
Abstract
Cerebral ischemia reperfusion (I/R) injury easily develops in ischemic stroke, resulting in more serious injury. Ferroptosis is involved in cerebral I/R injury, but the mechanism remains unclear. Prostaglandin E2 (PGE2) is potential to regulate ferroptosis. This study mainly explored the regulation effects of PGE2 on ferroptosis induced by cerebral I/R. We first detected PGE2 levels and ferroptosis status in 11 human brain tissues. Then, we induced a cerebral I/R animal model to examine ferroptosis status in cerebral I/R. We further injected a ferroptosis inhibitor to define the response of the PGE2 pathway to ferroptosis. Finally, we injected PGE2 and pranoprofen to explore the regulation of the cyclooxygenases 2 (COX-2)/PGE2 pathway on ferroptosis in cerebral I/R. We found that PGE2 release was correlated with the levels of reactive oxygen species, malondialdehyde, glutathione peroxidase 4, COX-2, and Spermidine/spermine N1-acetyltransferase 1. Ferroptosis can be induced by cerebral I/R, while inhibition of ferroptosis induced by cerebral I/R can inactivate PGE2 synthases, degrade enzyme, and parts of PGE2 receptors, and reduce cerebral infarct volume. In turn, PGE2 inhibited ferroptosis through the reduction of Fe2+, glutathione oxidation, and lipid peroxidation, while pranoprofen, one of the COX inhibitors, played an opposite role. In conclusion, PGE2 was positively correlated with ferroptosis, inhibition of ferroptosis induced by cerebral I/R can inactivate COX-2/PGE2 pathway, and PGE2 inhibited ferroptosis induced by cerebral I/R, possibly via PGE2 receptor 3 and PGE2 receptor 4.


Inhibition of ferroptosis inactivates the COX-2/PGE2 pathway. Cerebral ischemia reperfusion injury induces the secretion of PGE2. After the inhibition of ferroptosis by Fer-1, the expression of cyclooxygenases (COX-1 and COX-2) decreased, and PGE2 synthases cPGES, mPGES-1, and mPGES-2 were also reduced. At the same time, the PGE2 degradation enzyme 15-PGDH was also reduced. Changes in these enzymes ultimately result in the declination of PGE2. Besides, the expression of PGE2 receptors EP3 and EP4 is also inhibited, indicating that the function they mediate is also impaired. In conclusion, after cerebral ischemia reperfusion injury, the inhibition of ferroptosis inactivates the COX-2/PGE2 pathway
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke is an acute episode of focal dysfunction of the brain, retina, or spinal cord lasting longer than 24 h, or of any duration if imaging (CT or MRI) or autopsy show focal infarction or hemorrhage relevant to the symptoms [1]. It is the second leading cause of death and a major cause of long-term disability worldwide [2]. Stroke can be divided into hemorrhagic stroke and ischemic stroke, in which ischemic stroke accounts for about 80% [3]. At present, restoring blood perfusion to the ischemic area as soon as possible is still the main strategy to treat ischemic stroke, such as drug thrombolysis and mechanical intravascular thrombectomy [4]. Concomitantly, the recovery of cerebral blood flow easily leads to cerebral ischemia reperfusion (I/R) injury [5]. The mechanism of cerebral I/R involves oxidative stress, inflammation, and excitotoxicity as well as various forms of cell death, including necrosis, apoptosis, autophagy, and ferroptosis [6, 7]. The prevention and treatment of cerebral I/R injury is a major challenge in ischemic stroke.
Ferroptosis is a new type of programmed cell death defined by Dixon et al. in 2012 [8]. It is mainly characterized by cell death caused by excessive accumulation of lipid peroxides dependent on iron ions. Morphological changes of ferroptosis were mainly presented in mitochondria as mitochondrial shrinkage, increased mitochondrial membrane density, rupture, or even disappearance of mitochondrial cristae. Ferroptosis is a complex process. Strikingly, the glutathione peroxidase 4 (GPX4)-glutathione (GSH) pathway is the most important antioxidant pathway, mainly involving system Xc−, cysteine, glutamate, GPX4, and GSH. A large number of reports have shown that ferroptosis plays an important role in a lot of diseases including tumors [9, 10], kidney injury [11, 12], neurological diseases [13, 14], liver injury [15], and I/R [16, 17]. Intervention against ferroptosis has become an effective target for the treatment of these diseases.
According to current reports, ferroptosis is involved in cerebral I/R. Wang, P., et al. reported that mitochondrial ferritin is upregulated in the ischemic brains, and lack of mitochondrial ferritin aggravates brain damage and neurological deficits by increasing lipid peroxidation and reducing GSH [18]. Conversely, mitochondrial ferritin overexpression alleviates these changes. Guo, H., et al. found that Carthamin yellow could ameliorate ischemic stroke partly by mitigating ferroptosis through inhibiting Fe2+ and reactive oxygen species (ROS) accumulation, and reversing acyl‑CoA synthetase long‑chain family member 4 (Acsl4), transferrin receptor 1, GPX4, and ferritin heavy chain 1 (Fth1) protein expression levels in the brain and reversing the levels of GSH, superoxide dismutase, and malondialdehyde (MDA) in the serum [19]. Chen, W., et al. demonstrated that ferritin overexpression could mitigate hippocampal neuronal injury and death by ameliorating ROS production and GSH consumption [20]. Besides, the treatment of ferroptosis inhibitors ferrostatin-1 (Fer-1) and liproxstatin-1 can reduce the cerebral infarction volume and mitigate the neurological deficits caused by middle cerebral artery occlusion (MCAO) [21]. It is confirmed that ferroptosis plays an important role in cerebral I/R, but the clear mechanism still needs to be explored.
