
Abstract
Estrogen replacement therapy (ERT) is potentially beneficial for the prevention and treatment of postmenopausal cerebral ischemia but inevitably increases the risk of cerebral hemorrhage and breast cancer when used for a long period of time. Genistein, a natural phytoestrogen, has been reported to contribute to the recovery of postmenopausal ischemic stroke with reduced risks. However, the underlying mechanism of genistein-mediated neuroprotection remains unclear. We reported that genistein exerted significant neuroprotective effects by enhancing the expression of neuronal G protein-coupled estrogen receptor (GPER) in the ischemic penumbra after cerebral reperfusion in ovariectomized (OVX) mice, and this effect was achieved through GPER-mediated inhibition of nod-like receptor protein 3 (NLRP3) inflammasome activation. In addition, we found that peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) was the pivotal molecule that participated in GPER-mediated inhibition of NLRP3 inflammasome activation in OVX mice after ischemia/reperfusion (I/R) injury. Our data suggest that the neuronal GPER/PGC-1α pathway plays an important role in genistein-mediated neuroprotection against I/R injury in OVX mice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Postmenopausal stroke seriously threatens health and shortens life expectancy in women [1, 2]. How to effectively reduce cerebral ischemic injury in postmenopausal women and improve patient prognosis is an urgent scientific problem to be solved. A previous study suggested that the rising morbidity and disability rates of cerebral stroke in postmenopausal women are mainly due to estrogen deficiency, and estrogen replacement therapy (ERT) works as a promising preventive and therapeutic strategy [3,4,5]. However, a series of studies showed that ERT not only significantly increases the incidence of endometrial cancer and breast cancer in postmenopausal women, but also had a time window for its neuroprotective effect, which seriously impairs the clinical application of ERT for stroke prevention [6,7,8]. Therefore, estrogen-like substances are urgently needed and should receive more attention for postmenopausal stroke therapy.
Genistein (4′,5,7-trihydroxyisoflavone) is one of the most abundant phytoestrogens and can be extracted from plants such as soybeans and broad beans [9]. Recent studies support the benefits of genistein in protecting the brain from chronic or acute damage through involvement in inflammatory responses, apoptotic pathways, and oxidative stress [10]. Moreover, genistein may serve as an alternative therapy for postmenopausal patients with cerebral ischemia due to the lack of ERT-related side effects [11]. However, the underlying targets of genistein-mediated neuroprotection remain largely unknown.
G protein-coupled receptor 30 (GPER), an estrogen membrane receptor, is widely expressed in the nervous system [12]. Our previous studies showed that GPER activation alleviated ischemia/reperfusion (I/R) injury in OVX mice by inhibiting the microglial inflammatory response [13]. Inflammation plays a crucial role in ischemic stress by affecting the survival of neuronal cells, and inflammasomes have attracted increasing attention because of their ability to trigger inflammation [14, 15]. As the most widely reported inflammasome in the brain, the nod-like receptor protein 3 (NLRP3) inflammasome can be activated by endogenous stress and injury signals [16]. Activated NLRP3 can recruit pre-caspase-1 to form an inflammasome complex with apoptosis-associated speck-like protein (ASC) to activate caspase-1, which further promotes the release of inflammatory factors. Moreover, our latest study revealed that genistein alleviated neurological deficits after ischemia in reproductively senescent mice by inhibiting microglial NLRP3 expression [17]. However, there is still no conclusive evidence to verify whether the anti-inflammatory effect of genistein after I/R injury is mediated by GPER.
Peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α), a key transcriptional integrator, is involved in multiple physiological functions, especially the generation of proinflammatory mediators [18, 19]. In addition, PGC-1α might be activated by exogenous neuroprotective stimuli, such as exercise [20]. Notably, a recent study suggested a reduction in NLRP3 activation in microglia-specific PGC-1α-overexpressing mice [20]. Therefore, we hypothesized that genistein may indirectly regulate the activation of the NLRP3 inflammasome by acting on GPER and that PGC-1α may play an intermediate regulatory role in the GPER/NLRP3 pathway.
To confirm the role of GPER as a promoter of the neuroprotective effects of genistein, we first examined the temporal expression of GPER and assessed cerebral infarct size, neurobehavioral deficits, and apoptosis after the downregulation of neuronal GPER. Then, to investigate the role of the NLRP3 inflammasome in genistein-mediated neuroprotection, we examined inflammasome signaling in the ischemic penumbra after neuronal GPER downregulation, as well as inflammasome-mediated apoptosis. Furthermore, to explore the role of PGC-1α in mediating the GPER/NLRP3 pathway, we examined the activation of the NLRP3 inflammasome and ischemia-induced brain injury following the downregulation of neuronal PGC-1α. Our data reveal that neuronal GPER is a promising therapeutic target for postmenopausal cerebral ischemia.
Materials and Methods
Animals and Groups
Eight-week-old female C57BL/6J mice were provided by the Experimental Animal Center of the Fourth Military Medical University. The mice were housed at 22 ± 2 °C and 50 ± 1% relative humidity. All animal studies (including mouse euthanasia procedures) were conducted in accordance with the guidelines of the Animal Care Committee of the Fourth Military Medical University and in compliance with the guidelines of the AAALAC and IACUC. The mice were randomly assigned to different experimental groups, and all surgeries and behavioral tests were performed by investigators who were blinded to the groups.
Firstly, the expression of GPER in ischemic penumbra of OVX mice was measured at different time points (pre, 1 day, 3 days, 7 days, 14 days) after unilateral middle cerebral artery occlusion/reperfusion (MCAO/R). n=14 for each group: 7 were used for Western blot analysis and 7 for quantitative reverse transcription polymerase chain reaction (RT-qPCR). Subsequently, to verify the regulation of genistein (Gen) on GPER expression after reperfusion, OVX mice were randomly assigned to the following groups: Sham, MCAO, MCAO+Vehicle (vehicle was intraperitoneally injected at 6 h, 24 h, and 48 h after MCAO/R), and MCAO+Gen (10 mg/kg of genistein was intraperitoneally injected at 6 h, 24 h, and 48 h after MCAO/R). Vehicle is the dissolved solution of Genistein, consisting of 0.5% dimethyl sulfoxide (DMSO) and 99.5% mixed liquid (saline 50%, PEG300 40%, and tween-80 10%). n=21 for each group: 7 were used for Western blot analysis, 7 for RT-qPCR, and 7 for immunofluorescence staining.
Then, we used hSyn-GPER-shRNA to knockdown neuronal GPER to examine the effect of GPER on genistein-induced neuroprotection and NLRP3 inflammasome pathway during cerebral I/R. OVX mice were assigned into six groups: Sham, MCAO, MCAO+Vehicle, MCAO+Gen, MCAO+Gen+Sh, and MCAO+Gen+Sh-GPER. The first four groups were the same as described above. Mice in the MCAO+Gen+Sh and MCAO+Gen+Sh-GPER groups were injected with hSyn-shRNA (Sh) and hSyn-GPER-shRNA (Sh-GPER) 3 weeks before MCAO, respectively, and treated with genistein after reperfusion. 8 mice in each group were used for neurological score and infarct volume, 7 for terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling (TUNEL) staining, 14 for Western blot analysis (samples were collected in 3 days and 7 days after reperfusion, respectively), 7 for RT-qPCR, 7 for immunofluorescence, and 5 for caspase-1 activity and ELISA.
