
Abstract
Alzheimer’s disease (AD) is a neurodegenerative pathology characterized by progressive impairment of memory, associated with neurochemical alterations and limited therapy. The aim of this study was to evaluate the effects of inosine on memory, neuroinflammatory cytokines, neurotrophic factors, expression of purinergic receptors, and morphological changes in the hippocampus and cerebral cortex of the rats with AD induced by streptozotocin (STZ). Male rats were divided into four groups: I, control; II, STZ; III, STZ plus inosine (50 mg/kg); and IV, STZ plus inosine (100 mg/kg). The animals received intracerebroventricular injections of STZ or buffer. Three days after the surgical procedure, animals were treated with inosine (50 mg/kg or 100 mg/kg) for 25 days. Inosine was able to prevent memory deficits and decreased the immunoreactivity of the brain A2A adenosine receptor induced by STZ. Inosine also increased the levels of brain anti-inflammatory cytokines (IL-4 and IL-10) and the expression of brain-derived neurotrophic factor and its receptor. Changes induced by STZ in the molecular layer of the hippocampus were attenuated by treatment with inosine. Inosine also protected against the reduction of immunoreactivity for synaptophysin induced by STZ in CA3 hippocampus region. However, inosine did not prevent the increase in GFAP in animals exposed to STZ. In conclusion, our findings suggest that inosine has therapeutic potential for AD through the modulation of different brain mechanisms involved in neuroprotection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is characterized by progressive impairment of memory and other cognitive skills. It is a neurodegenerative disease that predominantly affects elderly people, with advanced age as the main risk factor; therefore, AD is a public health problem due to increased life expectancy [1,2,3]. Several mechanisms have been proposed to explain AD pathogenesis. Evidences have demonstrated that oxidative stress, dysfunction in cholesterol metabolism, neuroinflammation, low levels of brain-derived neurotrophic factor (BDNF), and alterations in cholinergic and purinergic signaling contribute to neurodegeneration and cognitive deficits [4,5,6,7,8,9,10,11].
BDNF and its high affinity to tyrosine kinase receptors (TrkB) play an important role in neuronal survival, cell differentiation, synaptic plasticity, and neuronal maintenance [11,12,13]. BDNF is a neurotrophin with the ability to increase long-term potentiation (LTP) which is directly associated with memory [14,15,16]. In fact, hippocampal and cortical post mortem samples of AD patients revealed a decrease in both BDNF and TrkB levels, indicating that deficits in BDNF signaling contribute to neuronal damage in this pathological condition [17,18,19,20,21].
AD is also associated with chronic inflammation in the central nervous system (CNS), characterized by an increase in the production of cytokines, chemokines, inflammatory factors, and infiltration of immune cells followed by neurodegeneration [2, 22]. The activation of microglia and astrocytes, accompanied by an increase in the production of pro-inflammatory cytokines, demonstrates a relationship between pro-inflammatory cytokine production and cognitive dysfunction [22]. Thus, astrogliosis and neuroinflammation appear to be integral components of AD onset and progression, which together with cholinergic neuronal loss are common findings in autopsy brain tissues of AD patients [2, 7, 23].
The participation of adenosine receptors in cognitive processes has been recognized over the years. Adenosine receptors such as A1 and A2A are located mainly at synapses and mediate the physiological actions of adenosine. The brain density of these receptors is affected by AD [24,25,26,27]. A1 loss appears to be among the factors related to cell death in the hippocampus, while an increase in A2A expression has been associated with neurodegenerative processes and memory deficits [28, 29]. On this note, the pharmacological modulation of adenosine receptors is an important target to be explored with the aim of preventing the action of signaling pathways involved in neurodegeneration in AD [27, 28].
Inosine, an endogenous purine nucleoside, is formed by deamination of adenosine by the enzyme adenosine deaminase and has been shown to have neuroprotective, anti-inflammatory, and antioxidant properties [30, 31]. Studies have suggested that the biological actions of inosine may be mediated through adenosine receptors or by the production of uric acid, an important peroxynitrite scavenger [32, 33]. Therefore, the aim of this study was to evaluate the effects of inosine on short-term memory, neuroinflammatory parameters, BDNF signaling, and expression of adenosine receptors in the hippocampus and cerebral cortex of rat model of AD. Besides, morphological changes, immunoreactivity of glial fibrillary acid protein (GFAP), and synaptophysin in the hippocampus also were evaluated.
Material and Methods
Chemicals
Inosine, streptozotocin (STZ), sodium citrate, butyrylcholine, hydrochloric acid, RIPA buffer, protease, and phosphatase inhibitors were purchased from Sigma Chemical Co. (St. Louis, MO, USA). All other reagents used in the detailed experiments were of analytical grade and the highest purity. TRIzol reagent and DNase I Amplification Grade were purchased from Invitrogen™ (Carlsbad, USA).
Animals
Adult male Wistar rats (60 days, 300–350 g) were provided by the Central Animal House of the Federal University of Pelotas. The animals were kept in cages under standard temperature (23 ± 1 °C), relative humidity (45–55%), and lighting conditions (12 h light/dark cycle) and with free access to standard rodent pelleted diet and water ad libitum. The Committee of Ethics and Animal Experimentation of the Federal University of Pelotas, Brazil, under protocol number CEEA 4808–2017, approved all animal procedures. The use of animals was in accordance with the Brazilian Guidelines for the Care and Use of Animals in Scientific Research Activities (DBCA), which is in agreement with the National Council of Control of Animal Experimentation (CONCEA).
Intracerebroventricular Injection of Streptozotocin
The animals were anesthetized with ketamine (75 mg/kg) and xylazine (10 mg/kg) for all surgical procedures. The head was placed in position in the stereotaxic apparatus, and a midline sagittal incision was made in the scalp in each animal. The stereotaxic coordinates for the lateral ventricle were measured accurately as anterio-posterior − 0.8 mm, lateral 1.5 mm, and dorso-ventral − 4.0 mm, relative to the bregma and ventral from the dura with the tooth bar set at 0 mm [9, 34]. Through a skull hole, the piston of a 28-gauge Hamilton® syringe of 10 μL attached to a stereotaxic apparatus was lowered manually into each lateral ventricle. The STZ groups received bilateral intracerebroventricular (ICV) injection of STZ (3 mg/kg, body weight) dissolved in citrate buffer (pH 4.4). The concentration was adjusted to deliver 5 μL/injection at the site. Animals in the control group received ICV injection of the same volume of citrate buffer.
Inosine Treatment
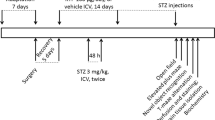
The animals were divided into four experimental groups (n = 10 each): I, control (C); II, STZ; III, STZ + inosine 50 mg/kg (STZ + Ino 50); and IV, STZ + inosine 100 mg/kg (STZ + Ino 100). The animals in groups II, III, and IV received bilateral ICV injection of STZ, while animals in group I received only citrate buffer. Three days after the surgical procedure, the animals in groups III and IV were treated with inosine (50 mg/kg or 100 mg/kg) and those in groups I and II received saline intraperitoneally (i.p.), as shown in Fig. 1. Inosine was dissolved in saline solution and administered everyday for 25 days. The body weight of the animals was evaluated weekly during the experimental period. Inosine dose was chosen based on previous studies indicating neuroprotection [35, 36].