Cyclooxygenases 2 (COX-2)/prostaglandin E2 (PGE2) pathway is closely related to cerebral I/R. The synthesis of PGE2 is regulated by a cascade of enzymes. Under the catalysis of phospholipase A2, phospholipids cleave to generate arachidonic acid, then secreted from cells. Arachidonic acid can be catalyzed by cyclooxygenase (COX), including COX-1 and COX-2, to manufacture prostaglandin G2 and prostaglandin H2. The transformation of prostaglandin H2 into PGE2 depends on PGE2 synthases cytosolic prostaglandin E synthase (cPGES), microsomal prostaglandin E synthase-1 (mPGES-1), and microsomal prostaglandin E synthase-2 (mPGES-2). At the same time, PGE2 can be reduced to 15-keto-PGE2 in the catalytic degradation of 15-hydroxyprostaglandin dehydrogenase (15-PGDH). Finally, PGE2 plays different roles by binding to different PGE2 receptors (EP) including EP1, EP2, EP3, and EP4. A report has confirmed that elevated levels of PGE2 during cerebral I/R are attributed to the increase of multiple enzymes during the synthesis phase [22]. Furthermore, COX-1, COX-2, mPGES-1, EP1, EP2, EP3, and EP4 have been shown to regulate cerebral I/R [23,24,25,26,27,28,29]. Noticeably, there is evidence of a link between the COX2/PGE2 pathway and ferroptosis. COX-2 is a ferroptosis marker gene, whose elevation is a hallmark of ferroptosis [30]. Besides, in a hemorrhagic stroke induced by oxyhemoglobin, PGE2 administration partially exacerbated lipid peroxidation and iron deposition and decreased GSH content and GPX4 [31]. While another report found that as a clinically approved, protective lipid species, PGE2 can act as a synergist to inhibit ferroptosis [32]. It seems that the role of PGE2 in ferroptosis is still debatable and needs to be confirmed by more comprehensive evidence.
In the present study, we searched for the correlation between PGE2 and ferroptosis by detecting the level of PGE2 and ferroptosis status in human brain tissues. Then, we used an MCAO model to simulate cerebral I/R and examined the status of ferroptosis comprehensively in cerebral I/R. We used exogenous injections of PGE2 and pranoprofen (Pra) to validate the regulatory role of the COX2/PGE2 pathway in ferroptosis. Furthermore, we used a ferroptosis inhibitor to identify the responded molecules in the COX2/PGE2 pathway to screen out potential targets in the COX2/PGE2 pathway that may regulate ferroptosis.
Materials and Methods
Patient Samples
The human brain tissues were obtained from the patients with cranial hypertension when decompression surgery is necessary from the Department of Neurosurgery, Xiangya Hospital, Central South University. This research was approved by the ethics committee of the Xiangya Hospital of Central South University and the human ethics identification number is 2021101119. These patients consisted of six males and five females, with an average age of 54.1 years. All participants involved in this study provided written informed consent.
Animals
All rats and procedures were approved by the Experimental Animal Center of Central South University (Changsha, China). All rats were housed on a 12-h light/dark cycle in a 25 °C 40–60% humidity-controlled facility and provided ad libitum access to food and water. Animals used in vivo studies were 250–300 g male SD rats at 8 weeks of age and were all healthy before experiments. The number of total animals is about 120 with 6 rats in each group.
Experimental MCAO Model
The rats were intraperitoneally anesthetized with pentobarbital sodium. An incision was opened in the middle of the neck. The right carotid artery and vagus nerve were gently dissected. The right common carotid and internal carotid arteries were temporarily ligated with arteriole clip. After ligation of the external carotid artery, a small incision was opened. A poly-l-lysine-coated monofilament was inserted from the incision, through the internal carotid artery, and finally to the middle cerebral artery. The nylon monofilament was pulled out for reperfusion after 2 h of blockage. The cortex of the infarcted hemisphere was collected for subsequent experiments.
Lateral Ventricle Injection
The anesthetized rats were placed on a stereotaxic frame (RWD Life Science, Shenzhen, China) in the horizontal prone position. Using a syringe pump (KDS legato 130, Holliston, MA, USA) and a Hamilton syringe, PGE2 or Fer-1 was infused into the right lateral ventricle at a flow rate of 0.5μL/min. Taking the bregma point as the origin, the stereotaxic coordinates of the lateral ventricle were lateral − 1.5 mm, anteroposterior − 1.2 mm, and dorsoventral − 4.5 mm. After being infused, the skull was sealed with bone wax.
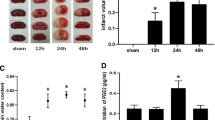
Measurement of Infarct Volume
Collect intact brain tissue and freeze it at − 20℃ for 20 min. Then, cut into 2-mm thick slices and stained with 2% 2,3,5-triphenyl tetrazolium chloride (TTC, Jincheng Biotech, China) at 37℃ from light. The infarct volume and the whole brain volume were determined with Image J software. Finally, the results were presented as (infarct volume/whole brain volume) × 100%.
Reverse‑Transcription‑Quantitative Polymerase Chain Reaction
After the tissue RNA was extracted with TRIZOL, 1 μg was reverse-transcripted. 7500 Real-Time PCR System (Applied Biosystems, Waltham, MA, USA) with a Two-Step SYBR® Prime Script™ RT‑qPCR kit (Takara, Shiga Prefecture, Japan) was used to perform PCR. The primers included COX-1, COX-2, cPGES, mPGES-1, mPGES-2, 15-PGDH, EP1, EP2, EP3, EP4, GPX4, Fth1, SAT1, glutaminase 2 (Gls2), and Acsl4. Their sequences are presented in Table 1. The specificity of all primers was confirmed by melt curves when first performed. After 95 °C for 30 s to pre-denaturation, the amplification was performed up to 40 cycles with 95 °C for 5 s and 60 °C for 34 s. Normalized by β‑actin, the relative quantitation of mRNA was obtained by the 2‑ΔΔCT method.