In addition, to explore whether PGC-1α is a key target in GPER-mediated inhibition of NLRP3 inflammasome activation, we used hSyn-PGC-1α-shRNA to knockdown neuronal PGC-1α. The OVX mice were divided into the MCAO+Gen+Sh (hSyn-shRNA was injected 3 weeks before MCAO, and genistein was intraperitoneally injected after reperfusion) and MCAO+Gen+Sh-PGC-1α (hSyn-PGC-1α-shRNA was injected 3 weeks before MCAO, and genistein was intraperitoneally injected after reperfusion) groups. 8 mice in each group were used for neurological score and infarct volume, 4 for TUNEL staining, 8 for Western blot analysis (samples were collected in 3 days and 7 days after reperfusion, respectively), 4 for RT-qPCR, 4 for immunofluorescence, and 5 for Duolink proximity ligation assay (PLA) immunoassy, caspase-1 activity and ELISA.
The protein and mRNA levels of GPER, NLRP3 inflammasome-related proteins, and PGC-1α were measured at 3 days after I/R. Caspase-1 activity and inflammatory cytokines were measured at 3 days after I/R. Infarct volume, TUNEL staining, and cleaved casepase-3 were analyzed at 7 days after reperfusion. Neurobehavioral outcomes were assessed 1 day before MCAO and 1 day, 3 days, and 7 days after reperfusion. A detailed experimental design can be seen in Fig. 2a.
Drugs Dilutions and Treatment
Genistein was purchased from Selleck Chemicals (S1342), dissolved in DMSO (D2650, sigma) to a concentration of 200mg/ml, then diluted to 1mg/ml (DMSO was 0.5%) with mixed liquid which consists of 50% saline, 40% PEG300 (202371, Sigma-Aldrich), and 10% tween-80 (P1754, Sigma-Aldrich). Mice were intraperitoneally injected with genistein or the same volume of vehicle at 6 h, 24 h, and 48 h after reperfusion. Combined with previous studies [17, 21], we first explored the effect of different doses of genistein on GPER expression in penumbra region after reperfusion, and set the concentration gradient of 1mg/kg, 5mg/kg, and 10mg/kg. The results showed that the expression of GPER induced by genistein increased in a time-dependent manner after reperfusion, and the expression level was significantly increased at 10mg/kg (Fig. S3). Therefore, the final dose selected for the experiment was 10mg/kg.
Ovariectomy
Ovariectomy was performed with bilateral dorsolateral incisions on the abdominal frontal wall to remove both ovaries [22]. The efficacy of ovariectomy was evaluated by vaginal cytology of mice for 7 consecutive days to confirm the cessation of the estrus cycle [23].
Focal Cerebral Ischemia/Reperfusion
MCAO was performed as described previously [24]. In brief, the mice underwent 1.5% isoflurane anesthesia, a small incision was made in the skin of the right neck, and the muscle was separated under a microscope to reveal the right common carotid artery. An incision is then made in the right external carotid artery, where silicon-coated monofilament (MSMC23B104PK100, RWD Life Science) was placed and passed forward through the internal carotid artery to block the middle cerebral artery. After 60 min, the silicon-coated suture was withdrawn, and the wound was sewed up. A heating pad was used throughout the operation, and the temperature of the anus was monitored, which was maintained at 36.5–37.5 ℃. A laser speckle contrast imager was used to record cortical blood flow, which estimated intravascular RBC movement from an image stack after a 6-ms exposure time and was programmed to generate 9 pixels (3×3) for comparison calculations. Each mouse was measured three times: 10 min before MCAO, during MCAO, and 10 min after MCAO. Only animals whose local cerebral blood flow was reduced by more than 80% of the pre-MCAO baseline level were included in further experiments. Representative laser speckle imaging is shown in Fig. S1. Those animals that died during or immediately after surgery (excluded from the study) had a mortality rate of about 10%. Sham-operated animals underwent the same anesthesia and surgery except for MCA occlusion.
The ischemic penumbra was defined and sampled as previously described [17]. In short, a coronal section was taken from the anterior tip of the frontal lobe (2 to 6 mm) (4-mm thick), followed by a longitudinal section (top to bottom) of the right hemisphere approximately 1 mm from the midline. Then, a cut was made at approximately 2 o’clock from the midpoint of the section, and the wedged tissue was the penumbra.
Measurement of Infarct Size
Mice were transcardially perfused with precooled normal saline immediately after anesthesia. The brain was removed and sliced into 1-mm coronal slices. These sections were then incubated at 37 °C in a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC, T8877, Sigma-Aldrich) for 15 min and fixed in 4% paraformaldehyde (PFA, 158127, Sigma-Aldrich) overnight. Image analysis software was used to compute the infarct size after the stained brain tissue was photographed. The infarct ratio was computed with the following formula: infarct volume = (contralateral hemisphere area –ipsilateral hemisphere noninfarct area)/contralateral area.
Assessment of Behavioral Deficits
Neurobehavioral outcomes were measured by the grid-walking test, the adhesive removal test, and the neurological deficit score. For the first two tests, mice were given 3 consecutive days of training before the operation to obtain a stable baseline value, and behavioral tests were performed at 1, 3, and 7 days after I/R.
As described in a previous study [25], the adhesive removal test can be used to evaluate somatosensory deficits. After the mice were acclimated in the test chamber for 5 min, tape (0.3×0.4 cm) was pasted on the left or right forepaws with equal pressure. The time until removal of the tape from each forepaw was assessed. The maximum testing time was 120 s. If this time was exceeded, it was recorded as 120 s.
The grid-walking test is used to evaluate the walking performance of mice. The grid apparatus (40×20 cm2; grid cell: 2×2 cm2; height: 50 cm) was located in a sound attenuated room. Performance was recorded for 60 s using a video camera located beside the apparatus at an angle of approximately 20 to 40°. A foot slip was recorded when one paw completely missed a bar, with the limb falling between the bars, or when the paw was correctly placed on the bar but slipped off during weight bearing. The total steps of the left forelimb and hind limb were counted, and the foot fault ratio was determined by dividing the number of foot slips of the left forelimb and left hind limb by the total number of left steps taken within 60 s.
The neurological deficit score was based on the previously described scoring system and was assessed blind at 7 days after I/R [26]. It includes six tests including spontaneous activity, symmetry of movements, symmetry of forelimbs, climbing wall of wire cage, reaction to touch on either side of trunk, and response to vibrissa touch. The scores indicate the following: for the first three tests, no movement (0), slight movement (1), slow movement (2), and normal movement (3); for the last three tests, no movement or response (1), weak movement or response (2), and normal movement or response (3). Each mouse’s score after completing the assessment was the sum of all six separate test scores. The neurological deficit score ranged from 3 to 18. The higher the score, the better the neurological outcome.
Immunofluorescence Staining and TUNEL Staining
After 3 or 7 days of reperfusion, the mice were anesthetized with isoflurane and then transcardially perfused with normal saline followed by 4% PFA. The brain was removed and fixed in 4% PFA at 4 °C overnight and then dehydrated in 30% sucrose until the tissue completely subsided. Then, 12-μm-thick coronal sections were prepared (0.5 mm prior to bregma). Five sections were randomly selected from each mouse for staining, washed three times using PBS, permeabilized with 0.3% Triton X-100-PBS for 1 h, blocked in 10% donkey serum for 1 h, and then incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used: mouse anti-NLRP3 (1:100, AG-20B-0014-C100, Adipogen), guinea pig anti-NeuN (1:300, ABN90, Sigma-Aldrich), rabbit anti-GPER (1:100, ab260033, Abcam), and rabbit anti-PGC-1α (1;100, ab191838, Abcam). Then, the sections were incubated with secondary antibodies at room temperature for 2 h in the dark, including Alexa Fluor 488-nm goat anti-guinea pig IgG (A-11073, Thermo Fisher Scientific), Alexa Fluor 594-nm goat anti-guinea pig IgG (A-11076, Thermo Fisher Scientific), Alexa Fluor 594-nm donkey anti-rabbit IgG (A-21207, Thermo Fisher Scientific), and Alexa Fluor 488-nm donkey anti-mouse IgG (A-21202, Thermo Fisher Scientific). DAPI (300 nM, C001, GeneCopoeia) was applied to visualize all cells. In addition, an in situ cell death detection kit (11684817910, Roche) was used to examine TUNEL staining. An Olympus fluorescence microscope was used to capture images, and the apoptosis index was calculated as the ratio of TUNEL and NeuN double-positive cells to NeuN-positive cells.