The induction of a rat model of AD induced by STZ (3 mg/kg) and intraperitoneal (i.p.) administration of inosine (50 or 100 mg/kg) for 25 days
Behavioral Procedure
Open-Field Test
Locomotor behavior was evaluated using an open-field apparatus after 27 days of STZ injection. The open-field test was performed in an apparatus consisting of a box with the floor of the arena divided into 16 equal squares (18 × 18 cm), placed in a sound-free room. The number of quadrants crossed over a period of 5 min was the parameter used to evaluate locomotor activity. This test was carried out to identify motor disabilities, which might influence memory behavioral tests. The apparatus was cleaned with 40% ethanol and dried after each individual animal session [9].
Object Recognition
Twenty-four hours after the open-field test, which was also used as habituation to the apparatus, the animals underwent an object recognition test to evaluate short-term memory. The task was performed on the 28th day after STZ injection. The animals were placed individually in a box with two identical objects (objects A and B) for 5 min for free exploration (training). After 2 hours, the animals were put back in the box for 5 min, and one of the previous objects (B) was replaced (object C). The time spent exploring the new and familiar objects was recorded. The results were calculated according to the recognition index = TC / (TA + TC) [9]. After this test, the animals were euthanized and each animal’s brain and blood were collected for analysis. The brain tissues were prepared and protein determination was performed according to each specific technique.
Western Blot Analysis of A1 and A2A Receptor Immunoreactivity
The A1 and A2A receptor immunoreactivity in the hippocampal and cortical membranes were evaluated by western blot analysis. Samples of the hippocampus and cerebral cortex were homogenized in ice-cold radioimmunoprecipitation assay buffer (RIPA buffer) with 1 mM protease and phosphatase inhibitors and centrifuged at 12.000 rpm at 4 °C for 10 min. The protein concentration was determined using a BCA Protein Assay Kit (Sigma-Aldrich, EUA). The diluted samples were separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham Biosciences, UK). After blocking, the membranes samples were incubated overnight at 4 °C with primary antibodies; A1 (dilution 1:800, Santa Cruz Biotechnology, CA, USA) and A2A (dilution 1:800, Santa Cruz Biotechnology, CA, USA) membranes were incubated with anti‐rabbit or anti‐mouse secondary antibodies (dilution 1:10.000, Santa Cruz Biotechnology, CA, USA) for 90 min at room temperature. The membranes were incubated with an enhanced chemifluorescent substrate (Amersham Biosciences) and analyzed with Amersham Imager 600 (GE Healthcare Life Sciences, EUA). The membranes were reprobed and tested for β-actin immunoreactivity, as a control for protein concentration.
RNA Extraction, cDNA Synthesis, and Quantitative Real-Time Polymerase Chain Reaction of BDNF and TrkB
Total mRNA was extracted from 50 to 100 mg of the hippocampus and cerebral cortex tissue, using TRIzol reagent followed by DNase treatment with DNase I Amplification Grade, to ensure minimum DNA contamination of the samples. The total RNA isolated was quantified, and its purity (260/280 and 260/230 ratios) was examined using a NanoVue spectrophotometer (GE, Fairfield, CT, USA). cDNA synthesis was performed using High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, UK) according to the manufacturer’s protocol. For reverse transcription, 1 μg of total RNA was used in a reaction volume of 20 μL. The amplification was performed with GoTaq® qPCR Master Mix (Promega, Madison, WI) using CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories Inc., CA, USA). The sequences of the primers used are listed in Table 1. The qPCR conditions were as follows: 10 min at 95 °C to activate the hot-start Taq polymerase, followed by 35 cycles of denaturation for 15 seconds at 95 °C, primer annealing for 60 seconds at 60 °C, and extension for 30 seconds at 72 °C (fluorescence signals were detected at the end of every cycle). Baseline and threshold values were automatically set using the Bio-Rad CFX Manager software. The number of PCR cycles required to reach the fluorescence threshold in each sample was defined as the Ct value, and each sample was analyzed in duplicate to obtain an average Ct. The 2 − ΔΔCT method was used to normalize the fold change in gene expression, using β-actin as a housekeeping gene.
Quantification of Cytokines in the Brain
Hippocampus and cerebral cortex tissues were homogenized in 10 mM Tris–HCl buffer (pH 7.4) on ice using a homogenizer. The homogenate was centrifuged at 14.000 × g for 30 min, and the supernatants were used for the analysis. The cytokines IL-4 and IL-10 were detected by an enzyme-linked immunosorbent assay (ELISA) using OptEIA kit (Pharmingen, San Diego, CA, USA), according to the manufacturer’s instructions. Standard curves allowed determination of cytokine concentrations in pg/mL. The absorbance was read at 450 nm using a Power Wave X microplate scanning spectrophotometer (BioTek Instruments, Inc.).
Biochemical Analysis in the Serum
Butirilcholinesterase (BuChE) activity was determined using a modification of the method of Ellman et al. (1961) and was expressed in µmol BuSCh/h/mg of protein [37]. The method is based on the formation of 5,5-dithiobis-acid nitrobenzoic measured at 412 nm, and the reaction was initiated by adding 0.8 mM butyrylthiocholine iodide (BuSCh). Serum cholesterol levels were determined using commercially available diagnostic kits supplied by Labtest® (Labtest, MG, Brazil).
Brain Morphological Parameters
Hematoxylin and Eosin (HE)
The brain specimens were fixed in 10% buffered formalin, processed, and included in paraffin, and subjected to histological cut in a microtome set to a thickness of 4 μm. The cuts placed on matte sheets were heated in an oven at 80 °C for 1 hour, deparaffinized in xylol, rehydrated in ethyl alcohol staggered, and washed in distilled water. The slides were placed in Harris hematoxylin dye for 5 min, washed in running water, differentiated in acid-alcohol for 1 min, washed again in running water, and dipped in 1% lithium carbonate. Subsequently, the slides were placed in eosin dye for 3 min, dehydrated in absolute alcohol, and placed in xylol for assembly with Entellan-type resin.
Immunohistochemistry for Synaptophysin
The brains were fixed in 10% buffered formalin, processed, and included in paraffin and subjected to histological sections in a microtome regulated to a thickness of 5 μm and placed on slides. The slides were heated in an oven at 75 °C for 2 hours, deparaffinized in xylol, and rehydrated in ethyl alcohol and then in distilled water for 5 min in PBS [38]. Antigen recovery of synaptophysin was performed in a water bath for 20 min at 95 °C in citrate buffer 20 mM (pH 6.0). Endogenous peroxidase activity was blocked with a 5% solution of hydrogen peroxide in methanol for 30 min, in the dark. Protein blocking for synaptophysin was performed with BSA diluted to 1% in PBS for 60 min. The sections were incubated overnight in a refrigerator at 2–8 °C, with the primary anti-synaptophysin antibody at dilution 1:6000 (Monoclonal Anti-Synaptophysin antibody produced in mouse, Sigma-Aldrich S5768). After this, HRP-labeled polymer conjugated (Envision + Dual Link system-HRP kit, Dakoref: K4061) was added and incubated for 30 min at room temperature. Diaminobenzidine (Liquid DAB + Substrate Chromogen System, Dako ref: K3468) was used to visualize the reactions staining. Diaminobenzidine (Liquid DAB + Substrate Chromogen System, Dako ref: K3468) was used to visualize the reactions staining, according to the manufacturer’s recommendations.
