
Abstract
Ischemic stroke (IS) is a common and serious neurological disease. Extensive evidence indicates that activation of the immune system contributes significantly to the development of IS pathology. In recent years, some long non-coding RNAs (lncRNAs), acting as competing endogenous RNAs (ceRNAs), have been reported to affect IS process, especially the immunological response after stroke. However, the roles of lncRNA-mediated ceRNAs in immune pathogenesis of IS are not systemically investigated. In the present study, we generated a global immune-related ceRNA network containing immune-related genes (IRGs), miRNAs, and lncRNAs based on experimentally verified interactions. Further, we excavated an IS immune-related ceRNA (ISIRC) network through mapping significantly differentially expressed IRGs, miRNAs, and lncRNAs of patients with IS into the global network. We analyzed the topological properties of the two networks, respectively, and found that lncRNA NEAT1 and lncRNA KCNQ1OT1 played core roles in aforementioned two immune-related networks. Moreover, the results of functional enrichment analyses revealed that lncRNAs in the ISIRC network were mainly involved in several immune-related biological processes and pathways. Finally, we identified 17 lncRNAs which were highly related to the immune mechanism of IS through performing random walk with restart for the ISIRC network. Importantly, it has been confirmed that NEAT1, KCNQ1OT1, GAS5, and RMRP could regulate immuno-inflammatory response after stroke, such as production of inflammatory factors and activation of the immune cells. Our results suggested that lncRNAs exerted an important role in the immune pathogenesis of IS and provided a new strategy to do research on IS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute ischemic stroke (IS), which results from the sudden disruption of blood flow and reduced oxygen levels in cerebral arteries, is a major cause of death and disability worldwide [1]. IS initiates a complex cascade of neurologic events that include excitotoxicity, oxidative/nitrative stress, calcium overload, inflammation, and apoptosis, which can lead to focal brain damage. Neuroinflammation—which comprises microglia activation, infiltration of circulating immune cells, and upregulation of proinflammatory cytokines and thrombotic/fibrinolytic factors—plays a critical role in stroke-induced brain injury [2,3,4]. Inhibiting microglia activation was shown to reduce infarct volume and attenuate apoptosis in a rat model of transient focal cerebral ischemia [5]; it has therefore been suggested that microglia activation following cerebral ischemia reflects the severity of brain injury [6]. Blood level of interleukin 6 (IL-6), a marker of systemic inflammation, is elevated in IS [7]; and a high plasma IL-6 level was found to be correlated with severe neurologic deficits and predicted early deterioration of brain function [8, 9]. However, the immuno-inflammatory mechanisms underlying IS are mostly unknown. Identifying novel gene signatures or biomarkers can provide etiologic insight into IS as well as potential therapeutic targets.
Recently, competing endogenous RNAs (ceRNAs) define a new regulatory mechanism between coding and non-coding RNAs, whereby RNA molecules containing microRNA (miRNA, ~20 nucleotides) response elements compete with each other by binding to a common miRNA [10]. Accumulating evidence demonstrates that long non-coding RNAs (lncRNAs, more than 200 nucleotides) regulate target gene expression by acting as ceRNAs for miRNAs and have been implicated in the development of various diseases [11]. For example, the lncRNA endogenous bornavirus-like nucleoprotein 3, pseudogene (EBLN3P), may act as a ceRNA to regulate dedicator of cytokinesis 4 (DOCK4) expression by competitively sponging the miRNA miR-144-3p, thereby modulating liver cancer progression [12]; XLOC_006390 may function as a ceRNA that negatively regulates the expression of miR-331-3p and miR-338-3p to promote cervical cancer tumorigenesis and metastasis [13]; and maternally expressed gene 3 (MEG3) is a ceRNA that competes with programmed cell death 4 (PDCD4) mRNA for direct binding to miR-21 in the regulation of ischemic neuronal death [14]. Even so, little is still known about the roles of lncRNA-mediated ceRNA in IS, and studies on it in IS are just beginning.
LncRNAs acting as ceRNAs also regulate the immuno-inflammatory response in various diseases. For instance, the lncRNA small nucleolar RNA host gene 15 (SNHG15) was shown to regulate the expression of programmed death ligand 1 (PD-L1) by inhibiting miR-141, which in turn promoted the resistance of stomach cancer cells to the immune response [15]. The lncRNA HIX003209 functioned as a ceRNA and enhanced inflammation by sponging miR-6089 via the Toll-like receptor 4 (TLR4)/nuclear factor (NF)-κB pathway in macrophages in rheumatoid arthritis [16]. lncRNAs acting as ceRNAs also participate in immuno-inflammation in the ischemic cascade. Knockdown of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), a ceRNA for miR-181c-5p, attenuated inflammatory injury after cerebral ischemia [17]. However, the role of lncRNAs acting as ceRNAs in the immune mechanisms of IS is poorly understood.