Duolink II Proximity Ligation Assay
The washing, permeabilizing, and blocking processes of the Duolink II in situ proximity ligation assay (PLA) immunoassay were similar to those of immunofluorescence staining. Briefly, brain tissue sections were incubated with mouse anti-NLRP3 (1:100, AG-20B-0014-C100, Adipogen) and goat anti-ASC (1:100, ab175449, Abcam) antibodies at 4 °C overnight. After being washed, the sections were then incubated with Duolink PLA Mouse MINUS (DUO92004, Sigma-Aldrich) and PLA Goat PLUS (DUO92003, Sigma-Aldrich) proximity probes at 37 ℃ for 2 h. Ligation and amplification were performed using the Duolink in situ detection reagent kit. DAPI was used to counterstain the nuclei. An Olympus fluorescence microscope was used to capture images, and red dots indicate interactions between NLRP3 and ASC.
Western Blotting
Western blotting was performed according to our previous studies [24]. The ischemic penumbra was dissected from mouse brain tissue and homogenized in RIPA lysis buffer (P0013B, Beyotime), which contained a whole proteinase inhibitor cocktail (1%, 78442, Thermo Fisher Scientific). Protein concentration was determined using BCA protein assay kit (23227, Thermo Fisher Scientific). The extracted samples were separated by 10% SDS-PAGE (P0012A, Beyotime) and electrically transferred to polyvinylidene difluoride membranes (PVDF, 88520, Thermo Fisher Scientific), which were then blocked with 5% nonfat milk (P0216, Beyotime) for 1 h at room temperature. The following primary antibodies were used for overnight incubation at 4 ℃: rabbit anti-GPER (1:1000, ab260033, Abcam), mouse anti-NLRP3 (1:1000, AG-20B-0014-C100, Adipogen), rabbit anti-pro-caspase-1 (1:1000, ab179515, Abcam), rabbit anti-cleaved-caspase-3 (1:1000, ab32042, Abcam), and rabbit anti-PGC-1α (1:1000, ab191838, Abcam) and rabbit anti-GAPDH (1:1000, 5174, Cell Signaling Technology). The membrane was incubated with a secondary anti-rabbit (1:5000, ab6721, Abcam) or mouse antibodies (1:5000, ab6789, Abcam) for 2 h at room temperature and the protein bands were visualized using Bio-Rad system.
Quantitative Reverse Transcription Polymerase Chain Reaction
According to the instructions, TRIzol reagent (Life Technologies, USA) was used to obtain total RNA in the ischemic penumbra. Then, a standard cDNA synthesis kit (Life Technologies, USA) was used for cDNA synthesis. The mRNA level of β-actin was used as an internal control. The primer sequences were as follows: GPER forward: 5′-AACAGAGCAGCGATCTGGAC-3′, reverse: 5′-GCAGAGTCCTTGGATGGCTT-3′; PGC-1α forward: 5′-GAAAGGGCCAAACAGAGAGA-3′, reverse: 5′-GTAAATCACACGGCGCTCTT-3′; GAPDH forward: 5′-AACAGAGCAGCGATCTGGAC-3′, reverse: 5′-GCAGAGTCCTTGGATGGCTT-3′; NLRP3 forward:5′-TCCTGGTGACTTTGTATGCGT-3′, reverse: 5′-TTCTCGGGCGGGTAATCTTC-3′; and Caspase-1 forward: 5′-AGATGGCACATTTCCAGGAC-3′, reverse: 5′-GATCTTCCAGCAGCAACTTC-3′. The reverse transcription reaction was performed in a 20-μl volume with 500 ng of total RNA at 16 °C for 30 min, 42 °C for 42 min, and 85 °C for 5 min. PCR cycling started with template denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 20 s, 72 °C for 20 s, and 78 °C for 20 s.
Cytoplasm/Nuclei Fractionation
Cytoplasm/nuclei fractionation was performed on brain tissues by using MinuteTM Cytoplasmic and Nuclear Fractionation kit (SC-003, Invent Biotechnologies). The tissues were added with cytoplasmic extraction buffer at the ratio of 1ul/mg and homogenized with ultrasonic grinder. Centrifuge the tube for 5 min at 15000rpm in a microcentrifuge at 4 °C. Transfer the supernatant (cytosol fraction) to a fresh pre-chilled 1.5-ml tube. Add appropriate amounts of nuclear extraction buffer to the pellet, repeat 15-s vortexing and 1-min incubation 5 times. Immediately transfer the nuclear extract to a pre-chilled filter cartridge with collection tube and centrifuge at 15000rpm in a microcentrifuge for 30 s. Discard the filter cartridge. Store nuclear extract at −80°C until use.
Detection of Caspase-1 Activity
Caspase-1 activity was analyzed with a Caspase 1 Activity Assay Kit (C1101, Beyotime). The assay is based on spectrophotometric detection of the chromophore p-nitroanilide (pNA) after cleavage from the labeled substrate acetyl-Tyr-Val-Ala-Asp p-nitroanilide (Ac-YVAD-pNA) catalyzed by caspase-1. The pNA light emission can be quantified using a spectrophotometer 405 nm, following the manufacturer’s instructions throughout the operation.
Evaluation of Inflammatory Cytokines
The penumbra tissues were homogenized by ultrasound and centrifuged at 10,000 g for 15 min, and the supernatant was collected and cryopreserved at −80 °C. ELISA kits purchased from Nanjing Jianchen Bioengineering Institute to measure levels of the inflammatory cytokines TNF-α (H052-1), IL-1β (H002), IL-6 (H007-1), and IL-18 (H015). There was no customized ELISA plate, and the experiment was carried out in strict accordance with the manufacturer’s protocols.
Transfection of AAV
AAV containing hSyn-PGC-1α-shRNA-GFP or hSyn-GPER-shRNA-GFP was purchased from GeneChem Co., Ltd. (Shanghai, China). The target sequence of PGC-1α was 5′-CCUGUUUGAUGACAGCGAA-3′, and the target sequence of GPER was 5′-CATGTACAGCAGCGTCTTC-3′. Transfection was performed by stereotactic injection. The injection coordinates were 1.5 mm lateral to the midsagittal line, 0.4 mm anterior to the bregma, and 1.5-mm deep in the dura mater. Three weeks after injection, the presence of AAV was determined by immunofluorescence analysis and Western blotting. The results are shown in Fig. S2.
Statistical Analysis
Statistical analyses were carried out using GraphPad Prism 7.0 software. All data are presented as the mean ± standard deviation (SD). Independent t tests were used to analyze differences between two independent groups. One-way ANOVA followed by Tukey post hoc analysis was used for multiple groups. Two-way repeated measures ANOVA analysis was used to assess differences between groups in the neurobehavioral tests. P<0.05 was considered statistically significant.