Negative controls were obtained performing the same protocol described above, with the omission of primary antibody, which was replaced by BSA. The slides were counterstained in Harris’ hematoxylin for 20 seconds and differentiated in 2% ammoniacal water for 20 seconds. The sections were dehydrated in absolute alcohol and placed in xylol for the assembly of the slides in Entellan-type resin. The images were obtained through a capture system using Leica DM6-B vertical digital research microscope and the Leica LAS X Life Science software. The quantification of synaptophysin was performed through optical density (OD) analysis using the software Image Pro Plus® 6.3 (Media 258 Cybernetics). Images of each region were captured per section.
Immunohistochemistry for GFAP
The brains were fixed in 10% buffered formalin, included in paraffin, and subjected to histological sections in a microtome regulated to a thickness of 3 μm and placed on slides. The slides were heated in an oven at 75 °C for 2 hours, deparaffinized in xylol, and rehydrated in ethyl alcohol and then in distilled water for 5 min in PBS. Antigenic recovery was performed in a water bath for 20 min at 94 °C in citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked with a 5% solution of hydrogen peroxide in methanol for 20 min in the dark. Protein blocking was performed with skimmed-milk powder diluted to 5% in PBS for 20 min.
The cuts were incubated overnight in a refrigerator at 2–8 °C, with the primary anti-GFAP antibody in 1:200 dilution. After incubation, the secondary IgGκ light chain HRP antibody conjugated at 1:200 dilution was applied and incubated for 1 hour and 30 min at room temperature, and the reaction was visualized with Liquid Dab (Dako, K3468) according to the manufacturer’s recommendations. After visualization, the slides were counterstained in Harris’ hematoxylin for 20 seconds and differentiated in 2% ammoniacal water for 20 seconds. The cuts were dehydrated in absolute alcohol and placed in xylol for the assembly of the slides in Entellan-type resin. The quantification of GFAP was performed through optical density (OD) analysis using an Olympus® BX 257 40 microscope coupled to a computer with the software Image Pro Plus® 6.3 (Media 258 Cybernetics). Images of each region were captured per section.
Statistical Analysis
Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for comparison of means using GraphPad Prism version 5.0 Program (Intuitive Software for Science, San Diego, CA, USA). P ≤ 0.05 was considered statistically significant in the analysis. All data are expressed as mean ± standard error (SEM).
Results
Effects in Body Weight of Animals
In Fig. 2, the animals treated only with STZ showed a reduction in body weight in relation to the control group during all experimental periods (P < 0.001). Inosine at 100 mg/kg alone was able to increase the body weight of the rats injected with STZ and not inosine at 50 mg/kg.

Effects of treatment with inosine (50 or 100 mg/kg) on the body weight of rats injected with STZ (3 mg/kg). **P < 0.01 and ***P < 0.001 when compared with the control group. #P < 0.05 and ##P < 0.01 when compared with the STZ group (n = 10 per group)
Inosine Prevents Memory Deficits Induced by STZ
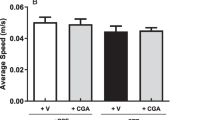
In the object recognition task, the percentage of exploratory preference of the new object in STZ group was significantly lower than that of the control group (P < 0.01), indicating memory impairment. Treatment with inosine 100 mg/kg increased the percentage of exploratory preference of the new object when compared with the STZ group, demonstrating that inosine is capable of restoring memory deficits induced by this experimental model (Fig. 3). As shown in Fig. 3, neither STZ nor STZ plus inosine treatment altered locomotor activity in the open-field test.

Effects of treatment with inosine (50 or 100 mg/kg) in locomotor activity (using the open-field apparatus) and short-time memory (using the object recognition test) of rats injected with STZ (3 mg/kg). **P < 0.01 when compared with the control group. ##P < 0.01 when compared with the STZ group (n = 10 per group)
Inosine Modulated the Brain Adenosine Receptors
Concerning the density of receptors in the hippocampus, our results showed an increase in immunoreactivity of A1 and A2A receptors in the STZ group (P < 0.05). Inosine (100 mg/kg) increased the levels of A1, and both inosine concentrations were effective in decreasing the immunoreactivity of the A2A receptor when compared to the STZ group (Fig. 4). In the cerebral cortex, only an increase in A2A density was observed in the STZ group (P < 0.05), which was prevented by inosine (50 mg/kg). Inosine (100 mg/kg) also increased the levels of A1 in the cerebral cortex of the rats (P < 0.05) (Fig. 4).

Effect of inosine (50 and 100 mg/kg) on the immunoreactivity (IR) of purinergic receptors A1 and A2A in hippocampal (A, C) and cortical membranes (B, D) of rats injected with STZ (3 mg/kg) evaluated by western blotting. *P < 0.05 and ***P < 0.001 compared with the control group. ##P < 0.01 and ###P < 0.001 when compared with the STZ group (n = 4–5 per group)
Inosine Modulates Expression of BDNF and TrkB Receptor
Expression levels of TrkB receptor was significantly decreased in the hippocampus of the STZ group (P < 0.01), while a trend of decreased levels of BDNF mRNA was observed in the hippocampus. Interestingly, inosine at both doses was capable of increasing the mRNA levels of TrkB and BDNF in the hippocampus (Fig. 5). No changes were observed in the mRNA levels of receptor TrkB in the cerebral cortex (Fig. 5). Though STZ did not reduce the levels of the BDNF mRNA in the cerebral cortex, inosine treatment at 100 mg/kg increased BDNF levels (Fig. 5).

Effects of inosine treatment (50 and 100 mg/kg) on the mRNA levels of tyrosine kinase receptor (TrkB) and brain-derived neurotrophic factor (BDNF) in the hippocampus and cerebral cortex of rats injected with STZ (3 mg/kg). ***P < 0.001 when compared with the control group. #P < 0.05, ##P < 0.01, and ###P < 0.001 when compared with the STZ group (n = 5–6 per group)
Inosine Prevent Alterations in Brain Levels of Anti-inflammatory Cytokines and Serum Biochemical Parameters
In Fig. 6, the results show that STZ reduced the levels of IL-4 and IL-10 in both the hippocampus and cerebral cortex of rats. In the hippocampus, treatment with inosine 100 mg/kg alone was capable of preventing these alterations. On the other hand, in the cerebral cortex only, the dose of inosine 50 mg/kg was effective in increasing the levels of both anti-inflammatory cytokines (Fig. 6). In the serum, STZ caused an increase in BuChE activity and total cholesterol levels when compared to the control group (P < 0.05) (Fig. 7). Inosine treatment at both doses (50 and 100 mg/kg) prevented these serum alterations (Fig. 7).