To address this point, we constructed a global immune-related ceRNA (GIRC) network based on experimentally validated miRNA–gene and miRNA–lncRNA interactions. A dysregulated IS immune-related ceRNA (ISIRC) network was excavated from the GIRC network using a computational approach based on gene, miRNA, and lncRNA expression profiles in patients with IS. By analyzing the topological properties of the networks, we identified the lncRNAs NEAT1 and KCNQ1OT1 as important regulatory components. Functional analyses revealed that lncRNAs that were dysregulated in IS were mainly associated with the immune response or inflammation. Thus, lncRNAs acting as ceRNAs play an important role in the immune pathogenesis of IS.
Material and Methods
Gene–miRNA–lncRNA Interaction Data
The miRNA–gene interactions were obtained from tarBase v7.0 [18], which contains experimentally validated miRNA–gene interactions curated from published experiments on 356 different cell types from 24 species. We downloaded 309,958 human miRNA–gene interaction pairs comprising 18,426 genes and 1033 miRNAs.
The miRNA–lncRNA interactions were obtained from starBase v3.0 [19], which contains interactions that have been experimentally validated (e.g., HITS-CLIP, PAR-CLIP, iCLIP, and CLASH). We downloaded 71,952 human miRNA–lncRNA interaction pairs, including 642 miRNAs and 3788 lncRNAs.
Immune-Related Genes (IRGs)
The IRGs were obtained from InnateDB [20] and ImmPort [21] databases. In total, 1040 IRGs were downloaded from InnateDB—a publicly available database containing information on genes involved in the innate immune response in humans—and 2483 were downloaded from ImmPort. We combined the 2 data sources and obtained a set of 2517 IRGs.
Gene, miRNA, and lncRNA Expression Profiles for IS
A search of lncRNA profile in IS was conducted in the Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo) with the following key words: (“lncRNA” and “acute ischemic stroke”), and the species was restricted as “Homo sapiens.” As a result, we could simply find out two datasets: GSE122709 based on GPL20795 platform and GSE140275 based on GPL16791 platform. The gene, miRNA, and lncRNA profiles under accession number GSE122709 [22] contained 5 IS patients and 5 age-, sex-, and vascular risk factor-matched controls, which were used for discovery dataset. Blood samples were collected from IS patients within 24 h and 7 days after symptom onset in aforementioned study [22]. For control samples, blood samples were collected only once. In the present study, we selected blood samples collected within 24h after symptom onset as the subsequent subjects, while lncRNA profile under accession number GSE140275 [23] was from 3 IS patients and 3 matched normal subjects and was used for validation dataset.
Human Gene and lncRNA Annotations
GENCODE (Release 35) annotation files that included the latest gene and lncRNA annotations in GTF format were used to standardize the symbols of the above-mentioned genes and lncRNAs [24].
Identification of the Differentially Expressed (DE)IRGs, DEmiRNAs, and DElncRNAs
We obtained RNA sequencing data for IS from the GEO dataset. In order to characterize RNA with dysregulated expression, DESeq package was used to detect DE genes (DEGs), DEmiRNAs, and DElncRNAs between IS and normal groups; |log2 fold change (FC)|>1.0 and adjusted P<0.05 were set as the cutoff values. The intersection of DEGs with IRGs yielded DEIRGs. We used the gplots package in R to generate volcano plots.
Construction of the GIRC Network
We constructed a GIRC network based on the theory that lncRNAs share common miRNA-binding sites with their target genes and function as miRNA sponges to regulate gene expression. For a given IRG–miRNA–lncRNA interaction, IRGs and lncRNAs shared common miRNAs, constituting a competing triplet. We constructed a GIRC network of all IRG–miRNA–lncRNA competing triplets that was visualized using Cytoscape software (https://cytoscape.org/), with the nodes in the network representing IRGs, miRNAs, and lncRNAs and edges representing their interactions.
Construction of an ISIRC
We excavated the ISIRC network from the GIRC network by hypergeometric test using the following formula:
For each lncRNA–gene pair, N is the total number of DEmiRNAs in IS, n is the number of miRNAs interacting with the gene, m is the number of miRNAs interacting with the lncRNA, and x is the number of miRNAs shared by the gene and lncRNA. P<0.05 was considered statistically significant. The ISIRC network was constructed and visualized using Cytoscape software.
Functional Enrichment and Protein–Protein Interaction (PPI) Analysis
To determine the potential functions of the DEIRGs and lncRNAs in the ISIRC network, clusterProfiler [25]—an R package of Bioconductor that allows statistical analysis and visualization of gene sets or clusters—was used to perform enrichment analyses of Gene Ontology (GO) functions and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. GO terms and KEGG pathways with a P value<0.05 were considered to be significantly enriched functional annotations. PPIs of DEIRGs in the ISIRC network were determined using STRING v11.0 [26] and visualized with Cytoscape software.
Random Walk with Restart to Prioritize DElncRNAs of IS
Random walk with restart was performed on the ISIRC network in order to prioritize DElncRNAs of IS by simulating a random walker and randomly moving from a set of source nodes to its network neighbors, as represented by the following equation:
where M is the column-normalized adjacency matrix of the ISIRC network; P0 is the initial probability vector, which is constructed so that a value of 1 was assigned to nodes representing genes known to be associated with disease, while 0 was assigned to other nodes; λ is the restart probability at each step of the random walk at source nodes; and Pt is a vector in which the ith element has the probability of being at node i at the time point t.