Results
Genistein Reversed the Downregulation of Neuronal GPER Expression Induced by I/R Injury at Day 3 After Reperfusion
We first measured the temporal expression of GPER in the ischemic penumbra after reperfusion in OVX mice. Western blot analysis revealed a time-dependent decrease in GPER expression after I/R compared with that in the sham group (Fig. 1a). GPER expression was prominently decreased at 1 day after I/R, reached the lowest point at 3 days, and remained reduced for at least 14 days (p < 0.0001, 1 day vs. pre; p < 0.0001, 3 days vs. pre; p < 0.0001, 7 days vs. pre; p = 0.0006, 14 days vs. pre; p = 0.0451, 1 day vs. 3 days; p = 0.0293, 7 days vs. 3 days; p < 0.0001, 14 days vs. 3 days). In addition, the mRNA level of GPER was also decreased in OVX mice after I/R and reached the lowest point at 3 days (p < 0.0001, 1 day vs. pre; p < 0.0001, 3 days vs. pre; p < 0.0001, 7 days vs. pre; p = 0.0133, 14 days vs. pre; p < 0.0001, 1 day vs. 3 days; p = 0.0005, 7 days vs. 3 days; p < 0.0001, 14 days vs. 3 days; Fig. 1b).

Genistein upregulated the expression of GPER in the ischemic penumbra after I/R. a Western blotting showed that GPER expression was downregulated after I/R. b The mRNA level of GPER was measured by RT-qPCR after I/R. *p < 0.05, ***p < 0.001, ****p < 0.0001 vs. the Pre group; #p < 0.05, ###p < 0.001, ####p < 0.0001 vs. the 3-day group. c Genistein increased the expression of GPER in the ischemic penumbra on day 3 after reperfusion. d Representative images of immunofluorescence staining with antibodies against GPER and NeuN in the ischemic penumbra. e GPER mRNA levels in the ischemic penumbra on day 3 after reperfusion in the presence of genistein treatment. f Quantitative analysis of the relative fluorescence intensity of GPER in neurons. Double immunofluorescence staining revealed that genistein increased the expression of GPER in neurons on day 3 after reperfusion. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The data are presented as the mean ± SD and analyzed by one-way ANOVA followed by Tukey’s post hoc, n=7
Next, we examined the effect of genistein on the expression of GPER after I/R injury. Genistein was administered intraperitoneally to OVX mice at 6 h, 24 h, and 48 h after I/R. Considering that the protein and mRNA levels of GPER decreased most significantly at 3 days after reperfusion, samples were collected at 3 days after reperfusion for analysis. As shown in Fig. 1c, genistein notably increased the expression of GPER in the penumbra compared with that in the vehicle group at 3 days after reperfusion (p = 0.0357, Gen vs. vehicle), it was still lower than that in the sham group (p = 0.0006, Gen vs. sham). Furthermore, the transcriptional level of GPER was increased with genistein treatment relative to vehicle group in OVX mice after cerebral I/R (Fig. 1e, p = 0.0082, Gen vs. vehicle). GPER has been indicated to be widely expressed in neurons, microglias, and astrocytes [27], and we then investigated the cell specificity of GPER using double immunofluorescence staining. The results showed that the percentage of GPER+NeuN+ cells relative to GPER+ cells in the penumbra region reached about 70%, indicating that GPER was mainly expressed in neurons (Fig. 1d and Fig. S4). In addition, genistein significantly increased the expression of GPER in neurons in the ischemic penumbra (Fig. 1f, p = 0.0119, Gen vs. vehicle). Accordingly, these results suggest that I/R stress can downregulate the protein and mRNA levels of GPER in neurons, and this effect is partially reversed by genistein administration.
GPER Plays an Important Role in Genistein-Mediated Defense Against Neurologic Deficits and Apoptosis After Ischemic Injury
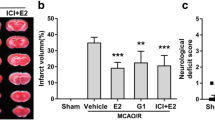
To determine whether GPER is involved in the neuroprotective effect of genistein on ischemia, we constructed an in vivo model of neuron-specific GPER knockdown using AAVs (Fig. S2a–2c). Then, for GPER downregulated mice, we hypothesized whether treatment with genistein can improve the prognosis of ischemia. The results revealed that the infarct volumes of genistein-treated OVX mice were notably decreased at 7 days after I/R, and this effect was blocked by GPER downregulation (Fig. 2b-c, p < 0.0001, Gen vs. vehicle; p = 0.0021, Gen+Sh-GPER vs. Gen+Sh). In addition, the adhesive removal test was used to assess sensomotor dysfunction, and the time to remove was decreased in the genistein administration group (Fig. 2d, p = 0.0245, Gen vs. vehicle). We also conducted a grid-walking test to examine the level of motor deficits, and the results revealed that genistein significantly increased the total steps and greatly reduced the foot fault ratio in OVX mice after I/R (Fig. 2e, p = 0.0060, Gen vs. vehicle; Fig. 2f, p = 0.0329, Gen vs. vehicle). Importantly, virus-mediated downregulation of neuronal GPER expression in the penumbra before MCAO exacerbated sensorimotor injury compared to the control virus group, even after postoperative treatment with genistein (Fig. 2d, p = 0.0353, Gen+Sh-GPER vs. Gen+Sh; Fig. 2e, p = 0.0019, Gen+Sh-GPER vs. Gen+Sh; Fig. 2f, p = 0.0498, Gen+Sh-GPER vs. Gen+Sh).

GPER participated in the neuroprotective effect of genistein against reperfusion injury. a Schematic diagram for the time frame of the experiment. b Representative images of TTC staining of mouse brains at 7 days after I/R. c The infract volumes, as measured by TTC staining. **p < 0.01, ****p < 0.0001. The data are presented as the mean ± SD and analyzed by one-way ANOVA followed by Tukey’s post hoc, n=8. d The adhesive removal test was performed to analyze somatosensory deficits before (Pre) and after reperfusion. e–f The total steps (e) and foot fault ratios (f) in the grid-walking test before (Pre) and after reperfusion. *p < 0.05, **p < 0.01, ****p < 0.0001. The data are presented as the mean ± SD and analyzed by two-way RM ANOVA, n=8
Our previous study showed that genistein had a significant antiapoptotic effect [17]. Here, we also investigated the impact of regulating GPER on the antiapoptotic effect of genistein. The data illustrated that genistein reduced the number of TUNEL-positive neurons in the penumbra at 7 days after reperfusion, while the reduction in apoptosis induced by genistein disappeared after the downregulation of GPER (Fig. 3a-b, p < 0.0001, Gen vs. vehicle; p = 0.0490, Gen+Sh-GPER vs. Gen+Sh). The protein level of cleaved caspase 3 in the penumbra, a well-known apoptotic executor, was significantly decreased in the genistein group at 7 days after reperfusion (Fig. 3a-b, p < 0.0001, Gen vs. vehicle). However, there was a noticeable increase in the GPER virus group compared to the control virus group, even though both groups were treated with genistein (Fig. 3c, p = 0.0282, Gen+Sh-GPER vs. Gen+Sh). Overall, these results suggest that GPER participates in genistein-mediated neuroprotection in the ischemic penumbra after cerebral reperfusion. It is worth noting that the apoptosis in the GPER virus group was worse than those in the control virus group, but there was still a statistical difference compared with that in the vehicle group (Fig. 3b, p = 0.0483, Gen+Sh-GPER vs. vehicle; Fig. 3c, p = 0.0478, Gen+Sh-GPER vs. vehicle), indicating that neuronal GPER was only partially involved in the anti-apoptotic effect of genistein, and there may be other molecular pathways.