Effects of treatment with inosine (50 or 100 mg/kg) on IL-4 and IL-10 levels in the hippocampus and cerebral cortex of rats injected with STZ (3 mg/kg). *P < 0.05, **P < 0.01, and ***P < 0.001 when compared with the control group. #P < 0.05 and ##P < 0.01 when compared with the STZ group (n = 4–5 animals per group)

Effects of treatment with inosine (50 or 100 mg/kg) on butyrylcholinesterase (BuChE) activity and cholesterol levels in the serum of rats injected with STZ (3 mg/kg). *P < 0.05 when compared with the control group. #P < 0.05 and ###P < 0.001 when compared with the STZ group (n = 4–5 per group)
Effects of Inosine in Morphological in Immunoreactivity for GFAP and Synaptophysin in Different Regions of the Hippocampus
Figure 8 shows the histological changes using hematoxylin and eosin staining in the CA1, CA3, and dentate gyrus (DG) regions of the hippocampus. In STZ group, in the CA1 and DG regions, there is a change in the morphology of the cells that make up the granular layer (see black arrows). In the control group, the nuclei were rounded and large with a lighter chromatin with visible nucleoli (see white arrows). In the STZ group, the heterogeneity of the cells was smaller, the cytoplasm was not apparent, and nuclei had elongated or fusiform morphology (see white arrows). In addition, the presence of “dark bodies” along the molecular layer, which are characterized by cells with a more basophilic nucleus, greater dyeing by hematoxylin, and an elongated morphology. These morphological changes in the molecular layer were attenuated by treatment with inosine 50 and 100 mg/kg in rats exposed to STZ.

Histopathological changes by hematoxylin and eosin (H&E) staining in the CA1, CA3, and dentate gyrus (DG) regions of the hippocampus of rats treated with inosine (50 and 100 mg/kg, i.p.) and injected with STZ (3 mg/kg) (100 × magnification). The square in the lower right field represents a magnification of 400 ×
Figure 9 shows the immunoreactivity for GFAP in the hippocampus of rats treated with inosine (50 and 100 mg / kg) and submitted to a sporadic model of Alzheimer’s type dementia (STZ 3 mg/kg). Image A shows the GFAP immunoreactivity between the different groups in the CA1, CA3, and DG regions. Image B shows a representation of the locations chosen for the acquisition of images in the respective regions. There was an increase in immunoreactivity of GFAP in CA1 (graph C), CA3 (graph D), and DG regions (graph E) in rats exposed to STZ. Inosine did not restore the increased GFAP immunoreactivity in AD rats.

(A) Immunoreactivity of glial fibrillary acid protein (GFAP, astrocyte marker) in the CA1, CA3, and dentate gyrus (DG) regions of the hippocampus (B) of rats treated with inosine (50 and 100 mg/kg, i.p.) and injected with STZ (3 mg/kg). Graphs represent the mean ± SEM of the cell density (optical density) of GFAP marked cells in the three analyzed regions: CA1 (C), CA3 (D), and DG (E). *P < 0.05 when compared with the control group.
Figure 10 shows the immunoreactivity for synaptophysin protein in the hippocampus of AD rats treated with inosine (50 and 100 mg/kg). Image A shows the synaptophysin immunoreactivity between the different groups in the CA1, CA3, and DG regions. Image B shows a representation of the locations chosen for the acquisition of images in the respective regions. In the CA1 and CA3 regions, we used the molecular layer and DG region the hilum was selected. Graph C shows that there was no significant difference in optical density between the groups compared to the control (P > 0.05). Graph D showed a reduction in optical density for synaptophysin in CA3 in the STZ group compared to the control (P = 0.006). Inosine at a dose of 100 mg/kg protected against the reduction of immunoreactivity for synaptophysin induced by STZ. In the DG, an increase in the optical density for synaptophysin was seen in the STZ groups treated with inosine 50 and 100 mg/kg (P = 0.0125).

(A) Immunoreactivity of synaptophysin in the CA1, CA3, and dentate gyrus (DG) regions of the hippocampus (B) of rats treated with inosine (50 and 100 mg/kg, i.p.) and injected with STZ (3 mg/kg). Graphs represent the mean ± SEM of the cell density (optical density) of GFAP marked cells in the three analyzed regions: CA1 (C), CA3 (D), and DG (E). *P < 0.05 and **P < 0.001 when compared with the control group. #P < 0.05 when compared with the STZ group.
Discussion
The STZ model is characterized by glucose hypometabolism and brain insulin resistance and represents a sporadic non-transgenic AD model. This model reproduced the molecular and behavioral characteristics of AD, such as memory deficits, neuroinflammation, oxidative stress, cholinergic alterations, and glia activation [39,40,41,42], and has been used for preclinical testing of pharmacology therapy for AD [9, 36, 43,44,45].
Using the behavioral task of object recognition, our results showed that the STZ group had less preference for the new object, confirming the memory deficits. Inosine 100 mg/kg was capable of preventing short-time memory dysfunctions induced by STZ. The effect of inosine in improving memory can be associated with the modulation of many neural pathways such as oxidative stress, ion pump activities, and cholinergic signaling [36]. In addition, in the present study, we also demonstrated that inosine is capable of modulating other mechanisms involved in memory deficits such as neuroinflammation, purinergic system, and BDNF signaling.
There are no known receptors specific for inosine; however, studies have suggested that this nucleoside is capable of interacting with adenosine receptors [46]. Adenosine receptors have many roles in the brain, such as in presynaptic and postsynaptic neuromodulatory activities and learning and memory [27, 46, 47]. Due to the inhibitory effects of A1 and excitatory effects of A2A, in neurodegenerative conditions, A1 has been described to have a neuroprotective effect, while A2A plays a crucial role in the neurodegeneration process [47]. Although the mechanism of action of A2A to improve the memory deficit it is not very clear, Pagnussat et al. [48] showed that activation of A2A decrease short-term memory in mice. In addition, treatment with A2A antagonist recovers memory and synaptic deficits in transgenic mouse model of DA [49]. On this note, an important finding of our study is the reduction of A2A expression in the hippocampus and cerebral cortex by inosine, which can be directly associated with memory improvement. Other studies have also demonstrated that inosine prevents the positive regulation of A2A in experimental models of autoimmune encephalomyelitis [32] and Parkinson’s disease [50].
