
Abstract
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease that leads to progressive cognitive, memory, and learning dysfunction that affects the aging population. Dexmedetomidine (Dex) might be beneficial for postoperative cognitive function in elderly patients. However, the exact mechanism underlying the protective role of Dex against cognitive impairment requires further elucidation. The present study aims to determine whether miR-129 is involved in the protective effect of Dex against Aβ1–42-induced hippocampal neuron apoptosis and cognitive impairment in mice. In our study, Y-shaped maze and water maze tests were conducted to evaluate the cognitive function of AD mice, while neuronal apoptosis was measured by Terminal Deoxynucleotidyl Transferase–Mediated dUTP Nick-End Labeling (TUNEL) staining. The findings showed that Dex administration resulted in the enhancement of miR-129 expression with declined hippocampal neuron apoptosis and attenuated cognitive impairment in Aβ1–42-injected mice. miR-129 targeted YAP1 and disrupted its interaction with JAG1, leading to a decline in hippocampal neuron apoptosis and attenuated cognitive impairment in Aβ1–42-injected mice. In conclusion, the miR-129/YAP1/JAG1 axis could potentially be the mechanism by which Dex protects AD mice from cognitive impairment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most prevalent neurodegenerative diseases that has placed significant burden on the health care system in both developed and developing countries [1]. AD strips people the patients of their independence due to the remarkable cognitive, behavioral, and psychological deficits it results [2]. The deposition of amyloid-beta (Aβ) peptides into amyloid plaques contributes to the development of cognitive dysfunction in AD [3]. Hippocampal atrophy has also been reported to be linked to cognitive malfunction in AD [4]. At present, there is no drug that can slow the progression of AD, much less permanently cure the disease.
Dexmedetomidine (Dex) is an α2-adrenoceptor agonist with high selectivity, which provides sedative, analgesic, and opioid-sparing clinical outcomes [5]. The anti-inflammatory and neuroprotective roles of Dex have been illustrated in previous animal studies [6]. Dex has been reported to prevent early postoperative cognitive decline in aging mice [7]. Dex provides neurocognitive protection for developing brain against isoflurane-induced injury, as observed in neonatal rats [8]. However, limited information is provided concerning the underlying mechanism behind the neurocognitive protection provided by Dex. microRNAs (miRNAs) are one of the important regulators participating in numerous biological processes responsible for growth and aging, which are therefore involved in neurodegenerative diseases such as AD [9]. From previous evidence, miR-129 was found to be implicated in the neuroprotective effect of Dex on hypoxic-ischemic brain injury in neonatal rats [10]. The hippocampal neurons in AD were observed to have a poor expression of miR-129 [11]. It is reasonable to decipher the mechanism responsible for neurocognitive protection of Dex from a perspective of miR-129. A miR-129-mRNA prediction in the StarBase database revealed the putative miR-129 binding sites in the YAP1 mRNA 3’UTR. Yes-associated protein 1 (YAP1), as a transcriptional regulator, promotes the growth of tissue and regeneration, whereas a signaling ligand, Jagged 1 (JAG1), cooperates with its cognate Notch receptor to play a role in neurogenesis and astrogenesis [12, 13]. On the other hand, the ability of miRNA to disrupt the interaction between YAP1 and JAG1 has been previously demonstrated [14, 15]. It has also been reported that the expression of YAP1 and JAG1 is up-regulated in AD patients [16, 17]. However, the combined regulatory role of the Dex/miR-129/YAP1/JAG1 axis in the apoptosis of hippocampal neurons and cognitive function in AD is yet to be thoroughly explored. This study aims to provide theoretical support for treatment of AD by investigating the relationship among Dex, miR-129, YAP1, and JAG1 and the related mechanisms.
Method
Compliance with Ethical Standards
Animal studies were performed with the approval of the institutional animal care and use committee of Linyi People’s Hospital (No. 20027) and in strict accordance with the Guide for the Care and Use of Laboratory animals published by the US National Institutes of Health.
Animal Models
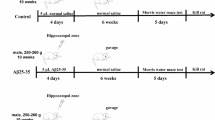
A total of 64 male NIH Swiss mice, aged 9 weeks and weighing 20 to 24 g, were purchased from Guangdong Experimental Animal Center, and 8 of them were used as normal controls, and the remaining 56 mice received treatment with Aβ1–42. In brief, Aβ1–42 dry powder (500 μg) was dissolved in 250 μL of saline containing 1% NH4OH, after which the Aβ1–42 solution was diluted to 2 g/L and incubation was carried out at 37 °C for 1 week to allow aggregation. The mice were weighed and anesthetized with 3% sodium pentobarbital (P3761, Sigma, San Francisco, CA, USA). The brain obtained from mice was fixed with flat skull head by stereotaxic apparatus to expose the bregma. Based on stereotactic atlas of the mouse brain, CAI injection site for bilateral hippocampus was located at 2 mm behind the bregma, 1.5 mm near the linear median, and 1.9 mm below the skull surface. Aβ1–42 was slowly injected with a micro-syringe. After the operation, bone wax was used for sealing purposes and the wound was sutured. Penicillin G8 × 105 U was intramuscularly injected for 3 days to prevent infection. Y-shaped maze and water maze tests were used to evaluate cognitive impairment in Aβ1–42-induced mice. Finally, 48 well-constructed AD mice were obtained, among which 16 AD mice were given injection of miR-129 agomir and agomir NC into the hippocampus area as described previously [10]. Likewise, 24 AD mice were given injection of Dex via tail vein, among which 16 AD mice were injected with miR-129 antagomir and antagomir NC into the hippocampus area, respectively.