GPER was involved in the antiapoptotic effect of genistein during I/R. a Representative TUNEL staining in the ischemic penumbra on day 7 after reperfusion. b The percentage of TUNEL-positive neurons in the ischemic penumbra. c Western blot analysis of cleaved caspase-3 expression in the ischemic penumbra 7 days after reperfusion. The upper panel shows cleaved caspase-3 and the corresponding GAPDH bands. The histogram in the lower panel shows the densitometric analysis results. *p < 0.05, **p < 0.01, ****p < 0.0001. The data are presented as the mean ± SD and analyzed by one-way ANOVA followed by Tukey’s post hoc, n=7
GPER Participated in Genistein-Mediated Neuroprotection by Suppressing the Synthesis and Activation of the NLRP3 Inflammasome
To uncover the relationship between GPER and the NLRP3 inflammasome, we first determined the effect of genistein treatment on NLRP3 protein levels after I/R. Western blot analysis indicated that genistein greatly decreased NLRP3 expression in the ischemic penumbra at 3 days after reperfusion, while the expression of NLRP3 was further increased after the downregulation of GPER (Fig. 4a, p < 0.0001, Gen vs. vehicle; p = 0.0259, Gen+Sh-GPER vs. Gen+Sh). Additionally, the mRNA level of NLRP3 was also decreased by genistein treatment and increased by GPER deficiency (Fig. 4b, p = 0.0013, Gen vs. vehicle; p = 0.0496, Gen+Sh-GPER vs. Gen+Sh). Furthermore, double immunofluorescence staining showed that the expression of neuronal NLRP3 was downregulated by genistein and upregulated by GPER deficiency (Fig. 4e-f, p = 0.0048, Gen vs. vehicle; p = 0.0497, Gen+Sh-GPER vs. Gen+Sh). NLRP3 can recruit pro-caspase-1 to form an inflammasome complex with ASC to activate caspase-1. We also measured the level of pro-caspase-1 and found that genistein could suppress the expression of pro-caspase-1 at 3 days after I/R, and this effect could be blocked by GPER downregulation (Fig. 4c, p = 0.0008, Gen vs. vehicle; p = 0.0205, Gen+Sh-GPER vs. Gen+Sh), which was consistent with the change in the mRNA level of pro-caspase-1 (Fig. 4d, p = 0.0019, Gen vs. vehicle; p = 0.0495, Gen+Sh-GPER vs. Gen+Sh).

GPER mediated the inhibitory effect of genistein on the formation of NLRP3 inflammasomes in neurons. a Western blot analysis of NLRP3 expression in the ischemic penumbra at 3 days after reperfusion. The upper panel shows NLRP3 and the corresponding GAPDH bands. The histogram in the lower panel shows the densitometric analysis results. b The mRNA level of NLRP3 was determined by RT-qPCR. c Western blot analysis of pro-caspase-1 expression in the ischemic penumbra at 3 days after reperfusion. The upper panel shows pro-caspase-1 and the corresponding GAPDH bands. The histogram in the lower panel shows the results of densitometric analysis. d The mRNA level of pro-caspase-1 was determined by RT-qPCR. e Coronal immunofluorescence images of the ischemic penumbra after staining with antibodies against NeuN and NLRP3. Neurons are marked with NeuN. f Quantitative analysis of the relative fluorescence intensity of NLRP3 in neurons. Double immunofluorescence staining revealed that genistein decreased the expression of NLRP3 in neurons on day 3 after reperfusion, and this inhibitory effect was offset by the downregulation of GPER. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The data are presented as the mean ± SD and analyzed by one-way ANOVA followed by Tukey’s post hoc, n=7
Given that the binding of ASC is critical for NLRP3 inflammasome activation, we next examined the interaction between these two proteins using the Duolink II proximity ligation assay. Duolink puncta (red) indicated the interaction of NLRP3 with ASC. Quantitative analysis indicated that genistein notably reduced the number of Duolink puncta compared with that in the vehicle group of OVX mice at 3 days after I/R and that GPER downregulation increased the number of Duolink puncta (Fig. 5a-b, p < 0.0001, Gen vs. vehicle; p = 0.0483, Gen+Sh-GPER vs. Gen+Sh). Activated caspase-1 cleaves the cytokine precursors pro-interleukin-1 beta (pro-IL-1β) and pro-IL-18 into their mature forms: IL-1β and IL-18 [28]. Thus, we examined caspase-1 activity, IL-1β, IL-18, and two other inflammatory cytokines (tumor necrosis factor alpha (TNF-α) and IL-6) in the ischemic penumbra at 3 days after cerebral reperfusion. As shown in Fig. 5c, genistein could inhibit caspase-1 activity, while downregulation of GPER reversed this inhibition (p = 0.0124, Gen vs. vehicle; p = 0.0475, Gen+Sh-GPER vs. Gen+Sh). The results revealed that the levels of IL-1β, IL-18, TNF-α, and IL-6 in the genistein group were strongly reduced (Fig. 5d, p = 0.0028, Gen vs. vehicle; Fig. 5e, p = 0.0005, Gen vs. vehicle; Fig. 5f, p < 0.0001, Gen vs. vehicle; Fig. 5g, p = 0.0002, Gen vs. vehicle), and as expected, GPER downregulation reversed the genistein-induced decrease in these inflammatory cytokines in OVX mice (Fig. 5d, p = 0.0025, Gen+Sh-GPER vs. Gen+Sh; Fig. 5e, p = 0.0061, Gen+Sh-GPER vs. Gen+Sh; Fig. 5f, p = 0.0006, Gen+Sh-GPER vs. Gen+Sh; Fig. 5g, p = 0.0003, Gen+Sh-GPER vs. Gen+Sh). In conclusion, we showed that GPER mediated the anti-inflammatory effect of genistein by indirectly inhibiting the activation of the NLRP3 inflammasome. However, the molecular pathway by which GPER inhibits the activation of NLRP3 inflammasomes is unknown.

Neuronal GPER was involved in genistein-mediated inhibition of NLRP3 inflammasome activation and the release of inflammatory cytokines. a The interaction (red particles) of NLRP3 with ASC was examined by Duolink analysis and DAPI (blue) counterstaining of nuclei. b The quantified results of the Duolink analysis. c Caspase-1 activity was measured by assay kit at 3 days after reperfusion in the ischemic penumbra. d–g The levels of the proinflammatory cytokines IL-6, IL-1β, TNF-α, and IL-18 in the ischemic penumbra were measured by ELISA at 3 days after reperfusion. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The data are presented as the mean ± SD and analyzed by one-way ANOVA followed by Tukey’s post hoc, n=7 (a, b), n=5 (c–g)
GPER Suppressed the Formation of the NLRP3 Inflammasome by Elevating the Expression of PGC-1α
Peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) is a key coregulator of gene transcription and is involved in many neurological diseases, such as neurodegeneration and neuroinflammation. To verify whether PGC-1α participates in GPER-mediated inhibition of the NLRP3 inflammasome in genistein-induced neuroprotection in cerebral I/R injury, we first measured the expression of PGC-1α in the genistein-treated I/R model. We found that PGC-1α expression was upregulated in both cytoplasm and nucleus after reperfusion, and further upregulated after genistein supplementation (Fig. 6a, p < 0.0001, Gen vs. vehicle; Fig. 6b, p < 0.0001, Gen vs. vehicle). However, PGC-1α was suppressed by GPER knockdown (Fig. 6a, p = 0.0003, Gen+Sh-GPER vs. Gen+Sh; Fig. 6b, p = 0.0039, Gen+Sh-GPER vs. Gen+Sh). Additionally, the mRNA level of PGC-1α was also increased in the genistein group and further decreased in the GPER-knockdown group (Fig. 6c, p < 0.0001, Gen vs. vehicle; p = 0.0003, Gen+Sh-GPER vs. Gen+Sh). Double immunofluorescence staining was performed and further showed that PGC-1α expression was increased in neurons treated with genistein and that GPER knockdown blocked the genistein-induced increase in PGC-1α in OVX mice (Fig. 6d-e, p < 0.0001, Gen vs. vehicle; p = 0.0088, Gen+Sh-GPER vs. Gen+Sh). These results suggest that PGC-1α may be an endogenous protective molecule and a regulated target of GPER in genistein neuroprotective effect.

