1000–1012. https://doi.org/10.1111/jcmm.17189
Stotland A (1853) Gottlieb RA (2015) Mitochondrial quality control: Easy come, easy go. Biochim Biophys Acta 1853(10 Pt B):2802–2811. https://doi.org/10.1016/j.bbamcr.2014.12.041
Wu M, Gu X, Ma Z (2021) Mitochondrial quality control in cerebral ischemia-reperfusion injury. Mol Neurobiol 58(10):5253–5271. https://doi.org/10.1007/s12035-021-02494-8
Yang M, He Y, Deng S, Xiao L, Tian M, Xin Y, Lu C, Zhao F, Gong Y (2021) Mitochondrial quality control: a pathophysiological mechanism and therapeutic target for stroke. Front Mol Neurosci 14:786099. https://doi.org/10.3389/fnmol.2021.786099
Clark MA, Shay JW (1982) Mitochondrial transformation of mammalian cells. Nature 295(5850):605–607. https://doi.org/10.1038/295605a0
Spees JL, Olson SD, Whitney MJ, Prockop DJ (2006) Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA 103(5):1283–1288. https://doi.org/10.1073/pnas.0510511103
McCully JD, Cowan DB, Pacak CA, Toumpoulis IK, Dayalan H, Levitsky S (2009) Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol 296(1):H94-h105. https://doi.org/10.1152/ajpheart.00567.2008
Ikeda G, Santoso MR, Tada Y, Li AM, Vaskova E, Jung JH, O’Brien C, Egan E, Ye J, Yang PC (2021) Mitochondria-rich extracellular vesicles from autologous stem cell-derived cardiomyocytes restore energetics of ischemic myocardium. J Am Coll Cardiol 77(8):1073–1088. https://doi.org/10.1016/j.jacc.2020.12.060
Doulamis IP, Guariento A, Duignan T, Kido T, Orfany A, Saeed MY, Weixler VH, Blitzer D, Shin B, Snay ER, Inkster JA, Packard AB, Zurakowski D, Rousselle T, Bajwa A, Parikh SM, Stillman IE, Del Nido PJ, McCully JD (2020) Mitochondrial transplantation by intra-arterial injection for acute kidney injury. Am J Physiol Renal Physiol 319(3):F403-f413. https://doi.org/10.1152/ajprenal.00255.2020
Nascimento-Dos-Santos G, de-Souza-Ferreira E, Linden R, Galina A, Petrs-Silva H (2021) Mitotherapy: unraveling a promising treatment for disorders of the central nervous system and other systemic conditions. Cells 10(7):1827. https://doi.org/10.3390/cells10071827
Zampieri LX, Silva-Almeida C, Rondeau JD, Sonveaux P (2021) Mitochondrial transfer in cancer: a comprehensive review. Int J Mol Sci 22(6):3245. https://doi.org/10.3390/ijms22063245
Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J (2012) Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18(5):759–765. https://doi.org/10.1038/nm.2736
Xing C, Lo EH (2017) Help-me signaling: non-cell autonomous mechanisms of neuroprotection and neurorecovery. Prog Neurobiol 152:181–199. https://doi.org/10.1016/j.pneurobio.2016.04.004
Acquistapace A, Bru T, Lesault PF, Figeac F, Coudert AE, le Coz O, Christov C, Baudin X, Auber F, Yiou R, Dubois-Randé JL, Rodriguez AM (2011) Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells 29(5):812–824. https://doi.org/10.1002/stem.632
Vallabhaneni KC, Haller H, Dumler I (2012) Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev 21(17):3104–3113. https://doi.org/10.1089/scd.2011.0691
Sun C, Liu X, Wang B, Wang Z, Liu Y, Di C, Si J, Li H, Wu Q, Xu D, Li J, Li G, Wang Y, Wang F, Zhang H (2019) Endocytosis-mediated mitochondrial transplantation: transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 9(12):3595–3607. https://doi.org/10.7150/thno.33100
Yamada Y, Ito M, Arai M, Hibino M, Tsujioka T, Harashima H (2020) Challenges in promoting mitochondrial transplantation therapy. Int J Mol Sci 21(17):6365. https://doi.org/10.3390/ijms21176365
Chang JC, Wu SL, Liu KH, Chen YH, Chuang CS, Cheng FC, Su HL, Wei YH, Kuo SJ, Liu CS (2016) Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl Res 170:40-56.e43. https://doi.org/10.1016/j.trsl.2015.12.003
Paliwal S, Chaudhuri R, Agrawal A, Mohanty S (2018) Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J Biomed Sci 25(1):31. https://doi.org/10.1186/s12929-018-0429-1