Y-Shaped Maze Test
Y-shaped maze was prepared with the use of a black organic medical board. The three arms of the maze (30 × 15 × 8) were placed at an equal angle (120°). There was a movable partition that displayed different geometries as visual markers in the center of each arm. These arms were randomly set as the novel arm, start arm, and other arm. The novel arm was separated by a partition in the first phase of the experiment and opened in the second phase. The start arm was the arm of the mouse input where the maze was located. The start and other arm were kept open throughout the experiment for mice to approach these arms freely. Subsequently, the wood chips were placed in the maze and mixed to prevent residual odors after each training or test. The mice were placed at the end of the start arm and allowed to move freely in the maze for 5 min. The arm entry was regarded as successful when the hind paws of mice were fully placed in the arm. Meanwhile, the camera that was positioned at 1.5 m above the maze recorded the whole process.
Water Maze Test
The pool was set at 120 cm in diameter and 60 cm high, and platform was 12 cm in diameter with temperature of 25 ± 2 °C. All mice were tested on a hidden platform for 5 days and regularly trained 4 times a day. The mice facing the pool wall were sequentially placed from the four designated mark points into the water and the time at which the mice found the platform within 1 min (escape latency) was recorded. If the mice successfully climbed onto the platform, the time was recorded and the mice remained there for 1 min. If the mice failed to reach the platform within 1 min, they were placed on the platform and allowed to stay for 30 s. This was the place navigation test.
On the other hand, the space exploration laboratory was conducted as follows: the mice were placed into the water facing the wall from any point of entry and the time at which the mice entered the original platform quadrant was recorded within 1 min. Moreover, the maze consisted of a circular pool (120 cm in diameter and 50 cm high) with opaque water (22 ± 3 °C). A circular anthocyanin platform (10 cm in diameter and 28 cm high) was put into a quadrant 1 cm below the water surface. These four tests were conducted in mice daily for 5 days, with results observed. The mice also put into the water from the midpoint of the edge of a different quadrant and allow them to swim for a maximum of 90 s to find a hidden platform, staying for 10 s. The mice that failed to find a hidden platform within the specified time were guided and stayed on the platform for 10 s (escape latency: 90 s). On the last day of the space probe test, the platform was removed, and the swimming time and distance in the quadrant along with the platform crossing time where the platform was previously located were recorded for 90 s. The experimental process was recorded and analyzed by a data acquisition system.
Isolation and Culture of Hippocampal Neurons
The entire brain tissue of 1-day-old mice was removed following disinfection with 75% ethanol. The hippocampi were bluntly separated for the removal of the blood vessels and meninges. Then, the hippocampi (0.4 mm diameter) were cultured with 0.25% (g/L) Trypsin and 0.04 (g/L) DNase for 12 min, terminated with horse serum (Solarbio, Beijing, China), and pipetted about 10 times with a tubularis to disperse the cells. The hippocampi underwent incubation with Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (Gibco, Grand Island, New York, USA), 5% horse serum (Solarbio), KCI 25 mmoI/L, HEPES 10 mmoI/L, penicillin 105 U/L, and streptomycin 0.1 g/L, and then were filtrate with a 75-μM nylon mesh. The colatuie was added with a 35-mm petri dish coated with poly-L-lysine coverslips and incubated in an incubator (37 °C, 5% CO2 + 95% O2 mixed gas and saturated humidity). The cell density was 105 cells/L. On the third day, the cells were cultured with 5 μmol/L cytarabine, and the medium was changed to complete culture medium after 24 h. After 7 days, the cultured cells were infected with lentivirus vector harboring miR-129, anti-miR-129, YAP1 expression vector (oe-YAP1), empty vector (oe-NC), two anti-YAP1 shRNA constructs (sh-YAP1-1 and sh-YAP1-2), and scramble shRNA (sh-NC) alone or in combination as required.
RT-qPCR Assay
The total RNA was extracted from hippocampal tissues and cells by Trizol reagent (Invitrogen, New York, California, USA). The complementary DNA (cDNA) was generated according to the instructions provided on the kit (K1622; Fermentas Inc., Ontario, CA, USA). miR-129 was quantified using TaqMan MicroRNA Assay and TaqMan® Universal PCR Master Mix, and results were normalized to U6 expression. Other mRNAs were quantified according to TaqMan Gene Expression Assays protocol (Applied Biosystems, Foster City, CA, USA), and results were normalized to GAPDH expression. The relative quantification method (2−△△CT method) was employed to calculate the relative transcription level: △△Ct = △Ct experimental group-△Ct control group, △Ct = Ct (target gene)-Ct (internal reference), and the relative transcription level of the target gene mRNA = 2−△△CT. Table 1 listed primer information.
Western Blot Analysis
The total protein of hippocampus tissues and cells was extracted using RIPA lysis buffer containing Phenylmethanesulfonyl fluoride (PMSF), and incubation was carried out on ice for 30 min, which was followed by centrifugation at 8000 rpm for 10 min to obtain the supernatant. The concentration of total protein was measured by a bicinchoninic acid (BCA) kit. A total of 50 μg protein was dissolved in 2 × sodium dodecyl sulfate (SDS) loading buffer and boiled at 100 °C for 5 min. Subsequently, the above samples were subjected to SDS-polyacrylamide gel electrophoresis (PAGE), and the protein was transferred to polyvinylidene fluoride (PVDF) membrane through the wet transfer method. The protein was blocked with 5% skim milk powder for 1 h at room temperature and incubated at 4 °C overnight with diluted primary antibody to YAP1 (ab56701), JAG1 (ab7771), cleaves-Caspase3 (ab13847), cleaves-Caspase8 (ab25901), Bax (ab32503), and GAPDH (ab9485) (Abcam, Cambridge, UK). Subsequently, the protein membrane was washed 3 times with Tween 20 (TBST) for 10 min each and incubated with horseradish peroxidase (HRP)-labeled secondary antibody goat anti-rabbit immunoglobulin G (IgG) H&L (HRP) (ab97051, Abcam) for 1 h. A and B liquids from ECL Fluorescence Detection Kit (BB-3501, Amersham, UK) were mixed in a dark room and added on the membrane to exposure imaging under a gel imager. Bio-Rad image analysis system (BIO-RAD, Hercules, Cal, USA) was used for photographing, and Quantity One v4.6.2 software was for analysis. The relative protein content was regarded as the ratio of the gray value of the corresponding protein band to the gray value of GAPDH protein band.