Western Blotting
Radio immunoprecipitation assay lysis buffer containing 1% PMSF was used to extract tissue protein. Then, a bicinchoninic acid kit was used to measure protein concentration. An equal amount of protein samples was mixed with 5 × SDS sample buffer and heated to 95℃ for 10 min to expose epitopes. All samples were electrophoresed on an SDS‑PAGE gel and transferred to a polyvinylidene fluoride membrane. After blocking in 5% non-fat dried milk, the membranes were incubated with primary antibodies against β‑actin (1:5000, Sigma‑Aldrich, Saint Louis, MO, USA), GPX4 (1:5000, Cell Signaling Technology, Danvers, MA, USA), COX-1 (1:200, Cayman Chemical Company, Ann Arbor, MI, USA), COX-2 (1:200, Cayman Chemical Company), EP3 (1:200, Cayman Chemical Company), and EP4 (1:200, Cayman Chemical Company) overnight at 4 °C. Secondary antibodies were prepared with 5% non-fat dried milk and incubated at room temperature for 2 h. All proteins were detected using the enhanced chemiluminescence method (Bio‑Rad Laboratories, CA, USA). Band density was measured by ImageJ software and normalized to the β‑actin.
PGE2 Measurement
PGE2 was measured according to the prostaglandin E2 (PG-E2) ELISA Kit (CUSABIO, Wuhan, China) instruction. Tissues were homogenized in PBS and stored overnight at − 20 °C. After two freeze–thaw cycles were performed to break the cell membranes, the homogenates were centrifuged to get supernatant. Add standard or sample per well. Then, add HRP-conjugate and antibody to each well. Mix well and then incubate for 1 h at 37 °C. Wash the plate three times with wash buffer. Add substrate A and substrate B to each well. Incubate for 15 min at 37 °C in the dark. Add stop solution to each well and determine the optical density of each well using a microplate reader set to 450 nm.
MDA and ROS Measurement
MDA and ROS were detected by the rat malondialchehyche (MDA) ELISA Kit instruction (FEIYA BIOTECHNOLOGY, Yancheng, China) and rat reactive oxygen species (ROS) ELISA Kit instruction (FEIYA BIOTECHNOLOGY), respectively. The tissues were mashed with the proper amount of normal saline and centrifuged at 3000 g for 10 min to take the supernatant. Add 50μL standard of different concentrations to standard wells. Add 10μL sample and 40μL sample diluent to a well. Add 100μL HRP-conjugate reagent to each well and incubate for 60 min at 37℃. Wash five times with wash solution. Add chromogen solution A and B to each well and incubate for 15 min at 37℃ protecting from light. Finally, add 50μL stop solution to each well and read the OD value at 450 nm. Draw a standard curve with the concentration and OD value of the standard well and calculate the concentration of the sample.
Fe 2+ Measurement
Fe2+ was detected by the Iron Colorimetric Assay Kit (BioVision, Palo Alto, CA, USA). A total of 100-mg tissues were homogenized in 500 µL iron assay buffer, then centrifuged at 16,000 g for 10 min and collected the supernatant. For iron standard curve, dilute iron standard and set a concentration gradient in a 96-well plate, and add 5 µL iron reducer to each standard well and bring the volume of all standard wells to 100 µL with iron assay buffer. Add 50 µL sample and 50 µL iron assay buffer to sample wells to detect ferrous iron (II). After incubating the plate for 30 min at 37 °C, add 100 µL iron probe to each well and incubate for 60 min at 37 °C protecting from light. Measure the absorbance at 593 nm in a microplate reader. A standard curve is drawn according to the concentration and OD value of the standard well, then the concentration of iron in the sample can be calculated out.
GSH Measurement
GSH, oxidized glutathione (GSSG), or total (GSH + GSSG) were detected by the Glutathione Fluorometric Assay Kit (GSH, GSSG, and Total) (BioVision). Homogenize 40 mg tissues with 100 µL glutathione assay buffer and take 60 µL of each homogenate to 20 µL PCA. Centrifuge 2 min at 13,000 g at 4 °C to collect the supernatant. Dissolve GSH standard and add PCA to stabilize the standard GSH stock solution. Diluent standard GSH stock with assay buffer and generate concentration gradient, then bring the volume to 90 µL with assay buffer. Add 20 µL 6 N KOH to 40 µL of PCA preserved samples, then spin 2 min at 13,000 g at 4 °C. Transfer 10 µL of the neutralized samples to a 96-well plate. To detect GSH, bring the sample volume to 90 µL with assay buffer. To detect total glutathione, bring the sample well to 80 µL with assay buffer and add 10 µL of reducing agent mix to the wells to convert GSSG to GSH. To detect GSSG, bring the sample well volume to 70 µL with assay buffer and add 10 µL of GSH Quencher to quench GSH, then add 10 µL of reducing agent mix to destroy the excess GSH Quencher and convert GSSG to GSH. Add 10 µL of OPA probe into the standard and sample wells, and incubate at room temperature for 40 min. Read samples and standards on a fluorescence plate reader equipped with Ex/Em = 340/420 nm. Draw a standard curve and get the glutathione amount in each sample.
Transmission Electron Microscopy
A brain mass of about 1*1*3 mm3 was taken from the core area of the infarct cortex and fixed with 2.5% glutaraldehyde and 1% osmium acid successively. After dehydration with different concentrations of acetone, the samples were embedded in acetone and embedding solution and solidified in the oven. The samples were cut into sections of 50–100 nm with an ultra-thin slicer (Leica, Wetzlar, Germany), and double-stained with 3% uranium acetate and lead nitrate. Finally, the samples were observed and captured by a Hitachi HT7 700 transmission electron microscopes.
Statistical Analysis
All the experiments were repeated 3–6 times and each group contains 6 rats at least. The quantitative data was confirmed the normality and homoscedasticity previously, then presented as the mean ± standard deviation and analyzed using analysis of variance (ANOVA) statistical analysis. Correlation analysis was performed using Pearson product-moment correlation. Statistical analysis was performed using GraphPad Prism 8 software (GraphPad Software, Inc., La Jolla, CA, USA). P-value was set to 0.05.