All candidate DElncRNAs could be ranked according to their corresponding probability in p1, and DElncRNAs with high scores were considered to be the most likely lncRNAs related to IS. We analyzed the statistical significance of scores of each candidate DElncRNA. Through comparing the scores of lncRNAs in the network following n iterations of that known IS-related immune genes shuffling, the statistical significance for rejection of the null hypothesis was determined. In order to strictly maintain the network topological properties, random sampling without replacement was performed when doing random disturbance, and the degree distribution was guaranteed the same between selection seed node and the real. In iterations, the times of each lncRNA score that was higher than the actual value was recorded as m. The statistical significance P value for each lncRNA was calculated by the ratio of m and n; in this study, n was set to 1000 times.
Results
Construction and Topological Analysis of a GIRC Network
To construct a GIRC network, we downloaded human miRNA–gene and miRNA–lncRNA interactions from tarBase and starBase databases, respectively, along with human IRGs from InnateDB and ImmPort databases. The GIRC network was established by integrating the above data (Fig. 1a). The network contained 5190 nodes (including 1539 IRGs, 409 miRNAs, and 3242 lncRNAs) and 64,207 edges. The fact that a large proportion of nodes were lncRNAs suggests that they play an essential role in the GIRC network. It was discovered that the network approximated the scale-free network topology (R2=0.8888) of a transcriptional regulatory network (Fig. 1b). The degree distribution of a scale-free network follows a power law, at least asymptotically. The degree of a node is the number of neighbors it has in a network, which is an important attribute for evaluating the importance of a node. We therefore analyzed the degree distribution of lncRNAs (Fig. 1c). The lncRNAs KCNQ1OT1, NEAT1, and XIST had the highest degrees. These 3 lncRNAs are thought to be involved in regulating the immuno-inflammatory response in different diseases [27,28,29]. Thus, the GIRC network can be used as a starting point for investigating the immune mechanisms of IS.

Construction and analysis of the GIRC network. a GIRC network. Blue, yellow, and red nodes represent IRGs, miRNAs, and lncRNAs, respectively. Lines represent interactions among IRGs, miRNAs, and lncRNAs. The pie chart shows the number of IRGs, miRNAs, and lncRNAs in the network. b Degree distribution of all nodes in the GIRC network. c Degree distribution of lncRNAs
Identification of IS-Associated DEIRGs, DEmiRNAs, and DElncRNAs
The mRNA, miRNA, and lncRNA profiles in the blood of patients with IS within 24 h after symptom onset were determined by HITS. Based on the pre-established threshold of |log2FC|>1.0 and adjusted P<0.05, we identified 2176 DEGs (750 upregulated and 1426 downregulated). As the immuno-inflammatory response plays a critical role in the development of IS [30], we focused our analysis on IRGs. The intersection of DEGs and IRGs yielded 358 DEIRGs (130 upregulated and 228 downregulated; Fig. 2a, b). We also identified 157 DEmiRNAs (131 upregulated and 26 downregulated; Fig. 2c) and 170 DElncRNAs (107 upregulated and 63 downregulated; Fig. 2d).

Identification of DEIRGs, DEmiRNAs, and DElncRNAs in IS. a Venn diagram of IRGs and DEGs. Orange and purple circles indicate IRGs and DEGs, respectively; their intersection represents overlapping genes (i.e., DEIRGs). b Volcano plots of DEIRGs, c volcano plots of DEmiRNAs, d volcano plots of DElncRNAs. DEIRGs, DEmiRNAs, or DElncRNAs that are upregulated and downregulated are represented by red and green, respectively. The top 10 upregulated and downregulated differentially expressed RNAs are shown to the right of the corresponding volcano plots
Excavation of an ISIRC Network from the GIRC Network
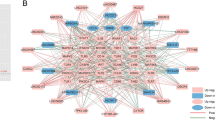
To investigate the relationship between DEIRGs, DEmiRNAs, and DElncRNAs and identify key lncRNAs in IS, we mapped 358 DEIRGs, 157 DEmiRNAs, and 170 DElncRNAs to the GIRC network. In total, 221 miRNA–gene interactions and 226 miRNA–lncRNA interactions were mapped. We then performed a significance test of the number of shared miRNAs between DEIRG and DElncRNA pairs using hypergeometric test. When a gene and lncRNA shared the same miRNA with P<0.05, an edge was added between them. A significantly dysregulated ISIRC network was constructed containing 173 nodes (including 81 genes, 30 miRNAs, and 62 lncRNAs) and 447 edges (Fig. 3a).

