
Abstract
Activated microglia have been reported to play an important role in Parkinson’s disease (PD). A more rapid cognitive decline has been associated with deposits of β-amyloid. In this study, the aim was to evaluate the role of brain β-amyloid and its relationship with activated microglia in PD patients with normal and impaired cognition. We studied 17 PD patients with normal cognition (PDn), 12 PD patients with mild cognitive impairment (PD-MCI), and 12 healthy controls (HCs) with [11C] Pittsburgh compound B (PIB) to assess the impact of β-amyloid deposition in the brain on microglial activation evaluated using the translocator protein 18-kDa (TSPO) radioligand [18F]-FEPPA. [11C] PIB distribution volume ratio was measured in cortical and subcortical regions. [18F]-FEPPA total distribution volume values were compared for each brain region between groups to evaluate the effect of PIB positivity while adjusting for the TSPO rs6971 polymorphism. Factorial analysis of variance revealed a significant main effect of PIB positivity in the frontal lobe (F(1, 34) = 7.1, p = 0.012). Besides the frontal (p = 0.006) and temporal lobe (p = 0.001), the striatum (p = 0.018), the precuneus (p = 0.019), and the dorsolateral prefrontal cortex (p = 0.010) showed significant group × PIB positivity interaction effects. In these regions, PD-MCIs had significantly higher FEPPA VT if PIB-positive. Our results indicate an interaction between amyloid-β deposition and microglial activation in PD. Further investigations are necessary to evaluate if amyloid deposits cause neuroinflammation and further neurodegeneration or if increased microglia activation develops as a protective response.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, presenting with progressive motor symptoms as well as cognitive impairment and dementia [1]. It has been suggested that the decline of cognition in PD may derive from a progressive dysfunction of different cortical regions related to the extension of the alpha-synuclein (α-syn) pathology to the cortex and/or the presence of neuritic plaques (β-amyloid deposition) [2] in association with neuroinflammatory mechanisms [3]. These neuroinflammatory mechanisms, such as activated microglia, are believed to play a critical role in the pathophysiology of PD [3]. Microglia are resident phagocytes of the central nervous system (CNS). They are ubiquitously distributed in the brain and get activated by neuronal cell death or protein aggregation. Upon activation, they express increased levels of the translocator protein 18 kDa (TSPO) on the outer layer of their mitochondria.
The development of biomarkers in positron emission tomography (PET) enables us to measure in vivo abnormal β-amyloid deposition and microglial activation using the well-established radiotracer [11C] Pittsburgh compound B (PiB) and [18F]-FEPPA, a second-generation 18-kDa TSPO radiotracer, respectively.
To date, a significant increase in [18F]-FEPPA total distribution volume (VT) has been observed in Alzheimer’s disease (AD) patients [4], and studies using the radioligand [11C]-PK11195, a first-generation biomarker of microglia, have detected neuroinflammation in primarily cortical regions in more severe cases of PD with dementia (PDD) [5].
While the presence of β-amyloid is a characteristic feature of AD [6], it has also been observed in PD [7]. To date, studies with [11C]-PIB in PD have shown a prevalence of elevated binding significantly lower [8] than what has been reported in cognitively normal elderly population [9]. Yet, increased amounts of amyloid have been observed in PDD, especially in patients with dementia with Lewy bodies (DLB) [10]. Further, the presence of amyloid has been associated with a more rapid cognitive deterioration in PD patients [11].
To this point, the role of microglial activation and its relation with amyloid accumulation remains unclear. Activated microglia are present around β-amyloid plaques in AD [12]; however, it is uncertain whether β-amyloid develops as a response to neuroinflammatory processes or vice versa, amyloid deposits cause further microglial activation [5].
In this dual-tracer PET study, the aim was to evaluate the role of brain β-amyloid and its relationship with activated microglia in PD patients with normal cognitive function (PDn) and mild cognitive impairment (PD-MCI).
Methods
Participants
Twenty-nine patients meeting the UK Brain Bank criteria for the diagnosis of idiopathic PD and twelve healthy age-matched controls participated in our study. All participants were assessed for their cognitive performance using the Montreal Cognitive Assessment (MoCA) and their level of depression using the Beck Depression Inventory—II (BDI-II). The diagnosis of MCI was made according to current clinical guidelines [13]. A cut-off score of 26 was used to distinguish PD patients with (PD-MCI) and without (PDn) cognitive impairment. Further, MCI diagnosis was established when cognitive decline was reported by either the patient or informant or observed by the clinician. Cognitive deficits were not sufficient to interfere significantly with functional independence, as reported by patients or their informant. In total, our cohort consisted of 17 PDn and 12 PD-MCI patients. PD patients were also assessed for motor severity of the disease using the motor subset of the Unified Parkinson Disease Rating Scale (UPDRS-III). Exclusion criteria for all participants were (1) any history of head injury, psychiatric, or other neurological diseases, (2) alcohol or drug dependency or abuse, (3) contraindications for MRI scanning, and (4) use of nonsteroidal anti-inflammatory drugs. All participants underwent PET and structural MRI scans. All participants provided written informed consent following full explanation of the study procedures. The study was approved by the Centre for Addiction and Mental Health Research Ethics Board (127/2015).