Results
PGE2 is Positively Correlated with Ferroptosis
As a new form of cell death, ferroptosis has been reported to be involved in a variety of diseases. PGE2 is considered to be an important inflammatory factor that is increased in a lot of diseases. Whether PGE2 is related to ferroptosis remains to be determined. We collected 11 brain tissue samples and examined their PGE2 levels and ferroptosis status to determine their correlation. After measuring the level of PGE2, as well as ROS and MDA, correlation analysis was carried out. As Fig. 1A and B, ROS and MDA are both positively correlated with PGE2, suggesting that PGE2 is probably also positively correlated with ferroptosis. In addition, we further examined ferroptosis marker genes. As shown in Fig. 1C, GPX4, and PGE2 were negatively correlated, while COX-2 (Fig. 1D) and SAT1 (Fig. 1E) were positively correlated with PGE2. These results suggest that there is a positive correlation between PGE2 and ferroptosis.

PGE2 is positively correlated with ferroptosis. A Correlation analysis of ROS and PGE2. B Correlation analysis of MDA and PGE2. C Correlation analysis of GPX4 mRNA level and PGE2. D Correlation analysis of COX-2 mRNA level and PGE2. E Correlation analysis of SAT1 mRNA level and PGE2. Correlation analysis was performed using Pearson product-moment correlation. n = 11
Ferroptosis Induced by Cerebral Ischemia Reperfusion
Previous reports have demonstrated the presence of ferroptosis in cerebral I/R, but the exact status of ferroptosis has not been reported. Therefore, we established a rat model of MCAO to detect the status of ferroptosis in cerebral I/R.
The accumulation of iron ions is an important characteristic of ferroptosis. Fe2+ can catalyze Fenton reaction to promote the generation of ROS. As shown in Fig. 2A, the concentration of Fe2+ increased 24 h after I/R. GSH is the most important antioxidant in ferroptosis, which is oxidized from reduced GSH to GSSG. We observed that both the concentration of GSH and the ratio of GSH/GSSG decreased 24 h after reperfusion, suggesting that the antioxidant capacity provided by GSH decreased after cerebral I/R (Fig. 2B and C). At the same time, after cerebral I/R, ROS accumulation was also observed (Fig. 2D). Lipid peroxidation is also a major characteristic of ferroptosis. MDA is one of the main products of lipid peroxidation degradation. It increased at 24 h after cerebral I/R (Fig. 2E). In an overview of Fe2+, ROS, GSH, and MDA, we found that 24 h after I/R may be a notable time point, as it is the only time point at which any of these substances changed.

Ferroptosis induced by cerebral ischemia reperfusion. A Fe2+ level in cerebral I/R. B GSH level in cerebral I/R. C The ratio of GSH/GSSG in cerebral I/R. D ROS level in cerebral I/R. E MDA level in cerebral I/R. F Representative electron microscope images of ferroptotic mitochondria in cerebral I/R (magnification, × 20,000). G Statistical analysis of ferroptotic mitochondria in cerebral I/R. H Western blot analysis of GPX4 protein level in cerebral I/R. I The mRNA levels of GPX4, SAT1, Fth1, Acsl4, and Gls2 in cerebral I/R. n = 6 animals per group. Data were presented as the mean ± standard deviation and ANOVA statistical analysis was performed. *P < 0.05 vs. sham group
The mitochondrion is sensitive to ferroptosis. Mitochondrial shrinkage and thickening of membrane density were observed in ferroptotic cells, accompanied by rupture or even disappearance of mitochondrial cristae. However, differences were observed in cerebral I/R. As presented in the electron microscope images (Fig. 2F and G), after cerebral I/R, the fracture or disappearance of the mitochondrial crest can indeed be observed, while the mitochondria were not shrunk, even expanded, and the thickening of mitochondrial membrane density can be hardly observed. We hypothesized that the characteristic changes were not observed because of severe brain edema caused by cerebral I/R.
We finally examined the expression of several ferroptosis marker genes after cerebral I/R. GPX4, the enzyme converts GSH into GSSG to resist ferroptosis, reduced including mRNA and protein continuously (Fig. 2H and I). Fth1, a subunit of ferritin, which can store Fe2+, was observed reduced from 24 h after reperfusion (Fig. 2I). SAT1, which could induce lipid peroxidation and ferroptosis [33], upregulated at 12 h after reperfusion (Fig. 2I). Acsl4, which can catalyze the esterification of arachidonoyl or adrenoyl to promote lipid peroxidation and induce ferroptosis, increased at 48 h after reperfusion (Fig. 2I). Gls2, which could promote ferroptosis by upregulating p53, upregulated at 24 h and 48 h after reperfusion (Fig. 2I). These molecules are involved in the iron pathway, GPX4-GSH pathway, and lipid peroxidation pathway, which can comprehensively present the status of ferroptosis induced by cerebral I/R.
These findings suggest that ferroptosis is activated after cerebral I/R in many ways simultaneously.
Inhibition of Ferroptosis Inactivates the COX-2/PGE2 Pathway and Alleviates Cerebral Infarction
Previous results have confirmed a positive correlation between PGE2 and ferroptosis. Increased PGE2 and induction of ferroptosis have been revealed in cerebral I/R at the same time. It is worth exploring how ferroptosis intervention might affect the COX-2/PGE2 pathway. So, we probed the enzymes and receptors in the COX-2/PGE2 pathway using the ferroptosis inhibitor Fer-1. 40 nmol/kg Fer-1 was injected into the lateral ventricle 2 h before the construction of the MCAO model, and samples were taken 24 h after reperfusion. We first verified the successful inhibition of Fer-1 on ferroptosis. As presented in Fig. 3A, Fer-1 successfully reversed the decrease of GPX4 and Fth1, and also decreased Acsl4 and SAT1. These effects confirm that ferroptosis is indeed involved in cerebral I/R and has been successfully inhibited by Fer-1. Besides, it reduced the volume of cerebral infarction in cerebral I/R, playing a neuroprotective role (Fig. 3B and C).

