
Abstract
Unilateral photothrombotic stroke caused tissue infarct in the mouse cerebral cortex. The injury of the cerebral cortex impaired the mouse motor activity, in particular the functional asymmetry in forelimb use. In the peri-infarct cortical tissue outside the infarct core cell apoptosis occurred at 4 and 7 days after PTS. The downregulation of acetylated α-tubulin, a marker of stable microtubules, showed the destruction of neurites, axons, and dendrites in injured neurons. However, the upregulation of GAP43 indicates the stimulation of neurite growth that was possibly aimed at the recovery of the cortical tissue in the damaged cerebral hemisphere. Application of MI-192, an inhibitor of histone deacetylases HDAC2 and HDAC3, demonstrated the neuroprotective activity in the mouse brain subjected to photothrombotic stroke. It reduced the volume of the PTS-induced infarction core in the mouse brain, partly restored the functional symmetry in the forelimb use, decreased the level of PTS-induced apoptosis and acetylation of α-tubulin characteristic for stable microtubules, and increased the expression of GAP-43 in the cerebral cortex of the damaged hemisphere. These data point to the involvement of HDAC2 and HDAC3 in the photothrombotic injury of the mouse brain not only in the infarction core but also outside it. The application of MI192 after PTS reduced or eliminated these negative effects and exerted the neuroprotective effect on the mouse brain. It may be a promising neuroprotector agent for anti-stroke therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the case of ischemic stroke, vessel occlusion rapidly, for some minutes, interrupts oxygen and glucose delivery, and causes mitochondrial failure, ATP depletion, generation of reactive oxygen species (ROS), lesion of cell membranes, drop of ionic gradients, depolarization, release of glutamate and K+, influx of Na+, Ca2+, and water, edema, and finally necrosis of neurons and glial cells. Glutamate and K+-mediated depolarization activate NMDA receptors in the neighboring cells that elicit massive Ca2+ influx that initiates excitotoxic death of the neighboring cells. ROS and nitric oxide diffuse and cause oxidative and nitrosyl stress in the adjacent cells. These pathogenic processes spread from the infarction core to the surrounding cells. As a result, the necrotic core is surrounded by the transitional zone (penumbra), in which the secondary cell damage, mainly apoptosis, occurs. Although rapid necrosis of neurons and glial cells in the infarction core cannot be prevented for such a short time, the penumbra cells may be potentially saved because their death develops more slowly, during several hours. Such “therapeutic window” (3–6 h) allows to restrict the penumbra expansion and to reduce the negative consequences of ischemic stroke [1,2,3].
At present, diverse potential anti-stroke drugs belonging two main groups, thrombolytics and neuroprotectors, have been tested. However, thrombolytic therapy is not able to protect the majority of patients from acute ischemic injury. Therefore, treatment should focus on methods that improve the survival of neurons around the ischemic area. The list of the studied potential neuroprotectors includes antagonists of glutamate receptors, calcium channel blockers, antioxidants, inhibitors of apoptosis, etc. [4,5,6,7,8,9]. However, most of them, even showing promising results in the experiments on cell cultures and animals, did not pass the clinical trials because of inefficiency or significant adverse effects. Therefore, a new conceptual framework for stroke treatment is needed. Particular attention should be paid to signaling mechanisms that regulate survival or death of neurons and glial cells.
Recent proteomic studies showed the overexpression of several dozen signaling and neuronal proteins in the penumbra after photothrombotic stroke (PTS) in the rat cerebral cortex. Some of them increased survival of penumbra cells, whereas others were proapoptotic [10,11,12]. The mechanisms of the upregulation of these proteins, signaling cascades, and transcription factors that control the expression of different genes remain unknown. Global regulation of transcriptional activity in the cell is carried out by epigenetic processes such as posttranslational covalent modifications of histones (acetylation and deacetylation, phosphorylation and dephosphorylation, etc.), or by methylation and demethylation of DNA [13]. Histone acetylation causes chromatin decondensation and facilitates binding of transcription factors and RNA polymerase II to gene promoters. This stimulates transcription and protein synthesis in the cell. In contrast, histone deacetylase (HDAC) induces chromatin condensation that inhibits protein synthesis. HDAC regulates neuronal survival and death in various neuropathological situations including cerebral ischemia [14,15,16,17]. For example, upregulation of HDAC2 was recently shown to be associated with apoptosis of penumbra cells at 4 or 24 h after PTS in the rat cerebral cortex. This suggests that selective inhibition of some HDACs may be a promising strategy for protection of the brain tissue subjucted to ischemic stroke [18]. Some non-selective HDAC inhibitors such as valproic acid or sodium phenylbutyrate have demonstrated the neuroprotective capability against ischemic stroke and neurodegeneration in animals [19,20,21]. However, these HDAC inhibitors are non-selective that may cause negative side effects. Using more selective HDAC inhibitors may, in principle, eliminate these limitations.
In the present work, we studied the potential protective effect of MI192, a new inhibitor of HDAC2 and HDAC3, on the PTS-induced mouse cerebral cortex. We showed that its application reduced the infarct volume, inhibited apoptosis, improved the impaired motor activity, and increased the expression of protein GAP43 associated with growth of new axons and restoration of neuronal connections in mice subjected to the unilateral photothrombotic stroke.