Genistein regulated the expression of PGC-1α through GPER in neurons. a Immunoblotting was used to measure the cytoplasmic protein level of PGC-1α after I/R in the penumbra. The relative optical density was calculated by dividing the density of the PGC-1α band by that of the corresponding GAPDH band. b Immunoblotting was used to measure the intranuclear protein level of PGC-1α after I/R in the penumbra. The relative optical density was calculated by dividing the density of the PGC-1α band by that of the corresponding histone band. c The mRNA level of PGC-1α was determined by RT-qPCR. d Representative immunofluorescence images showing staining with antibodies against NeuN and PGC-1α in the ischemic penumbra. e Quantitative analysis of the relative fluorescence intensity of PGC-1α in neurons. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The data are presented as the mean ± SD and analyzed by one-way ANOVA followed by Tukey’s post hoc, n=7
Next, we constructed a virus that specifically downregulated neuronal PGC-1α to observe the effect of PGC-1α downregulation on the synthesis and activation of the NLRP3 inflammasome in genistein-mediated neuroprotection against I/R injury (Fig. S2d–f). We found that neuronal PGC-1α downregulation increased the protein and mRNA levels of NLRP3 after genistein administration in OVX mice during I/R injury (Fig. 7a, p = 0.0039; Fig. 7d, p = 0.0002). Immunofluorescence staining further showed that neuron-specific PGC-1α downregulation could increase NLRP3 expression levels in neurons (Fig. 7b-c, p = 0.0022). In addition, pro-caspase-1 protein and mRNA levels were also increased in the PGC-1α virus group, which was consistent with the changes in NLRP3 (Fig. 7e, p = 0.0073; Fig. 7f, p = 0.0040). Furthermore, we confirmed that neuronal PGC-1α downregulation after treatment with genistein could increase the number of NLRP3-ASC complexes (Fig. 8a-b, p = 0.0013) and the activation of caspase-1 (Fig. 8c, p = 0.0005), promoting the release of inflammatory cytokines (Fig. 8d, p = 0.0022; Fig. 8e, p = 0.0005; Fig. 8f, p = 0.0003; Fig. 8g, p = 0.0023) in the penumbra at 3 days after I/R. All of these suggest that PGC-1α regulates the synthesis and activation of NLRP3 inflammasome and the release of downstream inflammatory cytokines.

Neuron-specific PGC-1α knockdown promoted the transcription and expression of NLRP3. a Western blot analysis showed that PGC-1α knockdown significantly improved the expression of NLRP3. b Representative immunofluorescence images showing staining with antibodies against NeuN and NLRP3 in the ischemic penumbra. c Quantitative analysis of the relative fluorescence intensity of NLRP3 in neurons. d The mRNA level of NLRP3 was determined by RT-qPCR. e Western blot analysis of pro-caspase-1 expression at 3 days after reperfusion in the ischemic penumbra. The upper panel shows pro-caspase-1 and the corresponding GAPDH bands. The histogram in the lower panel shows the densitometric analysis results. f The mRNA level of pro-caspase-1 was determined by RT-qPCR. **p < 0.01, ***p < 0.001. The data are presented as the mean ± SD and analyzed by independent t test, n=4

Neuron-specific PGC-1α knockdown promoted the activation of NLRP3 inflammasomes and the release of downstream inflammatory cytokines. a Duolink analysis showed the interaction (red particles) of NLRP3 with ASC and DAPI (blue)-counterstained nuclei. b Quantitative analysis of the interaction of NLRP3 with ASC. c Caspase-1 activity in the penumbra was examined by assay kit at 3 days after reperfusion. d–g The levels of the proinflammatory cytokines IL-6, IL-1β, TNF-α, and IL-18 in the penumbra were examined by ELISA at 3 days after reperfusion. **p < 0.01, ***p < 0.001. The data are presented as the mean ± SD and analyzed by independent t test, n=5
Moreover, we further verified whether regulation of PGC-1α affects the neuroprotective effect of genistein, and found that the infarct volumes in the PGC-1α knockdown group were significantly higher than those in the control virus group (Fig. 9a–b, p < 0.0001). We then assessed sensorimotor deficits following the downregulation of neuronal PGC-1α using the adhesive removal and grid-walking tests. The time taken to remove the adhesive tape was clearly increased in the PGC-1α-knockdown group (Fig. 9c, p = 0.0175). In addition, PGC-1α downregulation resulted in obvious decreases in the total steps and increases in the foot fault ratio in the grid-walking test after the restoration of circulation (Fig. 9d, p = 0.0029; Fig. 9e, p = 0.0495). TUNEL staining revealed that PGC-1α downregulation accelerated neuronal apoptosis at 7 days after reperfusion (Fig. 9f–g, p = 0.0234). Cleaved caspase-3 analysis also showed that PGC-1α downregulation exacerbated neuronal apoptosis at 7 days after I/R (Fig. 9h, p = 0.0040). These results indicate that PGC-1α in neurons contributes to the neuroprotective effect as an intermediate regulator and mediates the regulatory effect of GPER on the NLRP3 inflammasome, but do not rule out the existence of other neuroprotective pathways.

Neuron-specific PGC-1α knockdown exacerbated ischemic injury and apoptosis. a Representative TTC staining at 7 days after I/R. b The infract volumes, as shown by TTC staining. c The adhesive removal test was performed to analyze somatosensory deficits before (Pre) and after reperfusion. d–e The total steps (d) and foot fault ratios (e) in the grid-walking test before (Pre) and after reperfusion. f Representative TUNEL staining in the ischemic penumbra on day 7 after reperfusion. g The percentage of TUNEL-positive neurons in the ischemic penumbra. h Western blot analysis was performed to measure cleaved caspase-3 expression in the ischemic penumbra at 7 days after reperfusion. The upper panel shows cleaved caspase-3 and the corresponding GAPDH bands. The histogram in the lower panel shows the densitometric analysis results. *p < 0.05, **p < 0.01, ***p < 0.001. The data are presented as the mean ± SD. The data analyzed by independent t test (b, g, h) or two-way RM ANOVA (c–e). n=8 (a–e), n=4 (f–h)
Discussion
This study offers several new discoveries. First, genistein had significant anti-inflammatory and neuroprotective effects on the ischemic penumbra after I/R by upregulating neuronal GPER. Second, GPER knockdown counteracted the suppression of NLRP3 activation and inflammatory cytokine production during genistein-mediated neuroprotection against reperfusion injury. Third, neuronal PGC-1α functions as an important mediator between GPER and NLRP3 activation during I/R injury. These findings suggest that the GPER/PGC-1α/NLRP3 pathway is a promising target for alleviating cerebral I/R (Fig. 10).

Schematic diagram of the GPER/PGC-1α/NLRP3 pathway in genistein-mediated neuroprotection against reperfusion injury after ischemic stroke
Genistein is a natural isoflavone that is used as an alternative to postmenopausal hormone therapy for brain disease due to its safety and ability to effectively penetrate the blood–brain barrier (BBB) penetration [9, 10, 29]. Some preclinical studies support the beneficial effects of genistein in protecting against ischemia or other brain damage [30, 31]. It is thought that, like estrogen, genistein is a multipotent molecule that is involved in a variety of distinct mechanisms to strengthen brain recovery, including reducing oxidative stress, promoting growth factor signaling, and immunosuppression [32]. Additionally, these effects occur in endothelial cells, glia, and neurons to provide a synergistically beneficial profile to fight against brain injury [10]. However, the underlying mechanism of genistein-mediated neuroprotection against I/R injury is not fully understood, which is a serious question that remains to be solved.