In Situ Hybridization
A total of phosphate-buffered solution (PBS)–resuspended cell droplets (2–3) were placed on a glass slide coated with polylysine and dried in an incubator for 5 min. Cells were added with 1 μg/mL of proteinase K dilution, permeated at 37 °C for 30 min, and reaction was terminated with the addition of 0.1 mol/L glycine solution. The cells were cultured with 20 μL of pre-hybridization solution, and covered with siliconized cover slip and incubated in a wet box at 37 °C for 30 min. In addition, the cells were washed with 0.2 × saline sodium citrate (SSC) buffer three times at room temperature for 5 min each time, after which the cover slip was removed and the cells were also washed with 2 × SSC buffer that was preheated at 42 °C for 5 min each time as well as 0.5 × SSC buffer preheated at 37 °C once and 0.2 × SSC buffer for 15 min once respectively. Subsequently, the cells were successively incubated with blocking solution and rabbit anti-digoxigenin IgG at 37 °C for 30 min. The cells were cultured with HRP-goat anti-rabbit IgG and incubated in a wet box for 30 min. Next, the cells were developed with fresh 3,3′-diaminobenzidine (DAB) working solution for 20 min and terminated with distilled water. The results of positive signal of miR-129 (its base sequence: 5′-AAGTGACTGAAACGTAGCCT-3 probe) was observed under a microscope.
HE Staining
The experimental mice were anesthetized with 3% sodium pentobarbital, with the heart obtained from the mice, after which it was perfused with normal saline and 4% paraformaldehyde. The extracted hippocampal brain tissues were dehydrated with ethanol in a way of conventional gradient, trans-parented with xylene, impregnated with wax, and embedded with paraffin. Then the sections were stained with hematoxylin, washed with water, separated with hydrochloric acid, and immersed in tap water after the cells were dewaxed with xylene and dehydrated with graded ethanol (100–70%). In addition, the cells were stained with eosin, dehydrated with graded ethanol (70–100%), cleared with xylene, and finally sealed. The changes undergone by the hippocampal neurons were observed under a light microscope.
TUNEL Staining
The fluorescently labeled Terminal Deoxynucleotidyl Transferase–Mediated dUTP Nick-End Labeling (TUNEL) method was applied. According to the apoptosis kit, cells were stained, developed with DAB, and mounted. The cells and apoptosis percentage were observed in each section in 20 randomly selected high-power views, and the apoptotic index (AI) was AI (positive cells/total cells) × 100%.
Biochemical Studies
Extensive oxidative stress is well characterized in AD brains. Malondialdehyde (MDA) accumulation, reactive oxygen species (ROS) generation, and reductions in superoxide dismutase (SOD) and glutathione peroxidase (GSH-PX) activity are reflective of oxidative stress. Moreover, lactate dehydrogenase (LDH) leakage can reflect the neuron viability. As previously reported [18, 19], serum levels of MDA, ROS, SOD, GSH-PX, and LDH were measured. Briefly, the reaction mixture of total 1 mL consisted of 0.6 mL of PBS (0.5 M, pH 7.4), 0.1 mL PMS (10% w/v), 0.1 mL xanthine (1 mM), and 0.1 mL nitroblue tetrazolium (NBT, 57 mM) was incubated for 15 min at room temperature and reaction was initiated by the addition of xanthine oxidase (50 mU). The rate of reaction was measured by recording change in the absorbance at 550 nm.
Dual-Luciferase Reporter Gene Assay
The dual-luciferase reporter gene vector of YAP1 3′UTR and its mutant plasmids with mutation sites binding to miR-129 were Pmir YAP1-Grhl2-WT and Pmir YAP1-Grhl2-MUT. The reporter plasmid (miR-129 mimic plasmid) and its negative plasmid were respectively transfected to HEK293T cells, and after transfection 24 h the cells were lysed and centrifuged at 12,000 rpm for 1 min to collect the supernatant. The Dual-Luciferase Reporter Assay System (E1910, Promega, Madison, WI, USA).) was utilized to detect the luciferase activity. Each cell sample was added with 100 μL working solution of firefly luciferase to detect firely lueiferase. In addition, the samples were added with 100 μL working solution of renilla luciferase to detect renilla lueiferase with enzyme activity of renilla luciferase as an internal reference. The ratio of firefly luciferase activity and Renilla luciferase activity was considered as the relative luciferase activity.
Statistical Analysis
All data, representative of three independent experiments in triplicate, are analyzed by SPSS 21.0 software (IBM, Armonk, NY, USA), with p < 0.05 as a level of statistical significance. Measurement data were analyzed by normal distribution and homogeneity of variance and presented as the mean ± standard deviation. Unpaired designs between two groups were compared by unpaired t test, while differences among groups were determined by one-way analysis of variance (ANOVA), and Tukey’s post hoc test.