Methods
Chemicals
Polyvinylidene difluoride (PVDF) membranes, blocking buffer (TBS 1% Casein Blocker), and Сlarity Western ECL Substrate were purchased from Bio-Rad (Moscow, Russia). MI192 (N-(2-Aminophenyl)-4-[(3,4-dihydro-4-methylene-1-oxo-2(1H)-isoquinolinyl)methyl]-benzamide hydrochloride), an inhibitor of HDAC2/3 (Fig. 1), was obtained from Tocris, UK (ID 5647). All other reagents were purchased from Sigma-Aldrich-Rus (Moscow, Russia).

The structure of MI192
Animals and Ethical Approval
The experiments were performed on adult male outbred mice CD-1 (20–25 g) that were kept in standard cages in groups of 4–5 animals with free access to food and water at 12 light/12 dark cycle. The animal holding room was maintained at 22–25 °C, and an air exchange rate of 18 changes per hour. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All experimental procedures were carried out in accordance with the European Union guidelines 86/609/ЕЕС for the use of experimental animals and local legislation for ethics of experiments on animals. The animal protocols were evaluated and approved by the Animal Care and Use Committee of the Southern Federal University (Approval No 08/2016). No special randomization was performed to allocate subjects in the study.
Focal Photothrombotic Stroke in the Mouse Cerebral Cortex
The unilateral focal photothrombotic stroke is an ischemic stroke model. In PTS, local laser irradiation induces photoexcitation of the injected photosensitizing dye Rose Bengal (RB). Due to its physical properties, RB does not penetrate cells and remains in blood vessels. Administration of laser irradiation induces generation of highly reactive singlet oxygen that elicits platelet aggregation, vessel occlusion, and local thrombosis [12, 22, 23]. The PTS procedures in mice brain have been previously described [24]. Mice were anesthetized with chloral hydrate (300 mg/kg, intraperitoneally). After skin incision over the skull, the periosteum was removed. Then Rose Bengal was administrated intraperitoneally at a dose of 10 mg/g. In the following 5 min, the sensorimotor cortex (2 mm lateral to the bregma) [25] was irradiated by a diode laser (532 nm, Ø 1 mm, 15 min). The animals were euthanized by chloral hydrate overdose and decapitated at 4, 7, or 14 days after PTS. Control sham-operated mice were injected with a photosensitizer, but not irradiated by a laser [24].
Immunofluorescence Microscopy
Mice that were subjected to PTS were anesthetized by chloral hydrate and transcardially perfused with 10% formalin. After extraction, their brains were post-fixed overnight in 10% formalin and incubated 48 h in 20% sucrose in PBS at 4 °С. Brain frontal sections (20 μm thick; + 2 mm from bregma to − 4 mm) [25] were obtained using the vibratome Leica VT 1000 S (Germany). They were frozen in 2-methylbutane and stored at – 80 °С. After thawing, the slices were washed in PBS. Non-specific binding of antibodies was blocked by 5% BSA containing 0.3% Triton X-100 (1 h, room temperature). Then, the slices were incubated overnight at 4 °С in the same solution supplemented with the primary antibodies (all from Sigma-Aldrich): mouse anti-Neuron-Specific Enolase (NSE, SAB4200571; 1:1000), rabbit anti-GAP43 (SAB4300525; 1:1000). After triple 5-min washing in PBS, the slices were incubated 1 h with the fluorescence-labeled secondary antibodies anti-rabbit CF488A (SAB4600045, 1:1000) or anti-mouse CF555 (SAB4600302; 1:1000). Then the slices were mounted on glass slides in 60% glycerol in PBS. Negative control: omission of primary antibodies. Three representative fields from each brain region per animal were digitally captured using a 5-MP color CCD camera (Jenoptik, FRG) attached to an Eclipse FN1 microscope (Nikon, Japan) with epifluorescence using an excitation wavelength of 490 nm and emission wavelength of 525 nm and an excitation wavelength of 510 nm and emission wavelength of 565 nm filters (Nikon, Japan).
Quantitative fluorescence estimation of experimental and control preparations was carried out using 10–15 images acquired with the same camera settings. The average fluorescence intensity in the area occupied by the cells was determined in each image using the Image J software (http://rsb.info.nih.gov/ij/). The corrected total cell fluorescence intensity (CTCF) that is proportional to the protein expression level was calculated as:
where I is the corrected intensity of total cell fluorescence, Iid is the integrated fluorescence intensity, Ac is the area of selected cell, Ib is the mean background fluorescence [26]. Threshold values were determined once and remained constant during processing of all photos. The relative changes ΔI of the mean corrected cell fluorescence in the PTS-damaged hemisphere (Iexp) compared to that in the control cerebral cortex of sham-operated mice (ISO) were calculated as:
At least three representative fields were analyzed for each brain region. Seven rats were studied in each experiment. The intergroup comparisons were carried out using one-way ANOVA. The results are shown as mean ± SEM.