Inhibition of ferroptosis inactivates the COX-2/PGE2 pathway and alleviates cerebral infarction. A The mRNA levels of GPX4, SAT1, Fth1, and Acsl4 were measured treated with Fer-1(40 nmol/kg) in cerebral I/R. B Cerebral infarction images treated with Fer-1 (40 nmol/kg) in cerebral I/R. C Cerebral infarction volume statistics treated with Fer-1 (40 nmol/kg) in cerebral I/R. D PGE2 level measurement treated with Fer-1 (40 nmol/kg) in cerebral I/R. E Western blot analysis of COX-1 protein level treated with Fer-1 (40 nmol/kg) in cerebral I/R. F Western blot analysis of COX-2 protein level treated with Fer-1 (40 nmol/kg) in cerebral I/R. G The mRNA level of 15-PGDH was measured treated with Fer-1 (40 nmol/kg) in cerebral I/R. H The mRNA levels of cPGES, mPGES-1, and mPGES-2 were measured treated with Fer-1 (40 nmol/kg) in cerebral I/R. n = 6 animals per group. Data were presented as the mean ± standard deviation and ANOVA statistical analysis was performed. *P < 0.05 vs. sham + vehicle group, #P < 0.05 vs. I/R + vehicle group
Since we previously identified the correlation between ferroptosis and PGE2, we explored how the COX-2/PGE2 pathway would respond after inhibiting ferroptosis induced by cerebral I/R. We first examined the PGE2 release. Figure 3D showed that the elevation of PGE2 after cerebral I/R was partially reversed by Fer-1. We further examined all the molecules in the COX-2/PGE2 pathway, hoping to find the answer from the generation and degradation process of PGE2. In the first-generation stage, COX-1 and COX-2 were the key enzymes. As a constituent enzyme, COX-1 did not change significantly after cerebral I/R, while the inductive enzyme COX-2 increased significantly. Both the two enzymes were significantly reduced after the application of Fer-1 (Fig. 3E and F). In the next synthesis phase, the elevated mPGES-1 and mPGES-2 after cerebral I/R were partially restored, and the constituent enzyme cPGES also decreased (Fig. 3H). In addition, the PGE2 degrading enzyme 15-PGDH was further reduced after the applying of Fer-1 (Fig. 3G). In a word, after cerebral I/R, the inhibition of ferroptosis can completely inactivate the synthase and degrading enzyme in the COX-2/PGE2 pathway, and eventually, reduce PGE2.
PGE2 Prevents Ferroptosis Induced by Cerebral Ischemia Reperfusion
Cerebral I/R can induce ferroptosis, accompanied by an increase of PGE2. It is reported that PGE2 administration can aggravate ferroptosis in hemorrhagic stroke. However, another report proved that PGE2 can act as a synergist to inhibit ferroptosis. In other words, the role of PGE2 in ferroptosis induced by hemorrhagic stroke is unclear. Furthermore, the role of PGE2 in ferroptosis induced by cerebral I/R has not been explored.
To address whether the COX-2/PGE2 pathway inhibits or aggravates ferroptosis induced by cerebral I/R, we supplemented 40 μg/kg PGE2 to cerebral I/R by injecting it into the lateral ventricle. As presented in Fig. 4A and C, PGE2 supplementation attenuated the iron accumulation and ROS overproduction, as well as the reduction of GSH (Fig. 4B). At the same time, PGE2 supplementation can reduce ferroptotic mitochondria from the electron microscope images (Fig. 4F and G). Among ferroptosis marker genes, PGE2 restored the decrease of Fth1 and GPX4 (Fig. 4D and E). In general, PGE2 supplementation will inhibit ferroptosis induced by cerebral I/R.

PGE2 prevents ferroptosis induced by cerebral ischemia reperfusion. A Fe2+ level measurement treated with PGE2 (40 μg/kg) in cerebral I/R. B GSH level measurement treated with PGE2 (40 μg/kg) in cerebral I/R. C ROS level measurement treated with PGE2 (40 μg/kg) in cerebral I/R. D The mRNA level of GPX4 was measured treated with PGE2 (40 μg/kg) in cerebral I/R. E The mRNA level of Fth1 was measured treated with PGE2 (40 μg/kg) in cerebral I/R. F Representative electron microscope images of mitochondria treated with PGE2 (40 μg/kg) in cerebral I/R (magnification, × 20,000). G Statistical analysis of ferroptotic mitochondria treated with PGE2 (40 μg/kg) in cerebral I/R. n = 6 animals per group. Data are presented as the mean ± standard deviation and ANOVA statistical analysis was performed. *P < 0.05 vs. sham + vehicle group, #P < 0.05 vs. I/R + vehicle group
In addition, Pra, a cyclooxygenase inhibitor, was used to inhibit the COX-2/PGE2 pathway and explore its regulation on ferroptosis induced by cerebral I/R. The rats were intraperitoneally injected with 20 mg/kg Pra and then subjected to cerebral I/R. In contrast to the effect of PGE2, the addition of Pra further aggravated ferroptosis induced by cerebral I/R. Pra led to a further decrease of GSH level and increased ROS production and lipid peroxidation (Fig. 5A, B, and C). Electron microscope images showed that the damage of mitochondria was also intensified. The number of ferroptotic mitochondria increased while the mitochondria began to rupture and ablate. In addition, Pra intensified the decrease of GPX4 in ferroptosis induced by cerebral I/R (Fig. 5D).