Excavated and analysis of the dysregulated ISIRC network. a The dysregulated ISIRC network in IS. Blue, yellow, and red nodes represent DEIRGs, DEmiRNAs, and DElncRNAs, respectively. Lines represent interactions among DEIRGs, DEmiRNAs, and lDEncRNAs. b Degree distribution of the ISIRC network, c degree distribution of DEIRGs, d degree distribution of DEmiRNAs, e degree distribution of DElncRNAs, f node betweenness in the ISIRC network, g immune mechanisms regulated by lncRNAs in IS
To characterize the topological features of the ISIRC network, we calculated the degree, closeness centrality, and betweenness of the ISIRC network, respectively. The degree of all nodes in the ISIRC network approximated that of a scale-free network (R2=0.8693; Fig. 3b). We analyzed the degree distribution of genes, miRNAs, and lncRNAs (Fig. 3c–e) and the betweenness of nodes (Fig. 3f) in the ISIRC network. We ranked the topological features of all nodes and identified the top 10 in each dimension (Table 1). Two lncRNAs (NEAT1 and KCNQ1OT1) known to play important roles in the immune system appeared on each list. In acute brain injury caused by oxygen-glucose deprivation/reoxygenation (OGD/R), classically activated (M1) microglia release cytokines that exacerbate inflammatory damage; NEAT1 may suppress the polarization of microglia toward the M1 phenotype to reduce the extent of injury [31]. KCNQ1OT1 has been shown to promote OGD/R-induced neuronal injury at least part by acting as a ceRNA in a mouse model of IS induced by middle cerebral artery occlusion (MCAO) [32]. Thus, our results indicate that lncRNAs contribute to the pathogenesis of IS by regulating immune cells or inflammatory factors (Fig. 3g).
Functional Enrichment and PPI Analyses
To investigate the functions of DElncRNAs in the ISIRC network, we carried out GO and KEGG pathway enrichment analysis using the genes they regulated. We identified 328 GO terms (273 biological processes (BPs), 40 molecular functions (MFs), and 15 cellular components (CCs); Supplementary Table 1). These genes were slightly enriched in critical immune-related BPs such as leukocyte adhesion, negative regulation of immune process, positive regulation of cytokine production, and myeloid cell homeostasis (Fig. 4a). Neutrophils, lymphocytes, and monocytes accumulate in the vasculature during hyperacute stroke and may affect clinical outcome [33]. The enrichment of MFs was mainly associated with growth factor binding, and the most enriched CCs were related to the cell membrane. The KEGG pathway analysis identified 15 enriched pathways (Fig. 4b and Supplementary Table 2). Several key pathways were related to immune response or inflammation including Th17 cell differentiation, TNF signaling pathway, and human T-cell leukemia virus 1 infection. PR-957—an inhibitor of the immunoproteasome subunit low molecular weight polypeptide 7—is known to provide neuroprotection by inhibiting Th17 cell differentiation in MCAO mice, which indicated that Th17 cell differentiation might play a part in aggravating brain injury [34]. Our findings demonstrate that lncRNAs regulate multiple signaling pathways and have a variety of functions in IS.

Functional enrichment analysis and establishment of the PPI network. a GO analysis of DEIRGs regulated by lncRNAs in the ISIRC network; the degree of enrichment increases from blue to red. Larger circles indicate a more significant proportion of genes among GO function genes. b KEGG pathway analysis of DEIRGs regulated by DElncRNAs in the ISIRC network; the degree of enrichment increases significantly from blue to red. Larger circles indicate a larger proportion of genes among KEGG pathway genes. c PPI network of DEIRGs in the ISIRC network. Circles represent DEIRGs, and the size of the node represents the degree. Lines represent interactions between proteins encoded by DEIRGs, and the width of the line represents the combined score between DEIRGs. d Subnetwork of DEIRGs involved in the immune mechanisms of IS; brown circles represent experimentally validated DEIRGs
To clarify the interactions of proteins encoded by the genes regulated by DElncRNAs in the ISIRCs network, we constructed a PPI network based on the STRING database (Fig. 4c). Nine DEIRGs have been reported that participate in immune mechanisms underlying IS—namely MAFB, SOCS1, SPHK1, ULK1, LRRFIP1, PTCH1, TNFSF14, GAS6, and IL6ST. A subnetwork of 9 DEIRGs was extracted from the PPI network (Fig. 4d).