PET Data Acquisition
Each participant underwent a 125-min [18F]-FEPPA and a 90-min [11C]-PIB PET scan using a 3D High-Resolution Research Tomography (HRRT) scanner (CS/Siemens, Knoxville, TN, USA), which measures radioactivity in 207 slices with an inter-slice distance of 1.22 mm. The detectors of the HRRT are an LSO/LYSO phoswich detector, with each crystal element measuring 2 × 2 × 10 mm3. Before the acquisition of the emission scan, a transmission scan with a single-photon point source, 137Cs (t1/2 = 30.2 years, Eγ = 662 keV), was acquired. [18F]-FEPPA images were reconstructed into a series of 34 time frames, including 1 frame of variable length, followed by frames comprising 5 × 30 s, 1 × 45 s, 2 × 60 s, 1 × 90 s, 1 × 120 s, and 22 × 300 s. Prior to the start of the PET scans, a custom-fitted thermoplastic mask was made for each participant to minimize head movement. The radiosyntheses of [18F]-FEPPA [14] and [11C]-PIB [15] have been described in detail elsewhere. For [18F]-FEPPA, an intravenous saline solution of 5.03 ± 0.26 (mean ± SD) mCi was administered as a bolus. An automated blood sampling system (ABSS, model #PBS-101 from Veenstra Instruments, Netherlands) was used to measure arterial blood radioactivity continuously at a rate of 2.5 ml/min for the first 22.5 min of the PET scan. In addition, manual arterial samples were obtained at 2.5, 7, 12, 15, 30, 45, 60, 90, and 120 min after injection of the radiotracer. The arterial samples were used to generate an input function of unmetabolized radioligand in the plasma for kinetic analysis, as previously described [16].
For [11C]-PIB, an intravenous saline solution of 9.68 ± 0.83 mCi was administered as a bolus. Images were reconstructed into a series of 34 frames comprising 4 × 15 s, 8 × 30 s, 9 × 60 s, 2 × 180 s, 8 × 300 s, and 3 × 600 s.
MRI Acquisition
MR images for all the participants were acquired for coregistration with the corresponding PET images and the anatomical delineation of the regions of interest (ROIs). Proton density (PD)-weighted MR images were chosen for better identification of ROIs [17]. 2D oblique PD-weighted MR images were acquired with a General Electric Discovery 3.0 T MRI scanner (slice thickness = 2 mm, repetition time (TR) = 6000 ms, echo time (TE) = min full, flip angle = 90°, number of excitations (NEX) = 2, acquisition matrix = 256 × 192, and field of view = 22 cm).
ROI-Based PET Image Analysis
Both PET images, [18F]-FEPPA and [11C]-PIB, were preprocessed, and ROIs were automatically generated using in-house software, ROMI, as previously described [17]. To obtain a quantitative measure of [18F]-FEPPA uptake, data were analyzed using the 2-tissue compartment model (2TCM) with total distribution volume (VT) as the outcome measure, as previously validated [16]. VT is a ratio at equilibrium of the radioligand concentration in tissue to that in plasma (i.e., specific binding and non-displaceable uptake, which includes non-specifically bound and free radioligand in tissue). In addition, we measured the percentage of the coefficient of variation (%COV = 100% × standard error / mean), where standard error was estimated from the diagonal of the covariance matrix of nonlinear least-square fitting [16]. Gray matter ROIs known to be affected in PD and PD-MCI were examined, including the frontal, prefrontal, and temporal cortices, precuneus, and striatum. From the different ROIs, we included VT with %COV of ≤ 20, which assured less noisy data. Therefore, the effective sample size for each ROI varied between 37 and 41. To address the potential issues of bias from the volume loss in older participants, [18F]-FEPPA time activity data for all participants were corrected for the effect of partial volume error (PVE) using the Mueller-Gartner partial volume error correction algorithm as implemented in Bencherif et al. [18]. [11C]-PIB retention was measured using the Logan plot with the cerebellum as a reference region to yield distribution volume ratio (DVR), as this method has been shown to provide stable and robust results in several ROIs [19]. The average DVR of cortical regions and the striatum was calculated, and a cut-off of 1.2 was set to divide each brain region into PIB-positive or PIB-negative [20]. The kinetic analysis of the radioligands was performed using PMOD 3.6 modeling software (PMOD Technologies Ltd., Zurich, Switzerland). It is important to note that PIB positivity status was defined for each region separately; however, for the sake of simplicity, we refer as PIB-positive and PIB-negative groups within the manuscript.