BDNF through the activation of its receptor (TrkB) plays a crucial role in the nervous system by providing trophic support to neurons and by regulating synaptic transmission and plasticity, such as long-term potentiation (LTP), an important pathway associated with memory [51]. Evidences have documented that adenosine receptors can modulate brain actions of BDNF. The activation of the receptor A2A is required to trigger various TrkB-mediated BDNF actions in the brain [52] to sustain the normal BDNF levels and BDNF-induced potentiation of synaptic transmission in the hippocampus [53]. In regard to this, the upregulation in A2A expression in STZ group in the cerebral cortex and hippocampus could explain, at least in part, the normal levels observed in BDNF mRNA in this group. However, an important result of this study was the increase in the levels of BDNF and TrkB mRNA mainly in the hippocampus by inosine. Our results corroborating with previous studies that also showed that BDNF mRNA was increased 2 hours after oral single inosine administration [54]. Considering that inosine decreased the expression of A2A and increased the expression of A1, we can suggest that this effect in BDNF and TrkB mRNA may be associated with A1 adenosine receptors. This result correlates the observation by Muto et al. [54] where A1 receptor antagonist partially inhibited the inosine-induced increase of BDNF mRNA.
Previous studies also revealed that BDNF supplementation increased the expression of synaptophysin in the cultured hippocampal slices [55]. Synaptophysin is a protein associated with the regulation of synaptic vesicle endocytosis and synapse formation [56,57,58]. In addition, alterations in hippocampus synaptophysin has been an important mechanism associated with memory decline [59]. Corroborating with this evidences, here, we also showed that STZ induced a memory decline and a decrease in immunoreactivity in sinaptophysin in CA3 hippocampus region. On the other hand, animals treated with inosine improved memory and showed an increase in the immunoreactivity of synaptophysin in CA3 and DG hippocampus regions. The CA3 region contributes to rapid encoding to novel information, formation and storage of arbitrary associations and short-term memory, while the DG has a key role in hippocampal memory formation [60]. In this line, considering that alterations in presynaptic proteins contributed to cognitive dysfunction, the increase in immunoreactivity of synaptophysin induced by inosine may contribute to enhanced synaptic plasticity leading to memory improvement. In addition, the mechanism involved in the synaptophysin increase by inosine can be possibly associated to BDNF signaling.
The association between the inflammatory markers and cognitive decline has been documented in both animal models and patients with AD [61]. Previous studies have demonstrated a relationship between early cytokine production and cognitive dysfunction and glial activation, which, in turn, is accompanied by an increase in the production of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-12) [62,63,64,65]. In this study, we showed a decrease in the levels of anti-inflammatory cytokines IL-10 and IL-4 in the STZ group. In the brain, IL-10 is capable of promoting neuronal survival by blocking the effects of proapoptotic cytokines and limiting inflammation by reducing the synthesis of pro-inflammatory cytokines and suppressing cytokine receptor expression/activation [64]. In addition, IL-4 is very important to immunity and plays a critical role in brain functions, such as homeostasis, neurogenesis, memory, and learning [62, 63]. Thus, the increase caused by inosine in the levels of these brain anti-inflammatory cytokines is an important neuroprotective function that can explain, together with other results, the improve of memory and reduction in damage in the hippocampus visualized in histology analysis.
Corroborating with the results described above, we also observed that STZ induced an increase in BuChE activity and inosine in both doses prevented these peripheral alterations. BuChE is a nonspecific cholinesterase able to hydrolyze acetylcholine, as well as other choline esters. While acetylcholinesterase is located mainly in neurons, BuChE is mainly associated with glial cells. Acetylcholine receptors are prominently expressed in immune cells and it is involved in the control of the production of many pro- and anti-inflammatory cytokines [66, 67]. Studies have suggested that serum BuChE could be used as a possible marker of systemic inflammation [64] because an increase in this enzyme activity reduces the tissue acetylcholine levels, leading to disrupted cholinergic anti-inflammatory responses. Peripheral BuChE is an α-glycoprotein synthesized in the liver and its serum level can be correlated with several clinical conditions; for example, it is increased in patients with AD in plasma levels and in tissue resulting in low levels of acetylcholine [68, 69]. Thus, it is plausible that a reduction caused by inosine in BuChE activity in the serum could increase the acetylcholine levels and contribute to the anti-inflammatory activity of this nucleoside. The effects of inosine on cholinergic signaling have also been reported in other studies [36].
In addition, an increase in GFAP was observed in the hippocampus in the STZ group. In pathological conditions, astrocytes become reactive, leading to an upregulation of pro-inflammatory cytokines, which are associated with neuronal damage [70]. Astrocyte reactivity is characterized by morphological changes and overexpression of GFAP [71].
However, in our study, inosine was not able to prevent GFAP overexpression induced by STZ. It is important to note that immunohistochemistry and histological analysis were prioritized in the hippocampus since behavioral memory tests are dependent on this brain region.
Lastly, elevated cholesterol levels are correlated with a higher incidence of memory impairment and dementia [72]. In fact, studies have investigated the potential therapeutic effects of lipid-lowering agents such as statins in experimental models of AD [73]. The relationship between lipid metabolism, especially cholesterol levels and AD, has been associated with apolipoprotein E4 (apoE4). ApoE4 is an important genetic risk factor for this neurodegenerative disease as it increases brain inflammation [74]. Interestingly, our results showed that inosine at both doses (50 and 100 mg/kg) was capable of preventing the increase in serum cholesterol levels induced by STZ. The exact mechanism associated with inosine and lipid metabolism has not been described in the literature, but recently Lima et al. (2020) [75] also demonstrated that inosine decreases cholesterol levels in a hypercholesterolemic animal model.