Identification of lncRNAs Regulating the Immune Pathogenesis of IS
To identify IS-related lncRNAs, we applied the random walk with restart algorithm to the ISIRC network. We first selected the 9 above-mentioned genes as seed nodes and then used the algorithm to prioritize lncRNAs in the network. We obtained 17 lncRNAs (NEAT1, KCNQ1OT1, GAS5, RMRP, TPT1-AS1, TMPO-AS1, SNHG3, PDCD4-AS1, MIR17HG, LINC00174, ZSWIM8-AS1, SH3BP5-AS1, NUTM2B-AS1, LINC00894, ITPKB-IT1, ASH1L-AS1, and ADAMTSL4-AS1) that were statistically significant (P<0.05) after 1000 times random walk with permutation. As lncRNAs with high significance scores are connected to and have functions similar to neighboring seed nodes, these 17 lncRNAs were presumed to have immune-related regulatory functions in IS. NEAT1 and KCNQ1OT1 had the highest degrees in the ISIRC network, highlighting their importance in immune mechanisms of IS. We extracted the 2 lncRNA-related subnetworks from the ISIRC network (Fig. 5a, b). In order to further dissect the roles of candidate lncRNAs, we characterized the potential regulatory mechanism of them in immune pathogenesis of IS (Fig. 5c) along with the interactions among DEIRGs, DEmiRNAs, and candidate DElncRNAs (Supplementary Table 3). To increase the credibility of our results, we consulted the relevant literature. Four lncRNAs—namely NEAT1, KCNQ1OT1, GAS5, and RMRP—have been previously reported to be involved in the immuno-inflammatory response following IS through regulation of microglia, macrophages, or inflammatory factors (Table 2). The functions of 76% (13/17) of the lncRNAs implicated in IS have not been reported. So they might be new stars in the immune mechanism research of IS. To further verify the reliability of our results, we downloaded another lncRNA expression profile (GSE140275) of IS from GEO database. By analyzing the high-throughput expression profile, 1223 DElncRNAs were identified (Supplementary Table 4). There were 3 (namely GAS5, TPT1-AS1, ADAMTSL4-AS1) common shared DElncRNAs, which were statistically significant (P<0.05) based on a hypergeometric test, between 17 candidate DElncRNAs and GSE140275 (Supplementary Figure 1). These findings further enhanced the credibility of our results.

Analysis of IS-associated lncRNAs from the ISIRC network identified with the random walk approach. Subnetworks of the lncRNAs NEAT1 a and KCNQ1OT1 b extracted from the ISIRC network. c Candidate lncRNAs regulating immuno-inflammatory response after IS
Discussion
Increasing evidence has elaborated that IS involves the activation of immuno-inflammatory responses that is associated with the upregulation of proinflammatory cytokines such as C-reactive protein (CPR), IL-1 and IL-6, and which leads to neuronal cell death [4, 7]. Several in vitro and in vivo studies found that lncRNAs involved in the inflammatory processes may be potential diagnostic biomarkers or therapeutic targets in IS [17, 35, 36]. lncRNA-mediated ceRNA regulatory networks can provide insight into the molecular basis of various diseases; thus, constructing a ceRNA network of immune-related lncRNAs can be useful to clarify the immune mechanisms of IS. In our study, an integrated and computational approach was performed using experimentally verified interactions and expression profiles of genes, miRNAs, and lncRNAs to explore the immune-related ceRNAs in IS.
In this study, we generated a GIRC network based on the theory of ceRNAs that lncRNAs and genes compete for miRNA sponges. lncRNAs with the highest degrees in the immune-related global network (e.g., KCNQ1OT1, NEAT1, and XIST) are known to be involved in immune responses or inflammation in various diseases. For instance, KCNQ1OT1 level was elevated in the plasma of patients with diabetic nephropathy, and its reduction suppressed inflammation, oxidative stress, and pyroptosis of renal tubular epithelial cells via the sponging of miR-506-3p [37]. Inhibition of lncRNA NEAT1 in inflammatory bowel disease could suppressed the inflammatory response by modulating the intestinal epithelial barrier and through exosome-mediated polarization of macrophages [28]. XIST was found to function as a ceRNA by binding to miR-370-3p, and XIST knockdown alleviated lipopolysaccharide-induced cell injury by enhancing cell viability and inhibiting apoptosis and inflammatory cytokine production in acute pneumonia [29]. Consequently, construction of the network from a global view elucidated that lncRNAs modulated immuno-inflammatory processes is a commonly phenomenon in diverse diseases, which would provide a significant background for the immune molecular mechanism studies of IS.
We established the ISIRC network by mapping the DEIRGs, DRmiRNAs, and DElncRNAs to the global network and analyzed the functions of the DElncRNAs. The results of the GO analysis revealed that lncRNAs in the ISIRC network were mainly involved in immune-related BPs such as leukocyte adhesion, negative regulation of immune process, positive regulation of cytokine production, and myeloid cell homeostasis. Similarly, the KEGG pathway analysis of genes regulated by lncRNAs in the ISIRC network revealed several enriched immune-related pathways including Th17 cell differentiation, TNF signaling, and HTLV-1 infection. The results of the original dataset showed that inflammation might play an important role in the development of IS (22). Our findings also suggested that several significant immune or inflammation-related biological pathways were closely concerned with IS, which is partially similar to the results of the original dataset. These lncRNAs may thus have important roles in the immune mechanisms of IS.