D’Souza A, Burch A, Dave KM, Sreeram A, Reynolds MJ, Dobbins DX, Kamte YS, Zhao W, Sabatelle C, Joy GM, Soman V, Chandran UR, Shiva SS, Quillinan N, Herson PS, Manickam DS (2021) Microvesicles transfer mitochondria and increase mitochondrial function in brain endothelial cells. J Control Release 338:505–526. https://doi.org/10.1016/j.jconrel.2021.08.038
Veziroglu EM, Mias GI (2020) Characterizing extracellular vesicles and their diverse RNA contents. Front Genet 11:700. https://doi.org/10.3389/fgene.2020.00700
Ma H, Jiang T, Tang W, Ma Z, Pu K, Xu F, Chang H, Zhao G, Gao W, Li Y, Wang Q (2020) Transplantation of platelet-derived mitochondria alleviates cognitive impairment and mitochondrial dysfunction in db/db mice. Clin Sci (Lond) 134(16):2161–2175. https://doi.org/10.1042/cs20200530
Shi C, Guo H, Liu X (2021) Platelet mitochondria transplantation rescues hypoxia/reoxygenation-induced mitochondrial dysfunction and neuronal cell death involving the FUNDC2/PIP3/Akt/FOXO3a Axis. Cell Transplant 30:9636897211024210. https://doi.org/10.1177/09636897211024210
Sommer CJ (2017) Ischemic stroke: experimental models and reality. Acta Neuropathol 133(2):245–261. https://doi.org/10.1007/s00401-017-1667-0
Holloway PM, Gavins FN (2016) Modeling ischemic stroke in vitro: status quo and future perspectives. Stroke 47(2):561–569. https://doi.org/10.1161/strokeaha.115.011932
Goldberg MP, Choi DW (1993) Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci 13(8):3510–3524. https://doi.org/10.1523/jneurosci.13-08-03510.1993
Xie Q, Zeng J, Zheng Y, Li T, Ren J, Chen K, Zhang Q, Xie R, Xu F, Zhu J (2021) Mitochondrial transplantation attenuates cerebral ischemia-reperfusion injury: possible involvement of mitochondrial component separation. Oxid Med Cell Longev 2021:1006636. https://doi.org/10.1155/2021/1006636
Huang PJ, Kuo CC, Lee HC, Shen CI, Cheng FC, Wu SF, Chang JC, Pan HC, Lin SZ, Liu CS, Su HL (2016) Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transplant 25(5):913–927. https://doi.org/10.3727/096368915x689785
Li X, Li Y, Zhang Z, Bian Q, Gao Z, Zhang S (2021) Mild hypothermia facilitates mitochondrial transfer from astrocytes to injured neurons during oxygen-glucose deprivation/reoxygenation. Neurosci Lett 756:135940. https://doi.org/10.1016/j.neulet.2021.135940
Kalda A, Eriste E, Vassiljev V, Zharkovsky A (1998) Medium transitory oxygen-glucose deprivation induced both apoptosis and necrosis in cerebellar granule cells. Neurosci Lett 240(1):21–24. https://doi.org/10.1016/s0304-3940(97)00914-2
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20(1):84–91. https://doi.org/10.1161/01.str.20.1.84
Zhang Z, Ma Z, Yan C, Pu K, Wu M, Bai J, Li Y, Wang Q (2019) Muscle-derived autologous mitochondrial transplantation: a novel strategy for treating cerebral ischemic injury. Behav Brain Res 356:322–331. https://doi.org/10.1016/j.bbr.2018.09.005
Liu K, Guo L, Zhou Z, Pan M, Yan C (2019) Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc Res 123:74–80. https://doi.org/10.1016/j.mvr.2019.01.001
Yip HK, Dubey NK, Lin KC, Sung PH, Chiang JY, Chu YC, Huang CR, Chen YL, Deng YH, Cheng HC, Deng WP (2021) Melatonin rescues cerebral ischemic events through upregulated tunneling nanotube-mediated mitochondrial transfer and downregulated mitochondrial oxidative stress in rat brain. Biomed Pharmacother 139:111593. https://doi.org/10.1016/j.biopha.2021.111593
Pourmohammadi-Bejarpasi Z, Roushandeh AM, Saberi A, Rostami MK, Toosi SMR, Jahanian-Najafabadi A, Tomita K, Kuwahara Y, Sato T, Roudkenar MH (2020) Mesenchymal stem cells-derived mitochondria transplantation mitigates I/R-induced injury, abolishes I/R-induced apoptosis, and restores motor function in acute ischemia stroke rat model. Brain Res Bull 165:70–80. https://doi.org/10.1016/j.brainresbull.2020.09.018