Pra aggravates ferroptosis induced by cerebral ischemia reperfusion. A GSH level measurement treated with Pra (20 mg/kg) in cerebral I/R. B ROS level measurement treated with Pra (20 mg/kg) in cerebral I/R. C MDA level measurement treated with Pra (20 mg/kg) in cerebral I/R. D The mRNA level of GPX4 was measured treated with Pra (20 mg/kg) in cerebral I/R. E Representative electron microscope images of mitochondria treated with Pra (20 mg/kg) in cerebral I/R (magnification, × 20,000). F Statistical analysis of ferroptotic mitochondria treated with (20 mg/kg) in cerebral I/R. n = 6 animals per group. Data are presented as the mean ± standard deviation and ANOVA statistical analysis was performed. *P < 0.05 vs. sham + vehicle group, #P < 0.05 vs. I/R + vehicle group
EP3 and EP4 Are Sensitive to Ferroptosis
The function of PGE2 is achieved by binding to different receptors, including EP1, EP2, EP3, and EP4. After cerebral I/R, the application of Fer-1 inactivates the COX-2/PGE2 pathway and ultimately reduces the release of PGE2. Furthermore, we examined the response of the four EPs. As can be seen from the Fig. 6A, Fer-1 cannot affect EP1 and EP2, but it successfully reduced the elevated mRNA levels of EP3 and EP4 after cerebral I/R, as well as their protein levels (Fig. 6B and C). As EP3 and EP4 are sensitive to ferroptosis induced by cerebral I/R, they could be potential targets for ferroptosis. Maybe the inhibition of ferroptosis by PGE2 is also achieved by EP3 and EP4.

EP3 and EP4 are sensitive to ferroptosis. A The mRNA levels of EP1, EP2, EP3, and EP4 were measured treated with Fer-1 (40 nmol/kg) in cerebral I/R. B Western blot analysis of EP3 protein level treated with Fer-1 (40 nmol/kg) in cerebral I/R. C Western blot analysis of EP4 protein level treated with Fer-1 (40 nmol/kg) in cerebral I/R. n = 6 animals per group. Data are presented as the mean ± standard deviation and ANOVA statistical analysis was performed. *P < 0.05 vs. sham + vehicle group, #P < 0.05 vs. I/R + vehicle group
Discussion
Ferroptosis has become one of the research hotspots around the world. The research on ferroptosis is now mainly focused on the replenishing of the ferroptosis pathway and the role and mechanism of ferroptosis in the pathophysiological process of different diseases. Multiple reports have proved that ferroptosis is involved in cerebral I/R, maybe an efficient regulatory target. The COX-2/PGE2 pathway also plays an important role in cerebral I/R. Several enzymes in the pathway, such as COX-1, COX-2, mPGES-1, and EPs, have been experimentally demonstrated to be involved in cerebral I/R and are important regulatory targets [23, 27,28,29, 34,35,36]. Generally, PGE2 is involved in cerebral I/R as an inflammatory factor [37]. In this paper, we further investigated the correlation between cerebral I/R, ferroptosis, and the COX-2/PGE2 pathway. In brief, we have made several important discoveries. Firstly, cerebral I/R can induce ferroptosis, and ferroptosis is positively correlated with PGE2. Furthermore, inhibition of ferroptosis in cerebral I/R inactivated the COX-2/PGE2 pathway and alleviated cerebral infarction, while PGE2 inhibited ferroptosis induced by cerebral I/R. Finally, EP3 and EP4 are sensitive to ferroptosis and may mediate the above processes.
Unlike previous articles that directly focused on the regulation of ferroptosis in cerebral I/R, we first identified the comprehensive status of ferroptosis after cerebral I/R, which was an essential part. We found that after cerebral I/R, ferroptosis was induced across the board, not limited to a single pathway. Important characteristics of ferroptosis including iron ion accumulation, GSH depletion, and excessive production of lipid peroxides all have been observed after cerebral I/R. In particular, the morphological changes of mitochondria only showed the rupture or disappearance of the crest, while mitochondrial shrinkage and membrane density thickening were not found. This may be a special case of cerebral I/R because cerebral I/R can lead to severe brain tissue edema, and even make mitochondria expand instead, and membrane density is difficult to increase. At the same time, the ferroptosis marker genes involved in the iron metabolism (Fth1), the GSH-GPX4 axis (GPX4), and the lipid peroxidation (SAT1, Acsl4) all responded accordingly. Taken together, the change patterns of these genes are different. We hypothesized that this may be due to their different distances from the core process of lipid peroxidation in the ferroptosis pathway. GPX4 directly catalyzes the inhibition of GSH on lipid peroxidation, and SAT1 directly regulates the catalysis of 15-lipoxygenase (ALOX15) on lipid peroxidation formation. They are very close to the core process of lipid peroxidation, which may result in their changes being observed at the early stage. Similarly, the regulation of Fth1, Acsl4, and Gls2 are remote from the core process of lipid peroxidation, which may cause them to change at a later stage. It was noted that ferroptosis induced by cerebral ischemia reperfusion-induced occurred in a relatively narrow time window. In other words, the characteristics of ferroptosis were almost entirely observed at 24 h, but only partially at 12 and 48 h, indicating that ferroptosis peaked at 24 h. This reminds us that when ferroptosis is used as a target to treat cerebral ischemia reperfusion injury, the intervention time should be around 24 h or earlier. Compared with the clinical time window of cerebral ischemia reperfusion therapy, the time window is almost consistent. The clinical time window is best controlled in 6 h, depending on the severity of the disease can be extended to 24 h at the latest. Therefore, the time window of the two is relatively consistent, which may provide a potential clue for clinical treatment. Besides, we demonstrate that cerebral I/R induces ferroptosis and present a detailed picture of ferroptosis, considering that the GSH-GPX4 axis is the most important and well-studied inhibitory pathway in ferroptosis. Other inhibitory pathways, such as coenzyme Q10 (CoQ10)/ferroptosis suppressor protein 1 (FSP1) axis, GTP cyclohydrolase 1 (GCH1)-tetrahydrobiopterin (BH4)-phospholipid axis, FSP1-endosomal sorting complexes required for transport (ESCRT)-III pathway, and dihydroorotate dehydrogenase axis, have not been studied for a long time, and the mechanism is not clear enough [38,39,40,41]. Therefore, we did not detect those pathways, which may be a shortcoming of our paper.
We confirm the positive correlation between PGE2 and ferroptosis by correlation analysis. Since ferroptosis has become an important target for the treatment of cerebral I/R, PGE2 shows the potential for ferroptosis and thus improves neurological function.