Results
miR-129 Is Poorly Expressed in AD
RT-qPCR detection revealed a decreased expression of miR-129 in serum of AD patients (Fig. 1a). Furthermore, the AD model was established and used Morris water maze and Y-shaped maze tests to estimate the cognitive impairment of AD mice. The escape latency of AD mice increased significantly, and the mice spent decreased times and less time on crossing the platform with shorter distance of swimming in the target quadrant (Fig. 1b–e). In addition, a consistent conclusion was found in the Y-shaped maze test. but there was poorer alternation behavior and shorter time in the novel arm (Fig. 1f, g). These findings were indicative of the successful establishment of AD mice. The positive signal of hybridization in situ was mainly located in the nucleus of the neurons, and miR-129 expression was decreased in the hippocampal neurons of AD mice by RT-qPCR and in situ hybridization compared with normal mice (Fig. 1h, i).

miR-129 is poorly expressed in AD. a miR-129 expression in serum of AD patients determined by RT-qPCR. The escape latency (b), the number of crossing platforms (c), the target quadrant time (d), and distance (e) of AD mice assessed by water maze test. The alternative behavior (f) and the dwell time (g) of novel arm of AD mice measured by Y-shaped maze test. miR-129 expression in hippocampal tissues of AD mice detected by RT-qPCR (h) and in hippocampal tissues hybridization in situ (i). * p < 0.05 vs. control mice by unpaired t test. Results are expressed as mean ± standard deviation of three technical replicates (n = 8)
miR-129 Protects AD Mice Against Cognitive Impairment
The effect of miR-129 on the cognitive function in AD mice was investigated, for the purpose of which the mice were injected with miR-129 agomir into their tail vein, after which the behavioral changes and miR-129 expression were detected. RT-qPCR detection demonstrated that the expression of miR-129 was elevated in hippocampal neurons of AD mice treated with miR-129 agomir compared with the neurons of AD mice treated with agomir NC (Fig. 2a). By performing water maze test, a decreased escape latency (Fig. 2b), as fewer times were recorded of the mice crossing platform (Fig. 2c), the mice spending longer time crossing platform (Fig. 2d), and longer distance of swimming (Fig. 2e) in the target quadrant, which were all observed in AD mice injected with miR-129 agomir compared with AD mice injected with agomir NC. Likewise, Y-shaped maze test revealed that AD mice injected with miR-129 agomir presented with improved alternation behavior (Fig. 2f) and shorter time (Fig. 2g) in the novel arm. Altogether, miR-129 plays a positive role in improving the cognitive function of AD mice.

miR-129 protects AD mice against cognitive impairment. a miR-129 expression in hippocampal tissues of AD mice following miR-129 agomir injection determined by RT-qPCR. The escape latency (b), the number of crossing platforms (c), the target quadrant time (d), and distance (e) of AD mice following miR-129 agomir injection assessed by water maze test. The alternative behavior (f) and the dwell time (g) of novel arm of AD mice following miR-129 agomir injection measured by Y-shaped maze test. * p < 0.05 vs. control mice and # p < 0.05 vs. AD mice injected with agomir NC by one-way ANOVA. Results are expressed as mean ± standard deviation of three technical replicates (n = 8)
Dex Protects AD Mice Against Cognitive Impairment by Regulating miR-129 Expression
Dex is reportedly capable of attenuating cognitive dysfunction [7] by partially regulating miR-129 [10]. The next step of our study was to assert whether Dex confers its neuroprotection by regulating miR-129 in AD mice. For this purpose, AD mice were injected with Dex via tail vein followed by injection of miR-129 antagomir and antagomir NC into the hippocampus area. The results of water maze test showed that AD mice had decreased escape latency and spent more time crossing the platforms, with diminished time as well longer swimming distance in the target quadrant in water maze test after Dex injection. Moreover, AD mice showed better alternating behaviors and shorter time in the novel arm in Y-shaped maze test after Dex injection. The subsequent hippocampal injection with miR-129 antagomir resulted in the negation of the effects of Dex on the performance of AD mice in water maze and Y-shaped maze tests (Fig. 3a–f). Next, serum contents of MDA, ROS, SOD, GSH-PX, and LDH were determined to evaluate AD-like pathology in mice. The results identified an increase in the contents of LDH, ROS, and MDA in AD mice, while the contents of SOD and GSH-PX were decreased when comparable to the normal mice. However, decreased contents of LDH, ROS, MDA, elevated contents of SOD, and GSH-PX were showed in AD mice injected with Dex with or without miR-129 antagomir compared with AD mice. As expected, miR-129 antagomir negated the effects of Dex on serum contents of MDA, ROS, SOD, GSH-PX, and LDH in AD mice (Fig. 3g). RT-qPCR results demonstrated that AD mice presented with decreased expression of miR-129 compared with the normal mice. Following Dex injection, AD mice exhibited an elevated miR-129 expression, whereas subsequent hippocampal injection with miR-129 antagomir likewise abrogated the effects of Dex on the expression of miR-129 in AD mice (Fig. 3h). In conclusion, miR-129 inhibition negated the protective effects of Dex against cognitive impairment and oxidative stress, which suggested that Dex has the potential to improve the cognitive function in AD mice and protects against hippocampal neurons in AD mice by regulating miR-129.