Visualization of Apoptotic Cells
The “In Situ Cell Death Detection Kit, TMR red” (no. 12156792910, Roche) based on TUNEL (TdT-mediated dUTP-X nick-end labeling) method was used for detection and quantification of apoptotic cell death. The sections were incubated with the reagents from the Cell detection kit as recommended by the producer (red signal) and with Hoechst 33342 (10 μg/ml; blue signal), which visualizes the cell nuclei, at 37 °С. Apoptotic coefficient (AI) was calculated as: N = (Number of mistakes made by a limb / Number of steps taken by this limb)*100% x.
The analysis was performed on 3 images for each of 7 animals in the group. The data were expressed as mean ± SEM.
Determination of Infarction Size
To determine the infarction size in the mouse brain after PTS, the coronal sections were stained by 2,3,5-triphenyltetrazolium chloride (TTC, T8877). Mice were euthanized by overdose of chloral hydrate (600 mg/kg, intraperitoneally) and decapitated in guillotine; the brains were quickly isolated and placed into the preliminary cooled adult mouse brain matrix (J&K Seiko Electronic Co., Ltd). The matrix with the brain tissue was placed into a freezer (− 80 °С) for 3–5 min, and 1-mm-thick slices were made. These slices were stained 30 min in the dark with 1% TTC at 37 °С. The infarction area and the area of the hemisphere in each section were measured using Image J (http://rsb.info.nih.gov/ij/). The infarction volume was calculated as Vi = (VCL− Vni)/VCL, where VCL is the volume of the contralateral hemisphere, and Vni is the volume of ischemic area in the ipsilateral hemisphere. This formula takes into account the possible contribution of cerebral edema, which is characteristic for PTS [22, 23], in the calculated infarction size [27].
MI192 Administration
The potential protective effects of some inhibitors are better to reveal at several days after ischemic stroke. We studied such effects at 4 or 7 days, or sometimes later. MI192 (a selective inhibitor of HDAC2/3; ID 5647, Tocris) was dissolved in dimethyl sulfoxide (DMSO) and then diluted with 2% Tween 20 in phosphate-buffered saline (PBS). The final DMSO concentration was 5%. MI192 (40 mg/kg) was injected intraperitoneally into mice 12 h after PTS during 3 days, once a day. Control sham-operated mice were injected every day for 3 days with a DMSO solution in PBS supplemented with 2% Tween 20.
Western Blotting
Mice were euthanized by chloral hydrate overdose at 4 and 7 days after PTS, i.e., at days 1 and 3 after the last injection of MI192. After decapitation, their cerebral cortices were isolated on ice. The tissue pieces (about 200 mg) were homogenized on ice, quickly frozen in liquid nitrogen, and then stored at − 80 °C. After thawing and centrifuging, the cytoplasmic and nuclear fractions were isolated from homogenates using a CelLytic NuCLEAR Extraction Kit (Sigma-Aldrich) reagent kit. They were lysed in extraction and labeling buffer (E0655, Sigma-Aldrich) containing proteases and phosphatase inhibitors. The protein content in the tissue extracts was determined using Bradford reagent (B6916, Sigma-Aldrich). Samples with 10–20 μg of protein in 15 μl were subjected to the Laemmli electrophoretic separation in a polyacrylamide gel (7–10%) in the presence of sodium dodecyl sulfate using Mini-PROTEAN Tetracell (Bio-Rad). The protein standard Color Burst Electrophoresis Marker (C1992, Sigma-Aldrich) was used for quantitative evaluation. After separation, the proteins were electrotransferred onto an Immun-Blot PVDF Membrane polyvinyl difluoride membrane (162-0177, Bio-Rad) using the Trans-Blot® Turbo ™ Transfer System (Bio-Rad, USA). Then the membrane was washed in phosphate buffer and incubated 1 h in blocking buffer (TBS 1% Casein Blocker, 161-0782, Bio-Rad). After washing in PBS, the membrane was incubated overnight at 4 °C with the primary rabbit antibodies to anti-acetyl-histone H4 (06-598, Millipore, 1:250), anti-histone H3 (Acetyl-Lys9) (SAB4500347, 1:500), and mouse antibodies to anti-acetylated α-tubulin (T7451, Sigma, 1:250) and β-actin (A5441, 1:5000). After incubation, the membrane was washed in TTVS buffer (10 mM TRIS buffer pH 8, 0.1% Tween 20) and incubated 1 h at room temperature with secondary anti-rabbit IgG-peroxidase antibodies (A6154, Sigma-Aldrich; 1: 1000) and peroxidase labeled anti-mouse antibody (NIF825, Amersham; 1: 5000). Protein detection was performed using Clarity Western ECL Substrate (Bio-Rad). To analyze chemiluminescence, a Fusion SL system (VilberLourmat, France) and Vision Capt program were used.
Behavior Tests
The Cylinder test (OOO NPK Otkrytaya nauka; Moscow Oblast, Russia) was used to assess the asymmetry in the forelimb activity, i.e. the mice forelimb use in spontaneous vertical activity. At different time intervals after the PTS (4, 7, or 14 days), the mice were placed in a transparent plexiglass cylinder (15 × 40 cm) with a mirror that was placed at 45° under the cylinder base to monitor animal movements in any cylinder parts. The number of contacts of the front limb with the cylinder wall was recorded for 3 min using a high-resolution camera. The ratio of the left forelimb contacts that was controlled by the PTS-injured right sensorimotor cortex, was calculated as N = (a + 0.5c)/(a + b + d) × 100% [29], where a is the number of the left paw contacts with the cylinder wall, b is the number of one paw contacts with the cylinder wall, and then by the second paw, c is the number of simultaneous contacts of both paws, and d is the number of contacts of the right front limb with the wall of the cylinder.