When ferroptosis induced by cerebral I/R is inhibited, the response of the COX-2/PGE2 pathway is unknown. Using the ferroptosis inhibitor Fer-1, we found that inhibition of ferroptosis in cerebral I/R resulted in the inactivation of the COX-2/PGE2 pathway. Specifically, after inhibiting the cerebral I/R-induced ferroptosis, synthases in the COX-2/PGE2 pathway, including COX-1, COX-2, mPGES-1, mPGES-2, and cPGES, as well as the degrading enzyme 15-PGDH, were decreased, ultimately leading to a decrease in PGE2. Fer-1 has been used in the study of cerebral I/R and has been shown to improve neurological deficits and alleviate cerebral infarction [21], and our results also confirm this effect. PGE2 is one of the inflammatory factors produced in large quantities after cerebral I/R. Our study confirmed that Fer-1 can inactivate the COX-2/PGE2 pathway and reduce the release of PGE2, which may be one of the mechanisms of its protection against cerebral I/R.
The role of PGE2 in ferroptosis has only been studied in hemorrhagic stroke, and there is considerable disagreement. A report showed that PGE2 can synergize with N-acetylcysteine to inhibit ferroptosis in hemorrhagic stroke [32]. But then another report suggested that PGE2 administration alone could aggravate ferroptosis [31]. The direction in which PGE2 contributes to ferroptosis seems uncertain. The role of PGE2 in ferroptosis induced by cerebral I/R has not been investigated. To more fully determine the regulatory direction of the COX-2/PGE2 pathway on ferroptosis induced by cerebral I/R, we used PGE2 and Pra, the COX inhibitor, respectively, from both positive and negative aspects. On the one hand, in cerebral I/R, additional supplementation of PGE2 appears to inhibit ferroptosis. After the administration of PGE2, iron accumulation, GSH depletion, and overproduction of peroxides during ferroptosis were all alleviated. In addition, the decrease of ferroptosis marker genes Fth1 and GPX4 was partially recovered. In contrast, after Pra was applied, ferroptosis was exacerbated. The results of these two parts are consistent, which can fully prove that PGE2 can effectively inhibit ferroptosis induced by cerebral I/R. This conclusion is contrary to that of Li, Y., et al. in hemorrhagic stroke [31]. Maybe ferroptosis responds differently to PGE2 in different stroke subtypes.
The function of PGE2 depends on its binding to different receptors. Among its four receptors, EP3 and EP4 are sensitive to ferroptosis. So, the inhibition of PGE2 on ferroptosis induced by cerebral I/R is likely to be achieved through EP3 and EP4. In this paper, we failed to confirm it experimentally, which is a major shortcoming. In our next work, we will complete this part as soon as possible.
In this paper, we presented the status of ferroptosis induced by cerebral I/R in detail, which can provide some basic clues for further studies. If the status of ferroptosis is not determined in advance, it is easy to conduct research blindly and thus go astray. At the same time, clinical data revealed a positive correlation between PGE2 and ferroptosis, which may be a good starting point for follow-up studies. Thus, we further identified the potential association between the COX-2/PGE2 pathway and the ferroptosis pathway in cerebral I/R. Inhibition of cerebral I/R induced ferroptosis results in inactivation of the COX-2/PGE2 pathway, while the administration of PGE2 inhibits ferroptosis. This paper did not involve specific experiments on the severity of cerebral I/R such as neurological deficit and cerebral infarction after administration of Fer-1, PGE2, and Pra, mainly considering that these contents have been proved in previous studies, and we focused more on the mechanisms involved. Based on these mechanisms, specific molecules with research value can be extracted for further exploration. For example, EP3 and EP4, the receptors sensitive to ferroptosis, are likely to be closely related to ferroptosis. In addition, PGE2 regulates ferroptosis in the opposite direction in hemorrhagic stroke and ischemic stroke, suggesting that the role of PGE2 in ferroptosis in other diseases may not be easily predicted accurately and may need specific experiments to determine.
Availability of Data and Materials
Data and materials will be made available under reasonable request.
Change history
08 October 2022
A Correction to this paper has been published: https://doi.org/10.1007/s12035-022-03068-y
References
Sacco RL, Kasner SE, Broderick JP, Caplan LR, Connors JJ, Culebras A et al (2013) An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 44(7):2064–89
GBD (2019) Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18(5):439–458
Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S et al (2018) Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation 137(12):e67–e492
Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K et al (2019) Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 50(12):e344–e418
George PM, Steinberg GK (2015) Novel stroke therapeutics: unraveling stroke pathophysiology and its impact on clinical treatments. Neuron 87(2):297–309
Iadecola C, Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14(11):1363–1368
Datta A, Sarmah D, Mounica L, Kaur H, Kesharwani R, Verma G et al (2020) Cell death pathways in ischemic stroke and targeted pharmacotherapy. Transl Stroke Res 11(6):1185–1202
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072
Yamaguchi Y, Kasukabe T, Kumakura S (2018) Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol 52(3):1011–1022
Bai T, Lei P, Zhou H, Liang R, Zhu R, Wang W et al (2019) Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J Cell Mol Med 23(11):7349–7359
Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Niño MD et al (2017) Ferroptosis, but Not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol 28(1):218–229
Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF et al (2018) Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol 314(5):F702-f714
Lane DJR, Ayton S, Bush AI (2018) Iron and Alzheimer’s disease: an update on emerging mechanisms. J Alzheimers Dis 64(s1):S379-s395
Xie BS, Wang YQ, Lin Y, Mao Q, Feng JF, Gao GY et al (2019) Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci Ther 25(4):465–475
Wang L, Zhang Z, Li M, Wang F, Jia Y, Zhang F et al (2019) P53-dependent induction of ferroptosis is required for artemether to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. IUBMB Life 71(1):45–56