Dex protects AD mice against cognitive impairment by regulating miR-129 expression. A-D, The escape latency (a), the times of crossing platform (b), the target quadrant time (c), and swimming distance in the target quadrant (d) of AD mice injected with Dex with or without miR-129 antagomir evaluated by water maze test. The alternate behavior (e) and the dwell time (f) of novel arm of AD mice injected with Dex with or without miR-129 antagomir measured by Y-shaped maze test. g Serum contents of MDA, ROS, SOD, GSH-PX, and LDH in AD mice injected with Dex with or without miR-129 antagomir. h miR-129 expression in AD mice injected with Dex with or without miR-129 antagomir measured by RT-qPCR. * p < 0.05 vs. control mice and # p < 0.05 vs. AD mice by one-way ANOVA. Results are expressed as mean ± standard deviation of three technical replicates (n = 8)
miR-129 Targets YAP1 and Disturbs its Interaction with JAG1
The mechanism underlying protection conferred by miR-129 against cognitive dysfunction in AD mice was investigated. miR-129-mRNA prediction in the StarBase database revealed putative miR-129 binding sites in the YAP1 mRNA 3′UTR (Fig. 4a). Previous evidence showed the contribution of YAP1 [16] and JAG1 [17] to the development of AD. The activity of JAG1 was reported to be dependent of YAP1 [15]. To further confirm the relationship between miR-129 and YAP1, we performed dual-luciferase reporter gene assays and observed declined luciferase intensity of reporter vector containing YAP1-Grhl2-WT in the presence of miR-129 mimic when compared with reporter vector containing YAP1-Grhl2-MUT (Fig. 4b). Likewise, we performed RT-qPCR and Western blot analysis and determined a decline in YAP1 expression after infection with lentiviral vector harboring miR-129 and an increase in YAP1 expression after infection with lentiviral anti-miR-129 in hippocampal neurons (Fig. 4c, d). To demonstrate whether YAP1 regulated the expression of JAG1, YAP1 was over-expressed or knocked down in cultured hippocampal neurons by lentiviral transduction of oe-YAP1 or sh-YAP1 (Fig. 4e). The results of RT-qPCR and Western blot analysis showed that oe-YAP1 declined the JAG1 expression in hippocampal neurons, whereas sh-YAP1 contributed to an elevated expression of JAG1 (Fig. 4f, g). The above results showed that YAP1 regulated the expression of JAG1.

miR-129 targets YAP1 and disturbs its interaction with JAG1. a Putative miR-129 binding sites in the YAP1 mRNA 3’UTR. b Luciferase intensity of reporter vector containing YAP1-Grhl2-WT in the presence of miR-129 mimic when comparable to reporter vector containing YAP1-Grhl2-MUT. The expression of YAP1 determined by RT-qPCR (c) and Western blot analysis (d) in response to lentiviral transduction of miR-129 mimic or inhibitor, * p < 0.05 vs. NC mimic and # p < 0.05 vs. NC inhibitor by unpaired t test. e Verification of YAP1 over-expression and knockdown by RT-qPCR. The expression of JAG1 determined by RT-qPCR (f) and Western blot analysis (g) in response to lentiviral transduction of oe-YAP1 or sh-YAP1, * p < 0.05 vs. sh-NC and # p < 0.05 vs. oe-NC by unpaired t test. h The expression of miR-129, YAP1, and JAG1 measured by RT-qPCR in response to lentiviral transduction of miR-129 mimic, miR-129 inhibitor, and miR-129 inhibitor plus sh-YAP1, * p < 0.05 vs. NC mimic, # p < 0.05 vs. NC inhibitor, and $ p < 0.05 vs. miR-129 inhibitor + sh-NC by one-way ANOVA. Results are expressed as mean ± standard deviation of three technical replicates (n = 8)
To verify the regulation of miR-129 on the expression of YAP1 and JAG1, the hippocampal neurons were infected with lentiviral vector harboring miR-129, anti-miR-129 alone, or in combination with sh-YAP1. As shown in Fig. 4h, RT-qPCR and western blot analysis determined a decline in YAP1 and JAG1 expression secondary to infection with lentiviral vector harboring miR-129 and increases in YAP1 and JAG1 expression after infection with lentiviral anti-miR-129 in hippocampal neurons. Moreover, loss of function of miR-129 inhibitor was observed following lentiviral transduction of sh-YAP1, as evidenced by the decline in the expression of YAP1 and JAG1 in hippocampal neurons with lentiviral anti-miR-129 in combination with sh-YAP1. Altogether, miR-129 inhibited YAP1-meditaed promotion of JAG1 by targeting YAP1.
Dex Prevents Hippocampal Neurons Against Apoptosis Through the miR-129/YAP1/JAG1 Axis
RT-qPCR results demonstrated that the expression of YAP1 or JAG1 expression was increased in AD mice compared with the normal mice. Following Dex injection, AD mice exhibited declined YAP1 and JAG1 expression, whereas subsequent hippocampal injection with miR-129 antagomir likewise abrogated the effects of Dex on the expression of YAP1 and JAG1 in AD mice (Fig. 5a). Subsequently, the effects of Dex and miR-129 on hippocampal neuron apoptosis in AD were determined. For this purpose, apoptosis-related proteins such as cleaved Caspase 3, cleaved Caspase 8, and Bax underwent western blot analysis. We found that cleaved Caspase 3, cleaved Caspase 8, and Bax were lower in hippocampal neurons of AD mice compared with those of normal mice, but higher in hippocampal neurons of AD mice injected with Dex or Dex and miR-129 antagomir when compared with AD mice (Fig. 5b). Notably, the contribution of Dex to expression of Caspase 3, cleaved Caspase 8, and Bax was partially abrogated after subsequent miR-129 antagomir injection. Additionally, TUNEL staining results found that Dex injection attenuated hippocampal neuron apoptosis in AD mice, whereas function of Dex was lost partially after subsequent miR-129 antagomir injection (Fig. 5c). Coherently, Dex increased the expression of miR-129, which then led to a decrease in YAP1 and JAG1 expression, inhibiting the neuronal apoptosis.