In conclusion, our findings showed that inosine is capable of reestablishing memory deficits and modulating adenosine receptors, inflammatory and neurotrophic factors, synaptic proteins, and cholesterol levels. We can propose that inosine is an innovative and useful tool for AD therapeutics due to its multi-target action as shown in our results. Thus, inosine may be considered as a promising strategy of preventing neurodegeneration.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Yiannopoulou KG, Papageorgiou SG (2020) Current and future treatments in alzheimer disease: an update. J Cent Ner Sys Dis 12:1179573520907397. https://doi.org/10.1177/1179573520907397
Kempuraj D, Mentor S, Thangavel R, Ahmed ME, Selvakumar GP, Raikwar SP, Dubova J, Zaheer S, Iyer S, Zaheer A (2019) Mast cells in stress, pain, blood-brain barrier, neuroinflammation and Alzheimer’s disease. Front Cell Neurosci 13:54. https://doi.org/10.3389/fncel.2019.00054
Gray SC, Kinghorn KJ, Woodling NS (2020) Shifting equilibriums in Alzheimer’s disease: the complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural Regen Res 15:1208. https://doi.org/10.4103/1673-5374.272571
Hampel H, Messulam M, Cuello A, Farlow M, Giacobini E, Grossberg G, Khachaturian A, Vergallo A, Cavedo E, Synder P, Khachaturian Z (2018) The cholinergic system in the pathophysyology and tretament of Alzheimer’s disease. Brain 141:1917–1933. https://doi.org/10.1093/brain/awy132
Moore AM, Mahoney E, Dumitrescu L, De Jager PL, Koran M, Petyuk VA, Robinson R, Ruderfer D, Cox N, Schneider J, Bennet D, Jefferson A, Hohman T (2020) APOE ε4-specific associations of VEGF gene family expression with cognitive aging and Alzheimer’s disease. Neurobiol Aging 87:18–25. https://doi.org/10.1016/j.neurobiolaging.2019.10.021
Butterfield DA, Mattson MP (2020) Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol Dis 138:104795. https://doi.org/10.1016/j.nbd.2020.104795
Hampel H, Caraci F, Cuello AC, Caruso G, Nisticò R, Corbo M, Baldacci F, Toschi N, Garaci F, Chiesa PA, Verdooner SR, Akman-Anderson L, Hernández F, Ávila J, Emanuele E, Valenzuela PL, Lucía A, Watling M, Imbimbo BP, Vergallo A, Lista S (2020) A path toward precision medicine for neuroinflammatory mechanisms in Alzheimer’s disease. Front Immunol 11:456. https://doi.org/10.3389/fimmu.2020.00456
Stanciu GD, Luca A, Rusu RN, Bild V, BescheaChiriac SI, Solcan C, Bild W, Ababei DC (2020) Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules 10:40. https://doi.org/10.3390/biom10010040
Pacheco SM, Soares MSP, Gutierres JM, Gerzson MFB, Carvalho FB, Azambuja J, Shetinger MRC, Stefanello FM, Spanevello RM (2018) Anthocyanins as a potential pharmacological agent to manage memory deficit, oxidative stress and alterations in ion pump activity induced by experimental sporadic dementia of Alzheimer’s type. J Nutr Biochem 56:193–204. https://doi.org/10.1016/j.jnutbio.2018.02.014
Woods LT, Ajit D, Camden JM, Erb L, Weisman GA (2016) Purinergic receptors as potential therapeutic targets in Alzheimer’s disease. Neuropharmacology 104:169–179. https://doi.org/10.1016/j.neuropharm.2015.10.031
Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S (2008) New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev 59:201–220. https://doi.org/10.1016/j.brainresrev.2008.07.007
Shen T, You Y, Joseph C, Mirzaei M, Klistorner A, Graham SL, Gupta V (2018) BDNF polymorphism: a review of its diagnostic and clinical relevance in neurodegenerative disorders. Aging Dis 9: 523–536. https://doi.org/10.14336/AD.2017.0717
Tebano MT, Martire A, Chiodi V, Ferrante A, Popoli P (2010) Role of adenosine A2A receptors in modulating synaptic functions and brain levels of BDNF: a possible key mechanism in the pathophysiology of Huntington’s disease. Sci World J 10:1768–1782. https://doi.org/10.1100/tsw.2010.164
Jerónimo-Santos A, Batalha VL, Mueller CE, Baqi Y, Sebastião AM, Lopes LV, Diógenes MJ (2014) Impact of in vivo chronic blockade of adenosine A2A receptors on the BDNF-mediated facilitation of LTP. Neuropharmacology 83:99–106. https://doi.org/10.1016/j.neuropharm.2014.04.006
Lu Y, Christian K, Lu B (2008) BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurob Learn Mem 89:312–323. https://doi.org/10.1016/j.nlm.2007.08.018
Korte M, Kang H, Bonhoeffer T, Schuman E (1998) A role for BDNF in the late-phase of hippocampal long-term potentiation. Neuropharmacology 37:553–559. https://doi.org/10.1016/s0028-3908(98)00035-5
Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW (1991) BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 7:695–702. https://doi.org/10.1016/0896-6273(91)90273-3
Connor B, Young D, Yan Q, Faull RLM, Synek B, Dragunow M (1997) Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Mol Brain Res 49:71–81. https://doi.org/10.1016/s0169-328x(97)00125-3
Allen SJ, Wilcock GK, Dawbarn D (1999) Profound and selective loss of catalytic TrkB immunoreactivity in Alzheimer’s disease. Biochem Biophys Res Commun 264:648–651. https://doi.org/10.1006/bbrc.1999.1561
Ferrer I, Marín C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolota E, Martí E (1999) BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol 58:729–739. https://doi.org/10.1097/00005072-199907000-00007
Holsinger RD, Schnarr J, Henry P, Castelo VT, Fahnestock M (2000) Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer’s disease. Mol Brain Res 76:347–354. https://doi.org/10.1016/s0169-328x(00)00023-1
Counil H, Krantic S (2020) Synaptic activity and (neuro)inflammation in Alzheimer’s disease: could exosomes be an additional link? J Alzheimer’s Dis 74:1029–1043. https://doi.org/10.3233/JAD-191237
Kamphuis W, Middeldorp J, Kooijman L, Sluijs JA, Kooi EJ, Moeton M, Freriks M, Mizee M, Hol E (2014) Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol Aging 35:492–510. https://doi.org/10.1016/j.neurobiolaging.2013.09.035
Rebola N, Pinheiro PC, Oliveira CR, Malva JO, Cunha RA (2003) Subcellular localization of adenosine A1 receptors in nerve terminals and synapses of the rat hippocampus. Brain Res 987:49–58. https://doi.org/10.1016/s0006-8993(03)03247-5
Cunha RA (2005) Neuroprotection by adenosine in the brain: from A 1 receptor activation to A 2A receptor blockade. Purinergic Sig 1:111–134. https://doi.org/10.1007/s11302-005-0649-1
Canas PM, Porciúncula LO, Simões AP, Augusto E, Silva HB, Machado NJ, Gonçalvez N, Alfaro T, Gonçalves F, Araújo I, Real J, Coelho J, Andrade G, Alemida R, Chen J, Kofalvi A, Cunha R (2018) Neuronal adenosine A2A receptors are critical mediators of neurodegeneration triggered by convulsions. eNeuro 26: ENEURO.0385–18.2018. https://doi.org/10.1523/ENEURO.0385-18.2018
Blum D, Sandau U, Laurent C, Batalha V, Leboucher A, Hamdane M, Pasquier F, Boison D, Buée L, Lopes L (2013) Adenosine receptors and Alzheimer’s disease. In Adenosine 385–407. Springer New York. https://doi.org/10.1007/978-1-4614-3903-5_19