The Foot-Error test was used to estimate the asymmetry in use of limbs on a raised grid [30]. The mice had to coordinate movements of their paws in order to rest them on the iron mesh. An inaccurate limb position leads to slipping through the holes in the grid, i.e. to mistakes. All mice go from one end of the cage to another end five times. The movements were registered using a video camera and then analyzed separately for each limb in a slow motion. The ratio of slippage into the mesh of the left paw that corresponded to the injury of the right sensorimotor cerebral cortex during PTS to the total number of steps was calculated as: N = (Number of mistakes made by a limb / Number of steps taken by this limb)*100%.
Statistical Analysis
The one-way analysis of variance (ANOVA) was used. The data are presented as mean ± SEM. The statistical significance is p < 0.05.
Results
MI192 Effect on PTS-Induced Apoptosis and Infarct Volume in the Mouse Cerebral Cortex
TUNEL-positive apoptotic cells were not revealed in the cerebral cortex of sham-operated mice after administration of PBS solution of DMSO with 2% Tween 20. However, TUNEL-positive cells were observed in the ipsilateral cortex regions around the infarct core on days 4 and 7 after photothrombotic stroke (Fig. 2). The administration of MI192 (40 mg/kg) during 3 days after PTS caused approximately twofold decrease (p < 0.05) in the number of PTS-induced apoptotic cells at these time intervals after photothrombotic stroke (Fig. 2).

Effect of HDAC2/3 inhibitor MI192 on apoptosis in the mouse cerebral cortex. a Typical fluorescence images of the mouse cortical regions stained by TUNEL (red fluorescence) at 4 or 7 days after PTS in the absence or presence of MI192. Scale bar 100 μm. b Changes in the cortical apoptotic index (AI, %) in different mice groups: Ctrl, control sham-operated mice; PTS, the peri-infarct cortical regions at 4 or 7 days after photothrombotic stroke; MI192, the peri-infarct cortical regions at 4 or 7 days after photothrombotic stroke and following intraperitoneal administration of MI192 (40 mg/kg). *р < 0005 relative to sham-operated mice; #p < 0.05 relative to PTS group. n = 21 (3 fields from 7 mice in each group). One-way ANOVA; M ± SEM
These data correspond to the twofold decrease in the PTS-induced infarct volume in the mouse brain at these time intervals (Fig. 3).

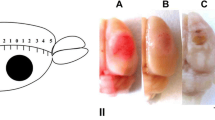
The changes in the infarct volume in the mouse brain at 4 and 7 days after photothrombotic stroke in the presence or absence of MI192. a The frontal sections of the mouse brain, stained with 2,3,5-triphenyltetrazolium chloride at 4 or 7 days after PTS scale bar 1 cm. b The changes in the infarct volume (mm3). One-way ANOVA, M ± SEM. Number of animals per group: n = 8, *p < 0.05
MI192 Effect on GAP-43 Expression in the Mice Cerebral Cortex After PTS
Growth-associated protein-43 (GAP43) is known to be involved in the recovery of the injured nervous system [31]. In the cerebral cortex of sham-operated mice, GAP43 co-localized with some neurons (Fig. 4a). Photothrombotic stroke in the mouse brain cortex increased the number of neurons in the peri-infarction cortical zone that express GAP43 at 4 and 7, but not 14 days (Fig. 4b). This can indicate the stimulation of the axon growth in the undamaged cortical tissue that leads to cerebral tissue recovery after PTS. Post-stroke administration of the HDAC2/HDAC3 inhibitor MI192 increased even more the level of GAP43 in these periods (Fig. 4b). Therefore, MI192 mediated the recovery processes after PTS. Such effects were not observed in the untreated contralateral cortex of the same animals.

GAP43 expression in cortical neurons of sham-operated and PTS-treated mice at 4 or 7 days after PTS with and without MI192 administration. а Typical fluorescence images. GAP43 (green fluorescence), NSE (neuronal marker, red fluorescence), and merged images. Scale bar, 50 μm. b The changes in the fluorescence intensity of all GAP-43-positive neurons (∆IGAP43) relative to that in the sham-operated group (ΔISO), *р < 0.05 relative to sham-operated mice; #p < 0.05 relative to PTS groups. n = 21 (3 fields from 7 mice in each group). One-way ANOVA; M ± SEM. * and #p < 0.05 relative to the sham-operated control and PTS groups, respectively
MI192 Effect on Acetylation of α-Tubulin, Histone H3, and H4 in the Mice Cerebral Cortex after PTS
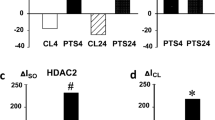
PTS induced the decrease in the acetylation of α-tubulin that is characteristic for stable microtubules [32] in the ipsilateral hemisphere of the mouse brain at 4 and 7 days. This indicated the destruction of microtubules in the deteriorated neurites, axon, and dendrites, not only in the infarction core, but also in the undamaged neighboring cortical regions. MI192 did not influence this effect (Fig. 5a). In the other experiment, MI192 significantly increased the acetylation of histone H4 (AcH4) in both PTS-treated and sham-operated mice (Fig. 5b), whereas PTS alone did not influence the acetylation of H4. Neither PTS nor MI192 influenced the acetylation of lysine 9 in histone H3 (AcH3K9) at 4 or 7 days after photothrombotic stroke (Fig. 5c).