Dex prevents hippocampal neurons against apoptosis through the miR-129/YAP1/JAG1 axis. a miR-129, YAP1, and JAG1 expression in hippocampal tissues of AD mice injected with Dex with or without miR-129 antagomir was measured by RT-qPCR. b YAP1, JAG1, Caspase 3, Caspase 8, and Bax protein expression in hippocampal tissues of AD mice injected with Dex with or without miR-129 antagomir was determined by western blot analysis. c Neuron apoptosis of hippocampal tissues of AD mice injected with Dex with or without miR-129 antagomir was detected by Tunel staining. The comparison of the data among groups was analyzed by ANOVA. * p < 0.05 vs. control mice and # p < 0.05 vs. AD mice by one-way ANOVA. Results are expressed as mean ± standard deviation of three technical replicates (n = 8)
Discussion
AD affects 10 % of the people over the age of 65 years old and is the most prevalent neurodegenerative disorder [20]. AD is characterized by the induction of oxidative stress and accumulation of amyloid beta (Aβ) resulting in neuronal damage and is closely linked to aging [21, 22]. In addition, the amyloidosis that was caused by Aβ aggregates has been proposed to be associated with the loss of neurons and decrease of cognitive function [23]. The present study emphasizes on the regulatory role of Dex in the treatment of AD, and we found evidence to supporting the hypothesis that Dex up-regulates the expression of miR-129 to suppress the apoptosis of hippocampal neurons via the inhibition of the expression of YAP1 and JAG1, improving the cognitive function in AD mice.
Initially, we demonstrated that miR-129 was down-regulated in AD patients, and exogenous miR-129 was beneficial to cognitive function of AD mice. Previous research identified that miR-129 is down-regulated in hippocampal neurons in epilepsy mice, as it suppressed cell apoptosis while promoting hippocampal neuron proliferation [24]. miR-196a depletion is shown in the hippocampus tissues of AD mice, while its recovery induces loss of LRIG3, therefore improving the cognitive function such as the capability of memory and inhibit the apoptosis of hippocampal neurons in AD mice [25]. Moreover, miR-34a could regulate NMDA (by miR-34a-5p) and AMPA (by miR-34a-3p) receptors to repressing the early AD pathology symptoms [26].
Subsequently, the protective effects of Dex against cognitive impairment and oxidative stress were reversed following the inhibition of miR-129 inhibition, which suggested that Dex has the potential to improve the cognitive function in AD mice and protects against hippocampal neurons in AD mice by regulating miR-129. Dex is capable of suppressing cognitive decline in AD patients after operation, through the inhibition of inflammation in microglia cells by c-Fos, NLRP3 and caspase-1 [27]. Dex inhibits astrocyte proptosis, thus providing protection to the glia cells [28]. Notably, we found that Dex promoted the expression of miR-129 to improve the cognitive impairment of AD mice. Several miRNAs were found to be involved in neuropathological changes seen in AD [29]. A study has shown that Dex promotes expression of miR-223-3p and down-regulates TIAL1 to protect the neurons [30]. Moreover, Dex could up-regulate the expression of miR-520-3p in osteosarcoma cell [31]. Dex also regulates the expression of miR-let-7b and COL3A1 protecting PC12 cells from the cytotoxicity with lidocaine, along with the neurons [32].
Furthermore, dual-luciferase reporter gene assay identified YAP1 as the target gene of miR-129. YAP1 was previously found to be highly expressed in the neurons of cerebral cortex following a traumatic brain injury [33]. There exists numerous evidence elaborating the interaction and function of miRNA and YAP1 in AD. For example, miR-622 up-regulation results in the inhibition of YAP1, which is the target point of miR-622 in glioma cells [34]. In addition, miR-186 is poorly expressed in hepatocellular carcinoma, but its increment could lead to a decrease in YAP1 expression in hepatocellular carcinoma cells [35]. However, up-regulation of YAP1 elevates the expression JAG1 to activate the signaling pathway of Notch in U251 cells [15]. High JAG1 expression in endothelium and smooth muscle cells suggests that the activation of Notch signal in the inflammatory cells could be the underlying mechanism in Buerger’s disease [36]. However, the activation of Notch induced by JAG1 suppresses the cell apoptosis while promoting the cell proliferation in various cancer [37]. miR-598 has been previously identified as a novel regulator in colorectal cancer metastasis by suppressing the expression of JAG1 and Notch signaling pathway [38].
In conclusion, the key findings from this study suggested that Dex suppressed the cell apoptosis of hippocampal neuron by promoting the expression of miR-129 and inhibiting YAP1/JAG1 axis, thus improving cognitive function in AD mice (Fig. 6). These findings demonstrated that miR-129 may serve as a promising diagnostic biomarker for AD. At present, the regulatory mechanism of the Dex/miR-129/YAP1/JAG1 axis remains scantly explored in the field of AD treatment, making it necessary to conduct additional studies to further elucidate the underlying mechanisms governing Dex and signaling pathway.