Rahman A (2009) The role of adenosine in Alzheimer’s disease. Cur Neuropharmacol 7:207–216. https://doi.org/10.2174/157015909789152119
Faivre E, Coelho JE, Zornbach K, Malik E, Baqi Y, Schneider M, Cellai L, Carvalho K, Sebda S, Figeac M, Eddarkaoui S, Caillierez R, Chern Y, Heneka M, Sergeant N, Muller C, Bueé L, Lopes L, Bi V (2018) Beneficial effect of a selective adenosine A2A receptor antagonist in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Front Mol Neurosc 11:235. https://doi.org/10.3389/fnmol.2018.00235
da Rocha LF, de Oliveira APL, Accetturi BG, de Oliveira MI, Domingos HV, de Almeida CD (2013) Anti-inflammatory effects of inosine in allergic lung inflammation in mice: evidence for the participation of adenosine A2A and A3 receptors. Purinergic Signal 9:325–336. https://doi.org/10.1007/s11302-013-9351-x
Ruhal P, Dhingra D (2018) Inosine improves cognitive function and decreases aging-induced oxidative stress and neuroinflammation in aged female rats. Inflammopharmacology 26:1317–1329. https://doi.org/10.1007/s10787-018-0476-y
Junqueira SC, dos Santos CI, Lieberknecht V, Cunha MP, Calixto JB, Rodrigues ALS (2017) Inosine, an endogenous purine nucleoside, suppresses immune responses and protects mice from experimental autoimmune encephalomyelitis: a role for A2A adenosine receptor. Mol Neurobiol 54:3271–3285. https://doi.org/10.1007/s12035-016-9893-3
Markowitz C, Spitsin S, Zimmernan V, Jacobs V, Udupa J, Hooper C, Koprowski H (2009) The treatment with multiple sclerosis with inosine. J Altern Complement Med 15:619–625. https://doi.org/10.1089/acm.2008.0513
Herman JP, Watson SJ (1987) The rat brain in stereotaxic coordinates (2nd edn) by George Paxinos and Charles Watson. Trends Neurosci 10:439–439
Dachir S, Shabashov D, Trembovler V, Alexandrovich AG, Benowitz L, Shohami E (2014) Inosine improves functional recovery after experimental traumatic brain injury. Brain Res 1555:78–88. https://doi.org/10.1016/j.brainres.2014.01.044
Teixeira FC, Gutierres JM, Soares MSP, de Mattos BDS, Spohr L, Do Couto CA, Bona N, Assmann C, Morsch V, Da Cruz I, Stefanello F, Spanevello RM (2020) Inosine protects against impairment of memory induced by experimental model of Alzheimer disease: a nucleoside with multitarget brain actions. Psychopharmacol 237:811–823. https://doi.org/10.1007/s00213-019-05419-5
Ellman G, Courtney K, Andres V, Feather-Stone R (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95. https://doi.org/10.1016/0006-2952(61)90145-9
Huf F, Bandiera F, Müller C, Gea L, Carvalho F, Rahmeier F, Reiter K, Tortorelli L, Gomez R, Fernandes M (2019) Comparative study on the effects of cigarette smoke exposure, ethanol consumption and association: behavioral parameters, apoptosis, glial fibrillary acid protein and S100b immunoreactivity in different regions of the rat hippocampus. Alcohol 77:101–112. https://doi.org/10.1016/j.alcohol.2018.08.009
Gulyaeva NV, Bobkova NV, Kolosova NG, Samokhin AN, Stepanichev MY (2017) Molecular and cellular mechanisms of sporadic Alzheimer’s disease: studies on rodent models in vivo. Biochemistry 82:1088–1102. https://doi.org/10.1134/S0006297917100029
Grieb P (2016) Intracerebroventricular streptozotocin injections as a model of Alzheimer’s disease: in search of a relevant mechanism. Mol Neurobiol 53:1741–1752. https://doi.org/10.1007/s12035-015-9132-3
Salkovic-Petrisic M, Hoyer S (2007) Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: an experimental approach. J Neural Transm Suppl 72:217–233. https://doi.org/10.1007/978-3-211-73574-9_28
Knezovic A, Osmanovic-Barilar J, Curlin M, Hof PR, Simic G, Riederer P, Salkovic-Petrisic M (2015) Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer’s disease. J Neural Transm 122: 577–592. 0.1007/s00702–015–1394–4
Gutierres JM, Carvalho FB, Schetinger MRC, Marisco P, Agostinho P, RodriguesM RubinMA, Schmatz R, da Silva CR, de P Cognato G, Farias JG, Signor C, Morsch VM, Mazzanti CM, Bogo M, Bonan CD, Spanevello R, (2014) Anthocyanins restore behavioral and biochemical changes caused by streptozotocin-induced sporadic dementia of Alzheimer’s type. Life Sci 96:7–17. https://doi.org/10.1016/j.lfs.2013.11.014
Oliveira J, Abdalla F, Dornelles GL, Adefegha S, Palma TV, Signor C, Berbardi JS, Bladissarelli J, Lenz LS, Magni LP, Rubin MA, Pillat MM, Andrade CM (2016) Berberine proctects against memory impairment and anxiogenic-like behavior in rats submitted to sporadic Alzheimer’s like dementia: involvement of acetylcholinesterase and cell death. Neurotoxiclogy 57:241–250. https://doi.org/10.1016/j.neuro.2016.10.008
Gerzson MF, Bona NP, Soares MS, Teixeira FC, Rahmeier FL, Carvalho FB, Cruz MF, Onzi G, Lenz G, Gonçalves R, Spanevello R, Stefanello FM (2020) Tannic acid ameliorates STZ-induced Alzheimer’s disease-like impairment of memory, neuroinflammation, neuronal death and modulates Akt expression. Neurot Res 37:1009–1017. https://doi.org/10.1007/s12640-020-00167-3
Gomez G, Sitkovsky MV (2003) Differential requirement for A2a and A3 adenosine receptors for the protective effect of inosine in vivo. Blood 102:4472–4478. https://doi.org/10.1182/blood-2002-11-3624
Stockwell J, Jakova E, Cayabyab FS (2017) Adenosine A1 and A2A receptors in the brain: current research and their role in neurodegeneration. Molecules 22:676. https://doi.org/10.3390/molecules22040676
Pagnussat N, Almeida AS, Marques DM, Nunes F, Chenet GC, Botton PHS (2015) Adenosine A2A receptors are necessary and sufficient to trigger memory impairment in adult mice. Br J Pharmacol 172:3831–3845. https://doi.org/10.1111/bph.13180
Silva A, Lemos C, Gonçalves F, Pilássova A, Machado N, Silva H, Canas P, Cunha R, Lopes J, Agostinho P (2018) Blockade of adenosine receptors recovers early deficits of memory and plasticity in the triple transgenic mouse modelo f Alzheimer’s disease. Neurobiol Dis 117:72–81. https://doi.org/10.1016/j.nbd.2018.05.024
El-Shamarka ME, Kozman MR, Messiha BA (2020) The protective effect of inosine against rotenone-induced Parkinson’s disease in mice; role of oxido-nitrosative stress, ERK phosphorylation, and A2AR expression. Naunyn-Schmiedeberg’s Arch Pharmacol 393:1041–1053. https://doi.org/10.1007/s00210-019-01804-1
Lu B, Nagappan G, Lu Y (2014) BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol 220:223–250. https://doi.org/10.1007/978-3-642-45106-5_9
Sebastião A, Assaife-Lopes N, Diógenes M, Vaz S, Ribeiro J (2011) Modulation of brain-derived neurotrophic fator (BDNF) actions in the nervous system by adenosine A (2A) receptors and the role of lipid rafts. Biochim Biophys Acta 1808:1340–1349. https://doi.org/10.1016/j.bbamem.2010.06.028