GPER is widely located in the hippocampus, cortex, striatum, and other nervous system regions [12, 33]. Previous studies on genistein have mainly focused on classic nuclear estrogen receptors [34, 35], while the effect of genistein on GPER has been less well studied. A recent study demonstrated that GPER participated in the anti-inflammatory effect of genistein on lipopolysaccharide (LPS)-induced microglial activation [36]. In this study, we observed that the downregulation of GPER expression in the ischemic penumbra induced by MCAO/R could be partially reversed by genistein in OVX mice. Importantly, the upregulated GPER was mainly expressed in neurons. However, the precise role of neuronal GPER in genistein-mediated neuroprotection has not been clarified, especially with regard to the specific downstream signaling pathways. To uncover the exact role of GPER in neurons, we constructed a neuron-specific GPER downregulation model in vivo and further examined the anti-inflammatory and antiapoptotic effects of genistein. The results revealed that neuronal GPER participates in genistein-induced neuroprotection against brain damage, and its underlying mechanism requires more attention.
Neuroinflammation also significantly contributes to neuronal death in reperfusion injury after ischemic stroke [37]. The NLRP3 inflammasome is a major inflammatory pathway that regulates caspase-1 activation and triggers the release of the proinflammatory cytokines IL-1β and IL-18 [28]. Our previous study confirmed that genistein could inhibit the NLRP3 inflammasome in microglia and exert anti-inflammatory effects. However, the correlation between GPER and the NLRP3 inflammasome and its potential key molecules, especially in neurons, are still poorly understood. Here, we showed that neuronal GPER upregulation induced by genistein could inhibit the activation of the NLRP3 inflammasome. Furthermore, we discovered that genistein dramatically decreased inflammatory cytokines, and this effect was reversed by the downregulation of GPER after I/R injury. The maturation of IL-1β and IL-18 is mainly regulated by the NLRP3 inflammasome, which is responsible for many inflammatory diseases [38]. We hypothesized that genistein plays an anti-inflammatory role by upregulating the transcription and expression of GPER, thereby indirectly inhibiting the activation of the NLRP3 inflammasome.
A recent study indicated that microglial PGC-1α plays a key role in protecting against cerebral ischemic damage by reducing NLRP3 activation and proinflammatory cytokine production [39]. PGC-1α is a key coregulator of gene transcription and is involved in the occurrence of a variety of neurological diseases, such as neurodegeneration and neuroinflammation [40]. Whether PGC-1α is involved in the signaling pathway by which GPER regulates the NLRP3 inflammasome remains unclear. To this end, we specifically designed an in vivo model of neuron-specific PGC-1α knockdown by AAV transfection. Surprisingly, we observed that PGC-1α expression in the penumbra was upregulated after ischemia reperfusion, but it was more significantly upregulated in the genistein administration group. Importantly, the downregulation of neuronal GPER antagonized the genistein-induced increase in PGC-1α. Moreover, we also found that neuronal PGC-1α downregulation increased the synthesis and activation of NLRP3 inflammasomes and exacerbated neurological dysfunction and apoptosis after I/R injury. These results strongly indicate that neuronal PGC-1 plays a pivotal role in genistein-mediated neuroprotection.
It should be pointed out that in our study, the downregulation of neuronal GPER only partially reversed the neuroprotective effect induced by genistein, suggesting that there may be other molecular pathways other than GPER, which is consistent with previous studies. For example, Donzelli et al. demonstrated that genistein exerts a neuroprotective effect by activating ERβ in cerebral ischemia models [34]. Studies have shown that genistein can reduce nerve damage by targeting oxidative stress, apoptosis, and inflammation [10]. There is increasing evidence that genistein is a pleiotropic neuroprotective molecule that acts on a variety of receptors, cell types, signaling pathways, and genes.
This pathway illuminates the underlying mechanism of GPER-induced inflammation in neurons during reperfusion injury after ischemic stroke. In conclusion, this study provides a new idea for the underlying mechanism behind the neuroprotective effect of genistein on cerebral ischemia/reperfusion injury. Neuronal GPER upregulation induced by genistein treatment can inhibit the activation of NLRP3 inflammasomes and result in anti-inflammatory and antiapoptotic effects on reperfusion by upregulating PGC-1α. Therefore, our study demonstrates that neuronal GPER enhances the endogenous defense system of neurons in the penumbra to fight against brain damage after I/R injury.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- ERT:
-
Estrogen replacement therapy
- GPER:
-
G protein-coupled estrogen receptor
- OVX:
-
Ovariectomized
- NLRP3:
-
Nod-like receptor protein 3
- PGC-1α:
-
Peroxisome proliferator-activated receptor-gamma coactivator 1α
- Genistein:
-
4′,5,7-Trihydroxyisoflavone
- I/R:
-
Ischemia/reperfusion
- MCAO:
-
Middle cerebral artery occlusion
- TTC:
-
2,3,5-Triphenyltetrazolium chloride
- PFA:
-
Paraformaldehyde
- PBS:
-
Phosphate-buffered saline
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- RT-qPCR:
-
Quantitative reverse transcription polymerase chain reaction
- PLA:
-
Proximity ligation assay
- ASC:
-
Apoptosis-associated speck-like protein
- BBB:
-
Blood-brain barrier
- LPS:
-
Lipopolysaccharide
References
Persky RW, Turtzo LC, Mccullough LD (2010) Stroke in women: disparities and outcomes. Curr Cardiol Rep 12(1):6–13. https://doi.org/10.1007/s11886-009-0080-2
Reeves MJ, Bushnell CD, Howard G, Gargano JW, Duncan PW, Lynch G, Khatiwoda A, Lisabeth L (2008) Sex differences in stroke: epidemiology, clinical presentation, medical care, and outcomes. Lancet Neurol 7(10):915–926. https://doi.org/10.1016/S1474-4422(08)70193-5
Alonso DLM, Egido JA, Fernandez C, Martinez-Vila E, Santos S, Morales A, Martinez E, Pareja A et al (2007) Risk of ischemic stroke and lifetime estrogen exposure. Neurology 68(1):33–38. https://doi.org/10.1212/01.wnl.0000250238.69938.f5
Grady D, Rubin SM, Petitti DB, Fox CS, Black D, Ettinger B, Ernster VL, Cummings SR (1992) Hormone therapy to prevent disease and prolong life in postmenopausal women. Ann Intern Med 117(12):1016–1037. https://doi.org/10.7326/0003-4819-117-12-1016
Engler-Chiurazzi EB, Brown CM, Povroznik JM, Simpkins JW (2017) Estrogens as neuroprotectants: estrogenic actions in the context of cognitive aging and brain injury. Prog Neurobiol 157:188–211. https://doi.org/10.1016/j.pneurobio.2015.12.008
Beral V (2003) Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 362(9382):419–427. https://doi.org/10.1016/s0140-6736(03)14065-2
Rossouw JE, Anderson GL, Prentice RL, Lacroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA et al (2002) Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 288(3):321–333. https://doi.org/10.1001/jama.288.3.321
Hodis HN, Mack WJ, Henderson VW, Shoupe D, Budoff MJ, Hwang-Levine J, Li Y, Feng M et al (2016) Vascular effects of early versus late postmenopausal treatment with estradiol. N Engl J Med 374(13):1221–1231. https://doi.org/10.1056/NEJMoa1505241
Mukund V, Mukund D, Sharma V, Mannarapu M, Alam A (2017) Genistein: Its role in metabolic diseases and cancer. Crit Rev Oncol Hematol 119:13–22. https://doi.org/10.1016/j.critrevonc.2017.09.004