Expression of acetylated α-tubulin, histone H4, and histone H3 at 4 and 7 days after PTS in the mouse brain. Immunoblotting of a acetylated α-tubulin (ac-α-Tub) expression in the cytoplasmic fractions of the cerebral cortex of sham-operated mice that were injected with PBS solution of DMSO with 2% Tween 20 at 24 h after PTS (Ctrl), and mice at 4 or 7 days after photothrombotic stroke (PTS) (4 days or 7 days, respectively); b, c Acetylated histone H4 (AcH4) and histone H3 acetylated on lysine 9 (acH3K9), respectively, in the nuclear brain fraction. MI192 is the group of mice subjected to PTS and following administration of MI192. n = 21 (3 fields from 7 mice in each group). One-way ANOVA. M ± SEM. * and #p < 0.05 relative to control and PTS groups, respectively
Behavior Impairment After PTS
Unilateral photothrombotic stroke in the mouse sensorimotor cortex impaired motor functions of animals (Tables 1 and 2). To assess the MI192 effect on the PTS-induced impairment of the mouse motor activity, the functional asymmetry in the forelimb use was studied by means of the Cylinder and Foot-Error tests. The number of touches to the cylinder wall by the left forelimb, the area of representation of which was in the PTS-damaged right cortex, decreased almost twofold at 4, 7, but not 14 days after PTS as compared with sham-operated animals (Table 1). Mice of these groups used mainly the right front paw for the vertical exploratory activity, whereas all control mice equally used both forelimbs during the first contact with the cylinder after the post and during the subsequent movements along the wall. MI192 treatment returned mice to the normal behavior (Table 1) possibly due to recovery of the PTS-damaged right brain hemisphere.
In the Foot-Error test, the mice move along the grid with holes. In the case of an error in the coordination of movements, the foot falls into the hole. PTS impaired the coordinated placement of limbs and significantly increased the number of errors as compared with the control mice. However, the mice treated with MI192 made significantly less errors at 4 and 7 but not 14 days after local infarct in the cerebral cortex (Table 2).
Discussion
The present experiments showed that unilateral photothrombotic stroke caused focal tissue infarct in the mouse cerebral cortex. At 4 and 7 days after PTS, this caused the functional asymmetry in the forelimb use and induced cell apoptosis in the peri-infarct tissue around the injured core. However, PTS induced opposite processes in the ipsilateral cortex outside the infarct core. The downregulation of acetylated α-tubulin, a marker of stable microtubules, indicated the destruction of neurites, axons, and dendrites in the damaged neurons. On the other hand, the upregulation of GAP43 showed stimulation of neurite growth and sprouting that was associated with the regeneration of injured axons.
Application of MI-192, an inhibitor of histone deacetylases HDAC2 and HDAC3, protected the mouse brain from negative consequences of photothrombotic stroke. MI-192 reduced the PTS-induced infarct volume, partly restored the functional symmetry in forelimb use, inhibited PTS-induced apoptosis, decreased the acetylation of α-tubulin that stabilizes microtubules, and caused overexpression of GAP-43, a marker of axon outgrowth and sprouting in the course of regeneration. The combination of all these effects suggests that MI192 may be a promising neuroprotective agent for stroke treatment. However, it should not be excluded that MI192 can exert some adverse effects like apoptosis promotion in the leukemia cell lines [33].
The obtained data suggest the involvement of HDAC2 and HDAC3 in the photothrombotic injury of the mouse brain not only in the infarction core, but also outside it. HDAC2 and HDAC3 are abundant in the brain. HDAC3 is the most prevalent in cortex and hippocampus [34,35,36]. These histone deacetylases are involved in various brain functions including responses to ischemic injury. HDAC2 plays the important role in the regulation of neuron death and survival [37, 38]. It localizes in the neuronal nuclei. The overexpression of HDAC2 in the cortical peri-infarct zone after photothrombotic stroke or middle cerebral artery occlusion (MCAO) was associated with the induction of apoptosis in the rodent brain [28, 38]. HDAC3 was also involved in the ischemic injury of the mouse brain. Unlike HDAC2, HDAC3 has both nuclear import and export signals, and therefore shuttles between the cytoplasm and the nucleus [35, 36]. Its inhibition was shown to decrease the cerebral infarct volume, attenuate oxidative stress, reduce apoptosis, and enhance autophagy after cerebral ischemia/reperfusion injury of mice. These effects were reversed by pretreatment of HDAC3 inhibitor RGFP966 [39].