We applied the random walk with restart algorithm to the ISIRC network and identified 17 lncRNAs that were closely associated with immuno-inflammation following IS. It was previously demonstrated that NEAT1 expression is positively correlated with National Institute of Health Stroke Scale score and the levels of proinflammatory factors such as CRP, TNF-α, IL-6, IL-8, and IL-22 and negatively correlated with that of the anti-inflammatory cytokine IL-10 in patients with IS [38]. Microglia are the main immune cells of the central nervous system and have diverse functions in the pathogenesis of various neurologic diseases [39]. NEAT1 was shown to regulate microglia activation and promoted neuronal apoptosis after cerebral ischemia/reperfusion (I/R) injury [31]. A portion role of lncRNA KCNQ1OT1 in IS also has been validated. KCNQ1OT1 was upregulated in patients with cerebral ischemia and directly interacted with and suppressed the expression of miR-140-3p [40]; conversely, KCNQ1OT1 knockdown attenuated inflammation, oxidative stress, and cell apoptosis induced by OGD/R in cerebral I/R injury [40]. Additionally, the severity of brain injury after IS was influenced by GAS5 and RMRP regulation of microglia activation [41, 42]. Though these 4 lncRNAs above-mentioned that have been validated by previous work are not specific for the IS, these results illustrate that our strategy for identifying lncRNAs involved in the immune regulatory of IS is reliable. However, there is little known about the functions of the other lncRNAs in the ISIRC network, which may thus be novel regulators of the immune mechanisms of IS. Our method also exists some limitations. Because of the inconsistent symbols of database, a great deal of genes and lncRNAs might be lost during the process of integrating the data, which may decrease our result. Another limitation of our study is that some of the identified lncRNAs may have been false positives because of the small number of samples from IS patients. In future studies, we will use more specimens to confirm the in vivo roles of lncRNAs in IS.
In conclusion, we identified 17 lncRNAs related to IS based on the assumptions of ceRNAs. The ISIRC network constructed in this study offers a comprehensive strategy for investigating the molecular basis of IS, and the identified lncRNAs may serve as novel therapeutic targets for IS treatment.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Deb P, Sharma S, Hassan KM (2010) Pathophysiologic mechanisms of acute ischemic stroke: an overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 17(3):197–218. https://doi.org/10.1016/j.pathophys.2009.12.001
Dziedzic T (2015) Systemic inflammation as a therapeutic target in acute ischemic stroke. Expert Rev Neurother 15(5):523–531. https://doi.org/10.1586/14737175.2015.1035712
Kawabori M, Yenari MA (2015) Inflammatory responses in brain ischemia. Curr Med Chem 22:1258–1277. https://doi.org/10.2174/0929867322666150209154036
Tuttolomondo A, Pinto A, Corrao S, Di Raimondo D, Fernandez P, Di Sciacca R, Arnao V, Licata G (2009) Immuno-inflammatory and thrombotic/fibrinolytic variables associated with acute ischemic stroke diagnosis. Atherosclerosis 203(2):503–508. https://doi.org/10.1016/j.atherosclerosis.2008.06.030
Sun M, Deng B, Zhao X, Gao C, Yang L, Zhao H, Yu D, Zhang F et al (2015) Isoflurane preconditioning provides neuroprotection against stroke by regulating the expression of the TLR4 signalling pathway to alleviate microglial activation. Sci Rep 5(1). https://doi.org/10.1038/srep11445
Emmrich JV, Ejaz S, Neher JJ, Williamson DJ, Baron J-C (2014) Regional distribution of selective neuronal loss and microglial activation across the MCA territory after transient focal ischemia: quantitative versus semiquantitative systematic immunohistochemical assessment. J Cereb Blood Flow Metab 35(1):20–27. https://doi.org/10.1038/jcbfm.2014.181
Worthmann H, Tryc AB, Goldbecker A, Ma YT, Tountopoulou A, Hahn A, Dengler R, Lichtinghagen R et al (2010) The temporal profile of inflammatory markers and mediators in blood after acute ischemic stroke differs depending on stroke outcome. Cerebrovasc Dis 30(1):85–92. https://doi.org/10.1159/000314624
Vila N, Castillo J, Dávalos A, Chamorro A (2000) Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke 31:2325–2329. https://doi.org/10.1161/01.str.31.10.2325
Acalovschi D, Wiest T, Hartmann M, Farahmi M, Mansmann U, Auffarth GU, Grau AJ, Green FR et al (2003) Multiple levels of regulation of the interleukin-6 system in stroke. Stroke 34:1864–1869. https://doi.org/10.1161/01.STR.0000079815.38626.44
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi Pier P (2011) A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell 146(3):353–358. https://doi.org/10.1016/j.cell.2011.07.014