Graphic abstract depicting the exact mechanism behind protection of Dex against cognitive impairment in AD mice. Dex protects AD mice against hippocampal neuron apoptosis and cognitive impairment by promoting the expression of miR-129 and inhibiting YAP1/JAG1 axis
References
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E (2011) Alzheimer's disease. Lancet 377(9770):1019–1031. https://doi.org/10.1016/S0140-6736(10)61349-9
Kosel F, Pelley JMS, Franklin TB (2020) Behavioural and psychological symptoms of dementia in mouse models of Alzheimer's disease-related pathology. Neurosci Biobehav Rev 112:634–647. https://doi.org/10.1016/j.neubiorev.2020.02.012
DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM (2002) Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science 295(5563):2264–2267. https://doi.org/10.1126/science.1067568
Mouiha A, Duchesne S, Alzheimer's Disease Neuroimaging I (2011) Hippocampal atrophy rates in Alzheimer's disease: automated segmentation variability analysis. Neurosci Lett 495(1):6–10. https://doi.org/10.1016/j.neulet.2011.02.065
Keating GM (2015) Dexmedetomidine: a review of its use for sedation in the intensive care setting. Drugs 75(10):1119–1130. https://doi.org/10.1007/s40265-015-0419-5
Gao J, Sun Z, Xiao Z, Du Q, Niu X, Wang G, Chang YW, Sun Y et al (2019) Dexmedetomidine modulates neuroinflammation and improves outcome via alpha2-adrenergic receptor signaling after rat spinal cord injury. Br J Anaesth 123(6):827–838. https://doi.org/10.1016/j.bja.2019.08.026
Qian XL, Zhang W, Liu MZ, Zhou YB, Zhang JM, Han L, Peng YM, Jiang JH et al (2015) Dexmedetomidine improves early postoperative cognitive dysfunction in aged mice. Eur J Pharmacol 746:206–212. https://doi.org/10.1016/j.ejphar.2014.11.017
Sanders RD, Xu J, Shu Y, Januszewski A, Halder S, Fidalgo A, Sun P, Hossain M et al (2009) Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology 110(5):1077–1085. https://doi.org/10.1097/ALN.0b013e31819daedd
Madadi S, Schwarzenbach H, Saidijam M, Mahjub R, Soleimani M (2019) Potential microRNA-related targets in clearance pathways of amyloid-beta: novel therapeutic approach for the treatment of Alzheimer's disease. Cell Biosci 9:91. https://doi.org/10.1186/s13578-019-0354-3
Zhou XM, Liu J, Wang Y, Zhang SL, Zhao X, Xu X, Pei J, Zhang MH (2018) microRNA-129-5p involved in the neuroprotective effect of dexmedetomidine on hypoxic-ischemic brain injury by targeting COL3A1 through the Wnt/beta-catenin signaling pathway in neonatal rats. J Cell Biochem 120:6908–6919. https://doi.org/10.1002/jcb.26704
Lau P, Bossers K, Janky R, Salta E, Frigerio CS, Barbash S, Rothman R, Sierksma AS et al (2013) Alteration of the microRNA network during the progression of Alzheimer's disease. EMBO Mol Med 5(10):1613–1634. https://doi.org/10.1002/emmm.201201974
Gao Y, Yang Y, Yuan F, Huang J, Xu W, Mao B, Yuan Z, Bi W (2017) TNFalpha-YAP/p65-HK2 axis mediates breast cancer cell migration. Oncogenesis 6(9):e383
Hu X, He W, Luo X, Tsubota KE, Yan R (2013) BACE1 regulates hippocampal astrogenesis via the Jagged1-Notch pathway. Cell Rep 4(1):40–49
Ren K, Li T, Zhang W, Ren J, Li Z, Wu G (2016) miR-199a-3p inhibits cell proliferation and induces apoptosis by targeting YAP1, suppressing Jagged1-Notch signaling in human hepatocellular carcinoma. J Biomed Sci 23(1):79. https://doi.org/10.1186/s12929-016-0295-7
Hao B, Chen X, Cao Y (2018) Yes-associated protein 1 promotes the metastasis of U251 glioma cells by upregulating Jagged-1 expression and activating the Notch signal pathway. Exp Ther Med 16(2):1411–1416. https://doi.org/10.3892/etm.2018.6322
Xu M, Zhang DF, Luo R, Wu Y, Zhou H, Kong LL, Bi R, Yao YG (2018) A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer's disease. Alzheimers Dement 14(2):215–229. https://doi.org/10.1016/j.jalz.2017.08.012
Guo HD, Tian JX, Zhu J, Li L, Sun K, Shao SJ, Cui GH (2015) Electroacupuncture suppressed neuronal apoptosis and improved cognitive impairment in the AD model rats possibly via downregulation of notch signaling pathway. Evid Based Complement Alternat Med 2015:393569–393569. https://doi.org/10.1155/2015/393569
Ahmad N, Umar S, Ashafaq M, Akhtar M, Iqbal Z, Samim M, Ahmad FJ (2013) A comparative study of PNIPAM nanoparticles of curcumin, demethoxycurcumin, and bisdemethoxycurcumin and their effects on oxidative stress markers in experimental stroke. Protoplasma 250(6):1327–1338. https://doi.org/10.1007/s00709-013-0516-9
Ahmad N, Ahmad R, Ahmad FJ, Ahmad W, Alam MA, Amir M, Ali A (2020) Poloxamer-chitosan-based Naringenin nanoformulation used in brain targeting for the treatment of cerebral ischemia. Saudi J Biol Sci 27(1):500–517. https://doi.org/10.1016/j.sjbs.2019.11.008
Kim K, Kim MJ, Kim DW, Kim SY, Park S, Park CB (2020) Clinically accurate diagnosis of Alzheimer's disease via multiplexed sensing of core biomarkers in human plasma. Nat Commun 11(1):119. https://doi.org/10.1038/s41467-019-13901-z