Kim JY, Park J, Chang JY, Kim SH, Lee JE (2016) Inflammation after Ischemic Stroke: The Role of Leukocytes and Glial Cells. Exp Neurobiol 25(5):241–251. https://doi.org/10.5607/en.2016.25.5.241
Xu S, Lu J, Shao A, Zhang JH, Zhang J (2020) Glial cells: role of the immune response in ischemic stroke. Front Immunol 11:294. https://doi.org/10.3389/fimmu.2020.00294
Oo TT, Pratchayasakul W, Chattipakorn N, Chattipakorn SC (2020) Potential Roles of myeloid differentiation factor 2 on neuroinflammation and its possible interventions. Mol Neurobiol 57(11):4825–4844. https://doi.org/10.1007/s12035-020-02066-2
Nakamura Y, Lo EH, Hayakawa K (2020) Placental mitochondria therapy for cerebral ischemia-reperfusion injury in mice. Stroke 51(10):3142–3146. https://doi.org/10.1161/strokeaha.120.030152
Babenko VA, Silachev DN, Zorova LD, Pevzner IB, Khutornenko AA, Plotnikov EY, Sukhikh GT, Zorov DB (2015) Improving the post-stroke therapeutic potency of mesenchymal multipotent stromal cells by cocultivation with cortical neurons: The role of crosstalk between cells. Stem Cells Transl Med 4(9):1011–1020. https://doi.org/10.5966/sctm.2015-0010
Bertero E, Maack C, O’Rourke B (2018) Mitochondrial transplantation in humans: “magical” cure or cause for concern? J Clin Investig 128(12):5191–5194. https://doi.org/10.1172/jci124944
Nasoni MG, Carloni S, Canonico B, Burattini S, Cesarini E, Papa S, Pagliarini M, Ambrogini P, Balduini W, Luchetti F (2021) Melatonin reshapes the mitochondrial network and promotes intercellular mitochondrial transfer via tunneling nanotubes after ischemic-like injury in hippocampal HT22 cells. J Pineal Res 71(1):e12747. https://doi.org/10.1111/jpi.12747
Hardeland R, Cardinali DP, Brown GM, Pandi-Perumal SR (2015) Melatonin and brain inflammaging. Prog Neurobiol 127–128:46–63. https://doi.org/10.1016/j.pneurobio.2015.02.001
Fiorina P, Lattuada G, Silvestrini C, Ponari O, Dall’Aglio P (1999) Disruption of nocturnal melatonin rhythm and immunological involvement in ischaemic stroke patients. Scand J Immunol 50(2):228–231. https://doi.org/10.1046/j.1365-3083.1999.00579.x
Atanassova PA, Terzieva DD, Dimitrov BD (2009) Impaired nocturnal melatonin in acute phase of ischaemic stroke: cross-sectional matched case-control analysis. J Neuroendocrinol 21(7):657–663. https://doi.org/10.1111/j.1365-2826.2009.01881.x
Bernardi P, Rasola A, Forte M, Lippe G (2015) The Mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev 95(4):1111–1155. https://doi.org/10.1152/physrev.00001.2015
Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED (2021) Cardiac energy metabolism in heart failure. Circ Res 128(10):1487–1513. https://doi.org/10.1161/circresaha.121.318241
Acknowledgements
All figures are created with BioRender.com.
Funding
This work was supported by the Senior Research Scholar Grant from the National Research Council of Thailand (SCC); Thailand Science Research and Innovation-Chaing Mai University (Fundamental Fund 2565 to SCC); the NSTDA Research Chair grant from the National Science and Technology Development Agency Thailand (NC); and the Chiang Mai University Center of Excellence Award (NC).
Author information
Authors and Affiliations
Contributions
NC and SCC: conception and funding acquisition; HTH: writing-original draft; TOO, NA, NC, and SCC: writing-reviewing and editing.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Publication
All authors have given final approval of this version and agreed to publish this article here.
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, H., Oo, T.T., Apaijai, N. et al. An Updated Review of Mitochondrial Transplantation as a Potential Therapeutic Strategy Against Cerebral Ischemia and Cerebral Ischemia/Reperfusion Injury. Mol Neurobiol 60, 1865–1883 (2023). https://doi.org/10.1007/s12035-022-03200-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-03200-y