Sheng Y, Ma J, Zhao J, Qi S, Hu R, Yang Q (2019) Differential expression patterns of specific long noncoding RNAs and competing endogenous RNA network in alopecia areata. J Cell Biochem 120(6):10737–10747. https://doi.org/10.1002/jcb.28365
Li H, Wang M, Zhou H, Lu S, Zhang B (2020) Long noncoding RNA EBLN3P promotes the progression of liver cancer via alteration of microRNA-144-3p/DOCK4 signal. Cancer Manag Res 12:9339–9349. https://doi.org/10.2147/cmar.S261976
Luan X, Wang Y (2018) LncRNA XLOC_006390 facilitates cervical cancer tumorigenesis and metastasis as a ceRNA against miR-331-3p and miR-338-3p. J Gynecol Oncol 29(6). https://doi.org/10.3802/jgo.2018.29.e95
Yan H, Rao J, Yuan J, Gao L, Huang W, Zhao L, Ren J (2017) Long non-coding RNA MEG3 functions as a competing endogenous RNA to regulate ischemic neuronal death by targeting miR-21/PDCD4 signaling pathway. Cell Death Dis 8(12):3211. https://doi.org/10.1038/s41419-017-0047-y
Dang S, Malik A, Chen J, Qu J, Yin K, Cui L, Gu M (2020) LncRNA SNHG15 contributes to immuno-escape of gastric cancer through targeting miR141/PD-L1. Onco Targets Ther 13:8547–8556. https://doi.org/10.2147/ott.S251625
Yan S, Wang P, Wang J, Yang J, Lu H, Jin C, Cheng M, Xu D (2019) Long non-coding RNA HIX003209 promotes inflammation by sponging miR-6089 via TLR4/NF-κB signaling pathway in rheumatoid arthritis. Front Immunol 10. https://doi.org/10.3389/fimmu.2019.02218
Cao D-w, Liu M-m, Duan R, Tao Y-f, Zhou J-s, Fang W-r, Zhu J-r, Niu L et al (2019) The lncRNA Malat1 functions as a ceRNA to contribute to berberine-mediated inhibition of HMGB1 by sponging miR-181c-5p in poststroke inflammation. Acta Pharmacol Sin 41(1):22–33. https://doi.org/10.1038/s41401-019-0284-y
Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos I-L, Maniou S et al (2015) DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res 43(D1):D153–D159. https://doi.org/10.1093/nar/gku1215
Li J-H, Liu S, Zhou H, Qu L-H, Yang J-H (2014) starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42(D1):D92–D97. https://doi.org/10.1093/nar/gkt1248
Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock REW et al (2013) InnateDB: systems biology of innate immunity and beyond—recent updates and continuing curation. Nucleic Acids Res 41(D1):D1228–D1233. https://doi.org/10.1093/nar/gks1147
Bhattacharya S, Andorf S, Gomes L, Dunn P, Schaefer H, Pontius J, Berger P, Desborough V et al (2014) ImmPort: disseminating data to the public for the future of immunology. Immunol Res 58(2-3):234–239. https://doi.org/10.1007/s12026-014-8516-1
Zhu W, Tian L, Yue X, Liu J, Fu Y, Yan Y (2019) LncRNA expression profiling of ischemic stroke during the transition from the acute to subacute stage. Front Neurol 10. https://doi.org/10.3389/fneur.2019.00036
Li S, Zheng H, Chen L, Xu C, Qu X, Qin Z, Gao J, Li J et al (2019) Expression profile and potential functions of circulating long noncoding RNAs in acute ischemic stroke in the Southern Chinese Han population. Front Mol Neurosci 12:290. https://doi.org/10.3389/fnmol.2019.00290
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D et al (2012) GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res 22(9):1760–1774. https://doi.org/10.1101/gr.135350.111
Yu G, Wang L-G, Han Y, He Q-Y (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J Integr Biol 16(5):284–287. https://doi.org/10.1089/omi.2011.0118
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT et al (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47(D1):D607–D613. https://doi.org/10.1093/nar/gky1131
Rochet E, Appukuttan B, Ma Y, Ashander LM, Smith JR (2019) Expression of long non-coding RNAs by human retinal Müller glial cells infected with clonal and exotic virulent toxoplasma gondii. Non-Coding RNA 5(4). https://doi.org/10.3390/ncrna5040048
Liu R, Tang A, Wang X, Chen X, Zhao L, Xiao Z, Shen S (2018) Inhibition of lncRNA NEAT1 suppresses the inflammatory response in IBD by modulating the intestinal epithelial barrier and by exosome-mediated polarization of macrophages. Int J Mol Med. https://doi.org/10.3892/ijmm.2018.3829
Zhang Y, Zhu Y, Gao G, Zhou Z (2019) Knockdown XIST alleviates LPS-induced WI-38 cell apoptosis and inflammation injury via targeting miR-370-3p/TLR4 in acute pneumonia. Cell Biochem Funct 37(5):348–358. https://doi.org/10.1002/cbf.3392
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA (2019) Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation 16(1):142. https://doi.org/10.1186/s12974-019-1516-2
Ni X, Su Q, Xia W, Zhang Y, Jia K, Su Z, Li G (2020) Knockdown lncRNA NEAT1 regulates the activation of microglia and reduces AKT signaling and neuronal apoptosis after cerebral ischemic reperfusion. Sci Rep 10(1):19658. https://doi.org/10.1038/s41598-020-71411-1