Ren C, Li D, Zhou Q, Hu X (2020) Mitochondria-targeted TPP-MoS2 with dual enzyme activity provides efficient neuroprotection through M1/M2 microglial polarization in an Alzheimer's disease model. Biomaterials 232:119752. https://doi.org/10.1016/j.biomaterials.2019.119752
Ren HX, Miao YB, Zhang Y (2020) An aptamer based fluorometric assay for amyloid-beta oligomers using a metal-organic framework of type Ru@MIL-101(Al) and enzyme-assisted recycling. Microchim Acta 187(2):114. https://doi.org/10.1007/s00604-019-4092-3
Ricke KM, Cruz SA, Qin Z, Farrokhi K, Sharmin F, Zhang L, Zasloff MA, Stewart AFR et al (2020) Neuronal protein tyrosine phosphatase 1B hastens amyloid beta-associated Alzheimer's disease in mice. J Neurosci 40(7):1581–1593. https://doi.org/10.1523/JNEUROSCI.2120-19.2019
Wu DM, Zhang YT, Lu J, Zheng YL (2018) Effects of microRNA-129 and its target gene c-Fos on proliferation and apoptosis of hippocampal neurons in rats with epilepsy via the MAPK signaling pathway. J Cell Physiol 233(9):6632–6643. https://doi.org/10.1002/jcp.26297
Yang K, Feng S, Ren J, Zhou W (2019) Upregulation of microRNA-196a improves cognitive impairment and alleviates neuronal damage in hippocampus tissues of Alzheimer's disease through downregulating LRIG3 expression. J Cell Biochem 120(10):17811–17821. https://doi.org/10.1002/jcb.29047
Xu Y, Chen P, Wang X, Yao J, Zhuang S (2018) miR-34a deficiency in APP/PS1 mice promotes cognitive function by increasing synaptic plasticity via AMPA and NMDA receptors. Neurosci Lett 670:94–104. https://doi.org/10.1016/j.neulet.2018.01.045
Li H, Zhang X, Chen M, Chen J, Gao T, Yao S (2018) Dexmedetomidine inhibits inflammation in microglia cells under stimulation of LPS and ATP by c-Fos/NLRP3/caspase-1 cascades. EXCLI J 17:302–311. https://doi.org/10.17179/excli2017-1018
Sun YB, Zhao H, Mu DL, Zhang W, Cui J, Wu L, Alam A, Wang DX et al (2019) Dexmedetomidine inhibits astrocyte pyroptosis and subsequently protects the brain in in vitro and in vivo models of sepsis. Cell Death Dis 10(3):167. https://doi.org/10.1038/s41419-019-1416-5
Miya Shaik M, Tamargo IA, Abubakar MB, Kamal MA, Greig NH, Gan SH (2018) The role of microRNAs in Alzheimer's disease and their therapeutic potentials. Genes (Basel) 9(4). https://doi.org/10.3390/genes9040174
Wang Q, Yu H, Yu H, Ma M, Ma Y, Li R (2019) miR2233p/TIAL1 interaction is involved in the mechanisms associated with the neuroprotective effects of dexmedetomidine on hippocampal neuronal cells in vitro. Mol Med Rep 19(2):805–812. https://doi.org/10.3892/mmr.2018.9742
Wang X, Xu Y, Chen X, Xiao J (2018) Dexmedetomidine inhibits osteosarcoma cell proliferation and migration, and promotes apoptosis by regulating miR-520a-3p. Oncol Res 26(3):495–502. https://doi.org/10.3727/096504017X14982578608217
Wang Q, She Y, Bi X, Zhao B, Ruan X, Tan Y (2017) Dexmedetomidine protects PC12 cells from lidocaine-induced cytotoxicity through downregulation of COL3A1 mediated by miR-let-7b. DNA Cell Biol 36(7):518–528. https://doi.org/10.1089/dna.2016.3623
Li D, Ji JX, Xu YT, Ni HB, Rui Q, Liu HX, Jiang F, Gao R et al (2018) Inhibition of Lats1/p-YAP1 pathway mitigates neuronal apoptosis and neurological deficits in a rat model of traumatic brain injury. CNS Neurosci Ther 24(10):906–916. https://doi.org/10.1111/cns.12833
Xu J, Ma B, Chen G, Wei D, Li L, Hu W (2018) MicroRNA-622 suppresses the proliferation of glioma cells by targeting YAP1. J Cell Biochem 119(3):2492–2500. https://doi.org/10.1002/jcb.26343
Ruan T, He X, Yu J, Hang Z (2016) MicroRNA-186 targets yes-associated protein 1 to inhibit Hippo signaling and tumorigenesis in hepatocellular carcinoma. Oncol Lett 11(4):2941–2945. https://doi.org/10.3892/ol.2016.4312
Tamai H, Kobayashi M, Takeshita K, Kodama A, Banno H, Narita H, Yamamoto K, Komori K (2014) Possible involvement of Notch signaling in the pathogenesis of Buerger's disease. Surg Today 44(2):307–313. https://doi.org/10.1007/s00595-013-0566-9
Li D, Masiero M, Banham AH, Harris AL (2014) The notch ligand JAGGED1 as a target for anti-tumor therapy. Front Oncol 4:254. https://doi.org/10.3389/fonc.2014.00254
Chen J, Zhang H, Chen Y, Qiao G, Jiang W, Ni P, Liu X, Ma L (2017) miR-598 inhibits metastasis in colorectal cancer by suppressing JAG1/Notch2 pathway stimulating EMT. Exp Cell Res 352(1):104–112. https://doi.org/10.1016/j.yexcr.2017.01.022
Acknowledgments
We acknowledge and appreciate our colleagues for their valuable efforts and comments on this paper.
Author information
Authors and Affiliations
Contributions
Chunzhi Li designed the study. Jun Zhao collated the data, carried out data analyses, and produced the initial draft of the manuscript. Chunzhi Li and Weiying Sun contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sun, W., Zhao, J. & Li, C. Dexmedetomidine Provides Protection Against Hippocampal Neuron Apoptosis and Cognitive Impairment in Mice with Alzheimer’s Disease by Mediating the miR-129/YAP1/JAG1 Axis. Mol Neurobiol 57, 5044–5055 (2020). https://doi.org/10.1007/s12035-020-02069-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02069-z