Schreihofer DA, Oppong-Gyebi A (2019) Genistein: mechanisms of action for a pleiotropic neuroprotective agent in stroke. Nutr Neurosci 22(6):375–391. https://doi.org/10.1080/1028415X.2017.1391933
Thangavel P, Puga-Olguin A, Rodriguez-Landa JF, Zepeda RC (2019) Genistein as potential therapeutic candidate for menopausal symptoms and other related diseases. Molecules 24(21). https://doi.org/10.3390/molecules24213892
Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, Prossnitz ER, Dun NJ (2007) Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol 193(2):311–321. https://doi.org/10.1677/JOE-07-0017
Zhang Z, Qin P, Deng Y, Ma Z, Guo H, Guo H, Hou Y, Wang S et al (2018) The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. J Neuroinflammation 15(1):206. https://doi.org/10.1186/s12974-018-1246-x
Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157(5):1013–1022. https://doi.org/10.1016/j.cell.2014.04.007
Sharma D, Kanneganti TD (2016) The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol 213(6):617–629. https://doi.org/10.1083/jcb.201602089
Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20(13). https://doi.org/10.3390/ijms20133328
Wang S, Wang J, Wei H, Gu T, Wang J, Wu Z, Yang Q (2020) Genistein attenuates acute cerebral ischemic damage by inhibiting the NLRP3 inflammasome in reproductively senescent mice. Front Aging Neurosci 12:153. https://doi.org/10.3389/fnagi.2020.00153
St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K et al (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127(2):397–408. https://doi.org/10.1016/j.cell.2006.09.024
Rius-Perez S, Torres-Cuevas I, Millan I, Ortega AL, Perez S (2020) PGC-1alpha, inflammation, and oxidative stress: an integrative view in metabolism. Oxid Med Cell Longev 2020:1452696. https://doi.org/10.1155/2020/1452696
Wrann CD, White JP, Salogiannnis J, Laznik-Bogoslavski D, Wu J, Ma D, Lin JD, Greenberg ME et al (2013) Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab 18(5):649–659. https://doi.org/10.1016/j.cmet.2013.09.008
Castello-Ruiz M, Torregrosa G, Burguete MC, Salom JB, Gil JV, Miranda FJ, Jover-Mengual T, Marrachelli VG et al (2011) Soy-derived phytoestrogens as preventive and acute neuroprotectors in experimental ischemic stroke: influence of rat strain. Phytomedicine 18(6):513–515. https://doi.org/10.1016/j.phymed.2011.02.001
Idris AI (2012) Ovariectomy/orchidectomy in rodents. Methods Mol Biol 816:545–551. https://doi.org/10.1007/978-1-61779-415-5_34
Mclean AC, Valenzuela N, Fai S, Bennett SA (2012) Performing vaginal lavage, crystal violet staining, and vaginal cytological evaluation for mouse estrous cycle staging identification. J Vis Exp (67):e4389. https://doi.org/10.3791/4389
Cai Y, Guo H, Fan Z, Zhang X, Wu D, Tang W, Gu T, Wang S et al (2020) Glycogenolysis is crucial for astrocytic glycogen accumulation and brain damage after reperfusion in ischemic stroke. iScience 23(5):101136. https://doi.org/10.1016/j.isci.2020.101136
Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P, Freret T (2009) The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc 4(10):1560–1564. https://doi.org/10.1038/nprot.2009.125
Garcia JH, Wagner S, Liu KF, Hu XJ (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation Stroke 26(4):627-634 635. https://doi.org/10.1161/01.str.26.4.627
Wang Z, Huang K, Yang X, Shen K, Yang L, Ruan R, Shi X, Wang M et al (2021) Downregulated GPR30 expression in the epileptogenic foci of female patients with focal cortical dysplasia type IIb and tuberous sclerosis complex is correlated with (18) F-FDG PET-CT values. Brain Pathol 31(2):346–364. https://doi.org/10.1111/bpa.12925
Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J et al (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 14(9):1590–1604. https://doi.org/10.1038/sj.cdd.4402194
Duan X, Li Y, Xu F, Ding H (2021) Study on the neuroprotective effects of Genistein on Alzheimer’s disease. Brain Behav 11(5):e2100. https://doi.org/10.1002/brb3.2100
Liang HW, Qiu SF, Shen J, Sun LN, Wang JY, Bruce IC, Xia Q (2008) Genistein attenuates oxidative stress and neuronal damage following transient global cerebral ischemia in rat hippocampus. Neurosci Lett 438(1):116–120. https://doi.org/10.1016/j.neulet.2008.04.058
Aras AB, Guven M, Akman T, Alacam H, Kalkan Y, Silan C, Cosar M (2015) Genistein exerts neuroprotective effect on focal cerebral ischemia injury in rats. Inflammation 38(3):1311–1321. https://doi.org/10.1007/s10753-014-0102-0
Rahman MM, Hongsprabhas P (2016) Genistein as antioxidant and antibrowning agents in in vivo and in vitro: a review. Biomed Pharmacother 82:379–392. https://doi.org/10.1016/j.biopha.2016.05.023
Hazell GG, Yao ST, Roper JA, Prossnitz ER, O’Carroll AM, Lolait SJ (2009) Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. J Endocrinol 202(2):223–236. https://doi.org/10.1677/JOE-09-0066
Donzelli A, Braida D, Finardi A, Capurro V, Valsecchi AE, Colleoni M, Sala M (2010) Neuroprotective effects of genistein in Mongolian gerbils: estrogen receptor-beta involvement. J Pharmacol Sci 114(2):158–167. https://doi.org/10.1254/jphs.10164fp
Schreihofer DA (2005) Transcriptional regulation by phytoestrogens in neuronal cell lines. Mol Cell Endocrinol 231(1–2):13–22. https://doi.org/10.1016/j.mce.2004.12.006
Du ZR, Feng XQ, Li N, Qu JX, Feng L, Chen L, Chen WF (2018) G protein-coupled estrogen receptor is involved in the anti-inflammatory effects of genistein in microglia. Phytomedicine 43:11–20. https://doi.org/10.1016/j.phymed.2018.03.039
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA (2019) Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation 16(1):142. https://doi.org/10.1186/s12974-019-1516-2
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD et al (2010) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11(12):1136–1142. https://doi.org/10.1038/ni.1960
Han B, Jiang W, Cui P, Zheng K, Dang C, Wang J, Li H, Chen L et al (2021) Microglial PGC-1alpha protects against ischemic brain injury by suppressing neuroinflammation. Genome Med 13(1):47. https://doi.org/10.1186/s13073-021-00863-5
Cheng CF, Ku HC, Lin H (2018) PGC-1alpha as a pivotal factor in lipid and metabolic regulation. Int J Mol Sci 19(11). https://doi.org/10.3390/ijms19113447
Funding
This work was supported by the National Natural Science Foundation of China (no. 81971226 to WGH; no. 81901079 to HYG), the Shaanxi Provincial Natural Science Foundation for Distinguished Young Scholars (no. 2021JC-33 to WGH), and the Surface Project of Shaanxi Provincial Natural Science Foundation (no. 2030JM-330 to SQW).
Author information
Authors and Affiliations
Contributions
H.G. and W.H. conceived and designed the study. S.W., Z.Z., J.W., L.M., J.Z., J.W., and Z.F. performed the experiments and data analysis. S.W., Z.Z., and J.W. prepared figures and the manuscript draft. H.G. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
All institutional and national guidelines for the care and use of laboratory animals were followed.
Consent to Participate
Not applicable.
Consent to Publish
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shiquan Wang, Zhen Zhang, and Jin Wang contributed equally to this article.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, S., Zhang, Z., Wang, J. et al. Neuronal GPER Participates in Genistein-Mediated Neuroprotection in Ischemic Stroke by Inhibiting NLRP3 Inflammasome Activation in Ovariectomized Female Mice. Mol Neurobiol 59, 5024–5040 (2022). https://doi.org/10.1007/s12035-022-02894-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02894-4