Histone deacetylase inhibitors are currently examined as potential therapeutic agents against brain injury [39,40,41,42,43]. In the present experiments, administration of the benzamidine derivative MI192, a selective inhibitor of HDAC2 and HDAC3, decreased the PTS-induced infarction size, reduced apoptosis, alleviated PTS-induced impairment of the motor behavior, and induced the upregulation of GAP43, which is associated with growth of axons and establishment of new interneuron connections. MI192 can penetrate the brain through the blood–brain barrier [43]. It can act on both HDAC2 and HDAC3. However, in the PTS-induced penumbra, these effects were possibly associated with the inhibition of HDAC2 rather than HDAC3, despite the similar IC50 values (30 and 16 nM, respectively) [33]. In fact, HDAC3 is known to deacetylate α-tubulin in the cytoplasm [44, 45]. However, MI192 did not influence the PTS-induced decrease in the acetylation of α-tubulin in the mouse brain. On the other hand, MI192 increased acetylation of histone H4, which was not stimulated by PTS. This effect was possibly due to prevention of the deacetylase activity of HDAC2 that, unlike HDAC3, deacetylates histones H2A, H2B, H3, and H4. The relocalization of HDAC3 from the nucleus to the cytoplasm was shown to reduce acetylation of α-tubulin in the cytoplasm and to elevate acetylation of lysine 9 in histone H3 (due to lack of HDAC3) [46]. In the present experiments, neither PTS nor MI192 influenced the acetylation of lysine 9 in histone H3 (AcH3K9) at 4 or 7 days after photothrombotic stroke. These data suggest that the effects of MI192 were associated with its inhibition of HDAC2 rather than HDAC3.
The response of the damaged ipsilateral mouse cerebral cortex to focal photothrombotic infarct included both damaging and recovery components. It included apoptosis and destruction of some neuronal cells. The cell destruction was evidenced by the downregulation of acetylated α-tubulin. α-Tubulin is a component of microtubules, which are abundant in axons and dendrites. Its acetylation after microtubule assembly stabilizes microtubules [32]. Therefore, the partial loss of acetylated α-tubulin indicates the destruction of neurons.
On the other hand, the upregulation of GAP43 at 4 and 7 days after PTS pointed to the recovery processes in the injured ipsilateral cortex. GAP43 is a nervous tissue-specific protein. It is a marker of postmitotic neurons. It is expressed at high levels in axonal growth cones and presynaptic nerve terminals during axonal growth and synapse formation in the course of embryonic development or axon regeneration [31, 47]. Its overexpression in the mouse peri-infarct cortex after photothrombotic stroke was possibly associated with outgrowth and sprouting of new axons and restoration of neuronal connections [43]. The application of MI192 like other non-specific HDAC inhibitors such as valproic acid, sodium phenylbutyrate, or trichostatin A was shown to induce growth and sprouting of axons in cultured neurons after oxygen and glucose deprivation [48].
In conclusion, unilateral photothrombotic stroke caused tissue infarct in the mouse cerebral cortex. The injury of the cerebral cortex impaired the mouse motor activity, in particular the functional asymmetry in the forelimb use. In the peri-infarct cortical tissue outside the infarct core cell apoptosis occurred at 4 and 7 days after PTS. The downregulation of acetylated α-tubulin, a marker of stable microtubules, showed the destruction of neurites, axons, and dendrites in injured neurons. However, the upregulation of GAP43 indicates the stimulation of neurite growth that was possibly aimed at the recovery of the cortical tissue in the damaged cerebral hemisphere.
Application of MI-192, an inhibitor of histone deacetylases HDAC2 and HDAC3, demonstrated the neuroprotective activity in the mouse brain subjected to photothrombotic stroke. It reduced the volume of the PTS-induced infarction core in the mouse brain, partly restored the functional symmetry in the forelimb use, decreased the level of PTS-induced apoptosis and acetylation of α-tubulin characteristic for stable microtubules, and increased the expression of GAP-43 in the cerebral cortex of the damages ipsilateral hemisphere. These data point to the involvement of HDAC2 and HDAC3 in the photothrombotic injury of the mouse brain not only in the infarction core but also outside it. The application of MI192 after PTS reduced or eliminated these negative effects and exerted the neuroprotective effect on the mouse brain. Thus, MI192 may be considered as a promising neuroprotector for anti-stroke therapy.