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X (2015) Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 59(2):298–308
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D et al (2019) Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ 26(11):2284–2299
Wang P, Cui Y, Ren Q, Yan B, Zhao Y, Yu P et al (2021) Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis 12(5):447
Guo H, Zhu L, Tang P, Chen D, Li Y, Li J et al (2021) Carthamin yellow improves cerebral ischemia-reperfusion injury by attenuating inflammation and ferroptosis in rats. Int J Mol Med 47(4):4885
Chen W, Jiang L, Hu Y, Tang N, Liang N, Li XF et al (2021) Ferritin reduction is essential for cerebral ischemia-induced hippocampal neuronal death through p53/SLC7A11-mediated ferroptosis. Brain Res 1752:147216
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL et al (2017) Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 22(11):1520–1530
Xu Y, Liu Y, Li K, Miao S, Lv C, Wang C et al (2020) Regulation of PGE(2) pathway during cerebral ischemia reperfusion injury in rat. Cell Mol Neurobiol. 41(7):1483–1496
Candelario-Jalil E, González-Falcón A, García-Cabrera M, Alvarez D, Al-Dalain S, Martínez G et al (2003) Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem 86(3):545–555
Peng Z, Li M, Tan X, Xiang P, Wang H, Luo Y et al (2020) miR-211–5p alleviates focal cerebral ischemia-reperfusion injury in rats by down-regulating the expression of COX2. Biochem Pharmacol 177:113983
Ikeda-Matsuo Y, Hirayama Y, Ota A, Uematsu S, Akira S, Sasaki Y (2010) Microsomal prostaglandin E synthase-1 and cyclooxygenase-2 are both required for ischaemic excitotoxicity. Br J Pharmacol 159(5):1174–1186
Ahmad AS, Yun YT, Ahmad M, Maruyama T, Doré S (2008) Selective blockade of PGE2 EP1 receptor protects brain against experimental ischemia and excitotoxicity, and hippocampal slice cultures against oxygen-glucose deprivation. Neurotox Res 14(4):343–351
Ahmad M, Saleem S, Shah Z, Maruyama T, Narumiya S, Doré S (2010) The PGE2 EP2 receptor and its selective activation are beneficial against ischemic stroke. Exp Transl Stroke Med 2(1):12
Ikeda-Matsuo Y, Tanji H, Narumiya S, Sasaki Y (2011) Inhibition of prostaglandin E2 EP3 receptors improves stroke injury via anti-inflammatory and anti-apoptotic mechanisms. J Neuroimmunol 238(1–2):34–43
Yang H, Li GP, Liu Q, Zong SB, Li L, Xu ZL et al (2021) Neuroprotective effects of Ginkgolide B in focal cerebral ischemia through selective activation of prostaglandin E2 receptor EP4 and the downstream transactivation of epidermal growth factor receptor. Phytother Res 35(5):2727–2744
Chen B, Chen Z, Liu M, Gao X, Cheng Y, Wei Y et al (2019) Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects. Brain Res Bull 153:122–132
Li Y, Wang J, Chen S, Wu P, Xu S, Wang C et al (2020) miR-137 boosts the neuroprotective effect of endothelial progenitor cell-derived exosomes in oxyhemoglobin-treated SH-SY5Y cells partially via COX2/PGE2 pathway. Stem Cell Res Ther 11(1):330
Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K et al (2018) N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol 84(6):854–872
Ou Y, Wang SJ, Li D, Chu B, Gu W (2016) Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A 113(44):E6806-e6812
Yin W, Badr AE, Mychaskiw G, Zhang JH (2002) Down regulation of COX-2 is involved in hyperbaric oxygen treatment in a rat transient focal cerebral ischemia model. Brain Res 926(1–2):165–171
Ikeda-Matsuo Y, Tanji H, Ota A, Hirayama Y, Uematsu S, Akira S et al (2010) Microsomal prostaglandin E synthase-1 contributes to ischaemic excitotoxicity through prostaglandin E2 EP3 receptors. Br J Pharmacol 160(4):847–859
Shimamura M, Zhou P, Casolla B, Qian L, Capone C, Kurinami H et al (2013) Prostaglandin E2 type 1 receptors contribute to neuronal apoptosis after transient forebrain ischemia. J Cereb Blood Flow Metab 33(8):1207–14
Liu Z, Liu Y, Zhou H, Fu X, Hu G (2017) Epoxyeicosatrienoic acid ameliorates cerebral ischemia-reperfusion injury by inhibiting inflammatory factors and pannexin-1. Mol Med Rep 16(2):2179–2184
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575(7784):688–692
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F et al (2020) GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci 6(1):41–53
Dai E, Meng L, Kang R, Wang X, Tang D (2020) ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun 522(2):415–421
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H et al (2021) DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593(7860):586–590
Acknowledgements
We thank the patients for granting permission to publish this information.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81571880, 81373147, 30901555, 30972870, 81360080) and the Natural Science Foundation of Hunan Province (2016JJ2157), the Fundamental Research Funds for the Central Universities of Central South University (2020zzts222). All the funding bodies funded in the study design, collection, analysis, interpretation of data, and writing the manuscript.
Author information
Authors and Affiliations
Contributions
Ying Liu and Jie Zhao designed and directed the project. Dun Yuan and Shun Yang collected clinical samples. Yunfei Xu and Kexin Li performed the experiments. Yunfei Xu, Kexin Li, Lin Zhou, Yao Zhao, Shuying Miao, and Caihong Lv analyzed the data. All authors discussed the results and contributed to the final manuscript. Yunfei Xu wrote the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This research was approved by the ethics committee of the Xiangya Hospital of Central South University, No: 2021101119. All participants involved in this study provided written informed consent. All procedures performed in studies involving animals were by the ethical standards of the institution or practice at which the studies were conducted by Experimental Animal Center of Central South University (Changsha, China), No: 2018sydw0222.
Consent to Participate
All participants involved in this study provided written informed consent.
Consent for Publication
All co-authors approved the final version of the manuscript and agreed to submit it to Molecular Neurobiology.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, Y., Liu, Y., Li, K. et al. COX-2/PGE2 Pathway Inhibits the Ferroptosis Induced by Cerebral Ischemia Reperfusion. Mol Neurobiol 59, 1619–1631 (2022). https://doi.org/10.1007/s12035-021-02706-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02706-1