Wang H-J, Tang X-L, Huang G, Li Y-B, Pan R-H, Zhan J, Wu Y-K, Liang J-F et al (2020) Long non-coding KCNQ1OT1 promotes oxygen-glucose-deprivation/reoxygenation-induced neurons injury through regulating MIR-153-3p/FOXO3 axis. J Stroke Cerebrovasc Dis 29(10):105126. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105126
Kollikowski AM, Schuhmann MK, Nieswandt B, Müllges W, Stoll G, Pham M (2020) Local leukocyte invasion during hyperacute human ischemic stroke. Ann Neurol 87(3):466–479. https://doi.org/10.1002/ana.25665
Guo Y, Chen X, Li D, Liu H, Ding Y, Han R, Shi Y, Ma X (2018) PR-957 mediates neuroprotection by inhibiting Th17 differentiation and modulating cytokine production in a mouse model of ischaemic stroke. Clin Exp Immunol 193(2):194–206. https://doi.org/10.1111/cei.13132
Wang J, Zhao H, Fan Z, Li G, Ma Q, Tao Z, Wang R, Feng J et al (2017) Long noncoding RNA H19 promotes neuroinflammation in ischemic stroke by driving histone deacetylase 1–dependent M1 microglial polarization. Stroke 48(8):2211–2221. https://doi.org/10.1161/strokeaha.117.017387
Wang Y, Luo Y, Yao Y, Ji Y, Feng L, Du F, Zheng X, Tao T et al (2019) Silencing the lncRNA Maclpil in pro-inflammatory macrophages attenuates acute experimental ischemic stroke via LCP1 in mice. J Cereb Blood Flow Metab 40(4):747–759. https://doi.org/10.1177/0271678x19836118
Zhu B, Cheng X, Jiang Y, Cheng M, Chen L, Bao J, Tang X (2020) Silencing of KCNQ1OT1 decreases oxidative stress and pyroptosis of renal tubular epithelial cells. Diabetes Metab Syndr Obes 13:365–375. https://doi.org/10.2147/dmso.S225791
Li P, Duan S, Fu A (2019) Long noncoding RNA NEAT1 correlates with higher disease risk, worse disease condition, decreased miR-124 and miR-125a and predicts poor recurrence-free survival of acute ischemic stroke. J Clin Lab Anal 34(2):e23056. https://doi.org/10.1002/jcla.23056
Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I (2018) Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173(5):1073–1081. https://doi.org/10.1016/j.cell.2018.05.003
Yi M, Li Y, Wang D, Zhang Q, Yang L, Yang C (2020) KCNQ1OT1 exacerbates ischemia–reperfusion injury through targeted inhibition of miR-140-3P. Inflammation 43(5):1832–1845. https://doi.org/10.1007/s10753-020-01257-2
Zhang H, Lu M, Zhang X, Kuai Y, Mei Y, Tan Q, Zhong K, Sun X et al (2019) Isosteviol sodium protects against ischemic stroke by modulating microglia/macrophage polarization via disruption of GAS5/miR-146a-5p sponge. Sci Rep 9(1):12221. https://doi.org/10.1038/s41598-019-48759-0
Li X, Sui Y (2020) Valproate improves middle cerebral artery occlusion-induced ischemic cerebral disorders in mice and oxygen-glucose deprivation-induced injuries in microglia by modulating RMRP/PI3K/Akt axis. Brain Res 1747:147039. https://doi.org/10.1016/j.brainres.2020.147039
Funding
This work was supported by grants from the National Natural Science Foundation of China (nos. 81820108014, 81771361, 82071407, 81801190, and 81901277), National Key Research and Development Project (no. 2018YFE0114400), and the Postdoctoral Foundation of Heilongjiang Province (no. LBH-TZ1019).
Author information
Authors and Affiliations
Contributions
All authors have participated in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. WL and CY are responsible of the part of the design of the work. LL, XS, and WT contributed to the part of acquisition and analysis of data. ZH and LX contributed to the part of interpretation of data. KX and BC are responsible of the creation of new software used in the work. LS, WJ, and NS have been involved in drafting the manuscript and revising it critically. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary table 1
The results of GO function enrichment based on DEIRGs regulated by DElncRNAs in the ISIRC network (XLS 124 kb)
Supplementary table 2
The results of KEGG enrichment analysis based on DEIRGs regulated by DElncRNAs in the ISIRC network (XLS 24 kb)
Supplementary table 3
The detailed interactions among DEIRGs, DEmiRNAs and candidate DElncRNAs (XLS 27 kb)
Supplementary table 4
The DElncRNAs in GSE140275 (XLSX 90 kb)
Supplementary Figure 1
Venn diagram of 17 candidate DElncRNAs and GSE140275; yellow ellipse indicates 17 candidate DElncRNAs, purple ellipse indicates DElncRNAs of GSE140275; overlap intersection indicates common shared DElncRNAs. (PNG 161 kb)
Rights and permissions
About this article

Cite this article
Li, S., Cao, Y., Zhang, H. et al. Construction of lncRNA-Mediated ceRNA Network for Investigating Immune Pathogenesis of Ischemic Stroke. Mol Neurobiol 58, 4758–4769 (2021). https://doi.org/10.1007/s12035-021-02426-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02426-6