References
Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67:181–198. https://doi.org/10.1016/j.neuron.2010.07.002
Manning NW, Campbell BC, Oxley TJ, Chapot R (2014) Acute ischemic stroke: time, penumbra, and reperfusion. Stroke 45:640–644. https://doi.org/10.1161/STROKEAHA.113.003798
Hankey GJ (2017) Stroke. Lancet 389:641–654. https://doi.org/10.1016/S0140-6736(16)30962-X
Majid A (2014) Neuroprotection in stroke: past, present, and future. ISRN Neurol 2014:515716. https://doi.org/10.1155/2014/515716
Rajah GB, Ding Y (2017) Experimental neuroprotection in ischemic stroke: a concise review. Neurosurg Focus 42(4):E2. https://doi.org/10.3171/2017.1.FOCUS16497
Patel Rajan AG, McMullen PW (2017) Neuroprotection in the treatment of acute ischemic stroke. Prog Cardiovasc Dis 59:542–548. https://doi.org/10.1016/j.pcad.2017.04.005
Karsy M, Brock A, Guan J, Taussky P, Kalani MY, Park MS (2017) Neuroprotective strategies and the underlying molecular basis of cerebrovascular stroke. Neurosurg Focus 42:E3. https://doi.org/10.3171/2017.1.FOCUS16522
Zhou Z, Lu J, Liu WW, Manaenko A, Hou X, Mei Q, Huang JL, Tang J et al (2018) Advances in stroke pharmacology. Pharmacol Ther 191:23–42. https://doi.org/10.1016/j.pharmthera.2018.05.012
Luo Y, Tang H, Li H, Zhao R, Huang Q, Liu J (2019) Recent advances in the development of neuroprotective agents and therapeutic targets in the treatment of cerebral ischemia. Eur J Med Chem 162:132–146. https://doi.org/10.1016/j.ejmech.2018.11.014
Demyanenko SV, Panchenko SN, Uzdensky AB (2015) Expression of neuronal and signaling proteins in penumbra around a photothrombotic infarction core in rat cerebral cortex. Biochem Mosc 80:790–799. https://doi.org/10.1134/S0006297915060152
Demyanenko S, Uzdensky A (2017) Profiling of signaling proteins in penumbra after focal photothrombotic infarct in the rat brain cortex. Mol Neurobiol 54:6839–6856. https://doi.org/10.1007/s12035-017-0736-7.
Uzdensky A, Demyanenko S, Fedorenko G, Lapteva T, Fedorenko A (2017) Photothrombotic infarct in the rat brain cortex: protein profile and morphological changes in penumbra. Mol Neurobiol 54:4172–4188. https://doi.org/10.1007/s12035-016-9964-5.
Kouzarides T, Berger SL (2006) Chromatin modifications and mechanisms. In: Allis CD, Jenuwein T, Reinberg D (eds) Epigenetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, pp. 191–209
Konsoula Z, Barile FA (2012) Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods 66:215–220. https://doi.org/10.1016/j.vascn.2012.08.001
Schweizer S, Meisel A, Märschenz S (2013) Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab 33:1335–1346. https://doi.org/10.1038/jcbfm.2013.93
Volmar CH, Wahlestedt C (2015) Histone deacetylases (HDACs) and brain functions. Neuroepigenetics 1:20–27. https://doi.org/10.1016/j.nepig.2014.10.002
Hu Z, Zhong B, Tan J, Chen C, Lei Q, Zeng L (2017) The emerging role of epigenetics in cerebral ischemia. Mol Neurobiol 54:1887–1905. https://doi.org/10.1007/s12035-016-9788-3
Demyanenko SV, Dzreyan VA, Neginskaya MA, Uzdensky AB (2019) Expression of histone deacetylases HDAC1 and HDAC2 and their role in apoptosis in the penumbra induced by photothrombotic stroke. Mol Neurobiol (in press)
Xuan A, Long D, Li J, Ji W, Hong L, Zhang M, Zhang W (2012) Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci 90:463–468
Ganai SA, Ramadoss M, Mahadevan V (2016) Histone deacetylase (HDAC) inhibitors - emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol 14:55–71
Wang ZY, Qin W, Yi F (2015) Targeting histone deacetylases: perspectives for epigenetic-based therapy in cardio-cerebrovascular disease. J Geriatr Cardiol 12:153–164. https://doi.org/10.11909/j.issn.1671-5411.2015.02.010.
Schmidt A, Hoppen M, Strecker JK, Diederich K, Schabitz WR, Schilling M, Minnerup J (2012) Photochemically induced ischemic stroke in rats. Exp Transl Stroke Med 4:13. https://doi.org/10.1186/2040-7378-4-13
Uzdensky AB (2018) Photothrombotic stroke as a model of ischemic stroke. Transl Stroke Res 9:437–451. https://doi.org/10.1007/s12975-017-0593-8
Demyanenko S, Neginskaya M, Berezhnaya E (2018) Expression of class I histone deacetylases in ipsilateral and contralateral hemispheres after the focal photothrombotic infarction in the mouse brain. Transl Stroke Res 9:471–483. https://doi.org/10.1007/s12975-017-0595-6
Paxinos G, Franklin KBJ (2013) Paxinos and Franklin’s the mouse brain in stereotaxic coordinates. Academic Press, Amsterdam
McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A (2014) Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13:1400–1412. https://doi.org/10.4161/cc.28401
Chelluboina B, Klopfenstein JD, Gujrati M, Rao JS, Veeravalli KK (2014) Temporal regulation of apoptotic and anti-apoptotic molecules after middle cerebral artery occlusion followed by reperfusion. Mol Neurobiol 49:50–65. https://doi.org/10.1007/s12035-013-8486-7
Demyanenko S, Berezhnaya E, Neginskaya M, Rodkin S, Dzreyan V, Pitinova M (2019, 2019) Сlass II histone deacetylases in the post-stroke recovery period-expression, cellular, and subcellular localization-promising targets for neuroprotection. J Cell Biochem. https://doi.org/10.1002/jcb.29266
Chen W, Qiao D, Liu X, Shi K (2017) Treadmill exercise improves motor dysfunction and hyperactivity of the corticostriatal glutamatergic pathway in rats with 6-OHDA-induced Parkinson’s disease. Neural Plast 2017:2583910–2583911. https://doi.org/10.1155/2017/2583910
Barth TM, Jones TA, Schallert T (1990) Functional subdivisions of the rat somatic sensorimotor cortex. Behav Brain Res 39:73–95
Carmichael ST, Archibeque I, Luke L, Nolan T, Momiy J, Li S (2005) Growth-associated gene expression after stroke: Evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol 193:291–311
Westermann S, Weber K (2003) Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol 42:938–947. https://doi.org/10.1038/nrm1260.