Tebano MT, Martire A, Potenza R, Pepponi R, Armida M, Domenici M, Schwarzchild M, Chen J, Popoli P (2008) Adenosine A(2A) receptors are required for normal BDNF levels and BDNF-induced potentiation of synaptic transmission in the mouse hippocampus. J Neurochem 104:279–286. https://doi.org/10.1111/j.1471-4159.2007.05046.x
Muto J, Lee H, Lee L, Uwaya A, Park J, Nakajima S, Nagata K, Ohno M, Ohsawa I, Mikami T (2014) Oral administration of inosine produces antidepressant-like effects in mice. Sci Rep 4:4199. https://doi.org/10.1038/srep04199
Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B (2001) Protein synthesis-dependent and independent regulation of hippocampal synapses by brain derived neurotrophic factor. J Biol Chem 276:37585–37593. https://doi.org/10.1074/jbc.M101683200
Tarsa L, Goda Y (2002) Synaptophysin regulates activity-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci 99:1012–1016. https://doi.org/10.1073/pnas.022575999
Kwon S, Chapman E (2011) Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70:847–854. https://doi.org/10.1016/j.neuron.2011.04.001
McMahon H, Bolshakov V, Hammer R, Siegelbaum S, Sudhof T (1996) Synaptophysin, a major synaptic vesicle protein, is not essential for neurotransmitter release. Proc Natl Acad Sci 93:4460–4764. https://doi.org/10.1073/pnas.93.10.4760
Sye C, Troncoso J, Kawas C, Peter M, Price D, Martin D (1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56:933–944. https://doi.org/10.1097/00005072-199708000-00011
Kesner R (2007) Behavioral functions of the CA3 subregion of the hippocampus. Leran Mem 14:771–778. https://doi.org/10.1101/lm.688207
Schwab C, McGeer PL (2008) Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J Alzheimer’s Dis 13:359–369. https://doi.org/10.3233/jad-2008-13402
Gadani SP, Cronk JC, Norris GT, Kipnis J (2012) IL-4 in the brain: a cytokine to remember. J Immunol 189: 4213–4219. 0.4049/jimmunol.1202246
Gambi F, Reale M, Iarlori C, Salone A, Toma L, Paladini C (2004) Alzheimer patients treated with an AChE inhibitor show higher IL-4 and lower IL-1β levels and expression in peripheral blood mononuclear cells. J Clin Psychopharmacol 24:314–321. https://doi.org/10.1097/01.jcp.0000125683.74595.2f
Strle K, Zhou JH, Broussard SR, Venters HD, Johnson RW, Freund GG, Dantzer R, Kelley K (2002) IL-10 promotes survival of microglia without activating Akt. J Neuroimmunol 122:9–19. https://doi.org/10.1016/s0165-5728(01)00444-1
Rossi C, Cusimano M, Zambito M, Finardi A, Capotondo A, Garcia-Manteiga JM, Comi G, Martino G, Muzio M (2018) Interleukin 4 modulates microglia homeostasis and attenuates the early slowly progressive phase of amyotrophic lateral sclerosis. Cell Death Dis 9:1–16. https://doi.org/10.1038/s41419-018-0288-4
De Jonge WJ, Ulloa L (2007) The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151:915–929. https://doi.org/10.1038/sj.bjp.0707264
Das UN (2007). Acetylcholinesterase and butyrylcholinesterase as possible markers of low-grade systemic inflammation. Med Sci Monit 13: RA214-RA221.
Rao AA, Sridhar GR, Das UN (2007) Elevated butyrylcholinesterase and acetylcholinesterase may predict the development of type 2 diabetes mellitus and Alzheimer’s disease. Med Hypotheses 69:1272–1276. https://doi.org/10.1016/j.mehy.2007.03.032
Mushtaq G, Greig H, N, A Khan J, A Kamal M, (2014) Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets 13:1432–1439. https://doi.org/10.2174/1871527313666141023141545
Li C, Zhao R, Gao K, Wei Z, Yin M, Lau L, Chui D, Yu A (2011) Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 8:67–80. https://doi.org/10.2174/156720511794604543
Ben Haim L, Carrillo-de Sauvage MA, Ceyzériat K, Escartin C (2015) Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cellu Neurosci 9:278. https://doi.org/10.3389/fncel.2015.00278
Salkovic-Petrisic M, Knezovic A, Hoyer S, Riederer P (2013) What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J Neural Transm 120:233–252. https://doi.org/10.1007/s00702-012-0877-9
Shepardson NE, Shankar GM, Selkoe DJ (2011) Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch Neurol 68:1239–1244. https://doi.org/10.1001/archneurol.2011.203
Ophir G, Amariglio N, Jacob-Hirsch J, Elkon R, Rechavi G, Michaelson DM (2005) Apolipoprotein E4 enhances brain inflammation by modulation of the NF-κB signaling cascade. Neurobiol Dis 20:709–718. https://doi.org/10.1016/j.nbd.2005.05.002
Lima D, Hacke ACM, Inaba J, Pessôa CA, Kerman K (2020) Electrochemical detection of specific interactions between apolipoprotein E isoforms and DNA sequences related to Alzheimer’s disease. Bioelectrochemistry 133:107447. https://doi.org/10.1016/j.bioelechem.2019.107447
Acknowledgements
The authors acknowledge the Conselho Nacional de Desenvolvimento Científico e Tecnológico and Fundação de Amparo à Pesquisa do Rio Grande do Sul. We would like to thank Editage (www.editage.com) for English language editing.
Funding
This study was funded by Fundação de Amparo à Pesquisa do Rio Grande do Sul (FAPERGS)—grant number: 19/2551–0001712-0). This study was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (CAPES—Finance code 001).
Author information
Authors and Affiliations
Contributions
Fernanda C. Teixeira, Jessié M. Gutierres, Mayara S.P. Soares: experimental design, animal treatment, memory behavior, biochemical analysis, statistical analysis, result interpretation, and manuscript preparation.
Eduardo B. Blödorn, William B. Domingues, Vinicius C. Farias: quantitative real-time polymerase chain reaction of BDNF and tyrosine receptor kinase B (TrkB).
Karine P. Reichert, Maria Rosa Chitolina: analysis of adenosine receptors.
Relber A. Gonçalves: cytokine analysis.
Adriana M. Zago, Fabiano B. Carvalho, Marilda C. Fernandes: histological analysis and immunoreactivity of glial fibrillary acid protein (GFAP) and synaptophysin.
Francieli M. Stefanello: experimental design and result interpretation.
Roselia M. Spanevello: experimental design, result interpretation, discussion, manuscript preparation, and funding.
Corresponding authors
Ethics declarations
Ethics Approval
The Committee of Ethics and Animal Experimentation of the Federal University of Pelotas, Brazil, under protocol number CEEA 4808–2017, approved all animal procedures. The use of animals was in accordance with the Brazilian Guidelines for the Care and Use of Animals in Scientific Research Activities (DBCA), which is in agreement with the National Council of Control of Animal Experimentation (CONCEA).
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflicts of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Teixeira, F.C., Soares, M.S.P., Blödorn, E.B. et al. Investigating the Effect of Inosine on Brain Purinergic Receptors and Neurotrophic and Neuroinflammatory Parameters in an Experimental Model of Alzheimer’s Disease. Mol Neurobiol 59, 841–855 (2022). https://doi.org/10.1007/s12035-021-02627-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02627-z