Boissinot M, Inman M, Hempshall A, James SR, Gill JH, Selby P, Bowen DT, Grigg R et al (2012) Induction of differentiation and apoptosis in leukaemic cell lines by the novel benzamide family histone deacetylase 2 and 3 inhibitor MI-192. Leuk Res 36:1304–1310. https://doi.org/10.1016/j.leukres.2012.07.002
Baltan S, Bachleda A, Morrison RS, Murphy SP (2011) Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl Stroke Res 2:411–423. https://doi.org/10.1007/s12975-011-0087-z
Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ (2007) Distribution of histone deacetylases 1-11 in the rat brain. J Mol Neurosci 31:47–58. https://doi.org/10.1007/BF02686117
Wagner FF, Weїwer M, Lewis MC, Holson EB (2013) Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics 10:589–604. https://doi.org/10.1007/s13311-013-0226-1
Krämer OH (2009) HDAC2: a critical factor in health and disease. Trends Pharmacol Sci 30:647–655. https://doi.org/10.1016/j.tips.2009.09.007
Lin YH, Dong J, Tang Y, Ni HY, Zhang Y, Su P, Liang HY, Yao MC et al (2017) Opening a new time window for treatment of stroke by targeting HDAC2. J Neurosci 37:6712–6728. https://doi.org/10.1523/JNEUROSCI.0341-17.2017
Zhao B, Yuan Q, Hou JB, Xia ZY, Zhan LY, Li M, Jiang M, Gao WW et al (2019) Inhibition of HDAC3 ameliorates cerebral ischemia reperfusion injury in diabetic mice in vivo and in vitro. J Diabetes Res 2019:8520856–8520812. https://doi.org/10.1155/2019/8520856
Gibson CL, Murphy SP (2010) Benefits of histone deacetylase inhibitors for acute brain injury: A systematic review of animal studies. J Neurochem 115:806–813. https://doi.org/10.1111/j.1471-4159.2010.06993.x
Fessler EB, Chibane FL, Wang Z, Chuang DM (2013) Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery. Curr Pharm Des 19:5105–5120
Yusoff SI, Roman M, Lai FY, Eagle-Hemming B, Murphy GJ, Kumar T, Wozniak M (2019) Systematic review and meta-analysis of experimental studies evaluating the organ protective effects of histone deacetylase inhibitors. Transl Res 205:1–16. https://doi.org/10.1016/j.trsl.2018.11.002
Seo YJ, Kang Y, Muench L, Reid A, Caesar S, Jean L, Wagner F, Holson E et al (2014) Image-guided synthesis reveals potent blood-brain barrier permeable histone deacetylase inhibitors. ACS Chem Neurosci 5:588–596. https://doi.org/10.1021/cn500021p
Bacon T, Seiler C, Wolny M, Hughes R, Watson P, Schwabe J, Grigg R, Peckham M (2015) Histone deacetylase 3 indirectly modulates tubulin acetylation. Biochem J 472:367–377. https://doi.org/10.1042/BJ20150660
Li X, Liu X, Gao M, Han L, Qiu D, Wang H, Xiong B, Sun SC et al (2017) HDAC3 promotes meiotic apparatus assembly in mouse oocytes by modulating tubulin acetylation. Development 144:3789–3797. https://doi.org/10.1242/dev.153353
Yang X, Wu Q, Zhang L, Feng L (2016) Inhibition of histone deacetylase 3 (HDAC3) mediates ischemic preconditioning and protects cortical neurons against ischemia in rats. Front Mol Neurosci 9:131. eCollection 2016. https://doi.org/10.3389/fnmol.2016.00131
Skene JH (1989) Axonal growth-associated proteins. Annu Rev Neurosci 12:127–156
Hasan MR, Kim JH, Kim YJ, Kwon KJ, Shin CY, Kim HY, Han SH, Choi DH et al (2013) Effect of HDAC inhibitors on neuroprotection and neurite outgrowth in primary rat cortical neurons following ischemic insult. Neurochem Res 38:1921–1934. https://doi.org/10.1007/s11064-013-1098-9
Funding
This study was funded by the Russian Science Foundation (grant no. 18-15-00110).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Demyanenko, S.V., Nikul, V.V. & Uzdensky, A.B. The Neuroprotective Effect of the HDAC2/3 Inhibitor MI192 on the Penumbra After Photothrombotic Stroke in the Mouse Brain. Mol Neurobiol 57, 239–248 (2020). https://doi.org/10.1007/s12035-019-01773-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-01773-9