For the [11C]-PIB activity, we decided to focus on the non-PVE-corrected DVR to avoid potential inaccuracies resulting from uncertainties in image registration and segmentation as previously reported [21]. However, for exploratory purposes, we investigated our findings after applying PVE-corrected DVRs; please see the “Results with PVE-Corrected DVR” section.
DNA Extraction and Polymorphism Genotyping
Genomic DNA was obtained from peripheral leukocytes by using high salt-extraction methods [22]. The TSPO polymorphism rs6971 was genotyped as previously reported [23]. Based on their genotype, the participants were classified as high-, mixed-, and low-affinity binders (HAB, MAB, and LAB, respectively), as described elsewhere [23]. [18F]-FEPPA is not quantifiable in LABs [23]; therefore, we did not include any LAB participants in our study.
Statistical Analysis
Analysis for Demographic Data
Normality assumptions of data were assessed using the Kolmogorov–Smirnov test. Significant differences in means of normally distributed continuous demographical as well as clinical variables were tested by using factorial analysis of variance (ANOVA). If variables did not satisfy the assumption, the Kruskal–Wallis test was applied. Difference in proportions (i.e., gender, handedness, TSPO genotype, and symptom-dominant side) was assessed by chi-squared tests.
Analysis for PET Data
For each brain region, [18F]-FEPPA VT values were compared between the three groups to evaluate the effect of PIB positivity using ANOVA. For this analysis, TSPO rs6971 polymorphism was added as a covariate to control the effect of different binding affinities. In a secondary analysis, our PD sample was split into two sub-groups with a shorter (< 5 years) and longer disease duration (> 5 years). To assess the effect of disease duration on VT values, we conducted a 2 × 2 factorial ANOVA with PIB positivity (PIB positive vs. PIB negative) and disease duration (short vs. long) as between-factors and TSPO polymorphism as a covariate in cortical as well as subcortical brain regions. Partial correlation was performed to explore the relationship between microglial activation (as measured by [18F]-FEPPA VT) and β-amyloid (measured by [11C]-PIB DVR) in each disease duration sub-group while correcting for the TSPO polymorphism. Partial correlation was also used to explore relations between [18F]-FEPPA VT and clinical characteristics while controlling for the TSPO genotype. Correlations between clinical measures and [11C]-PIB DVR were investigated using Pearson’s correlation tests. All statistical analyses were performed using SPSS Statistics version 20.0, with a significance level set at the threshold of p < 0.05. Bonferroni’s correction was applied to counteract the problem of multiple comparisons for the number of assessed ROIs (p < 0.007).
Results
Demographic and Clinical Characteristics
Demographical and clinical measures are displayed in Table 1. The factorial ANOVA as well as the Kruskal–Wallis test showed that there were no significant differences in all demographical and clinical measures and PET parameters between groups. As expected, PD-MCIs had significantly lower MoCA scores than HCs and PDns (p < 0.001). Chi-squared tests showed that there were no significant differences in the composition of gender and handedness across groups or in the composition of symptom-dominant side between PDns and PD-MCIs. There was a significant difference in the composition of TSPO genotype between groups (p = 0.021). There were also no significant differences in UPDRS III, disease duration, or total levodopa equivalent daily dose (LEDD) between PDns and PD-MCIs.
Overall, a total of 8 participants were PIB-positive in the HC group, 8 PIB-positive in the PD-Ns, and 4 PIB-positive in the PD-MCI group.
The Influence of β-Amyloid Deposition on Microglial Activation
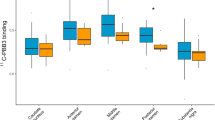
The factorial ANOVA analyses revealed a significant main effect of PIB positivity in the frontal lobe (F(1, 34) = 7.1, p = 0.012) (Fig. 1). There was a significant interaction between PIB positivity and subject groups in the frontal (p = 0.006) and temporal lobes (p = 0.001), striatum (p = 0.018), precuneus (p = 0.019), and dorsolateral prefrontal cortex (DLPFC) (p = 0.010) (Fig. 1 and Fig. 2). In these regions, the PD-MCI group had significantly higher [18F]-FEPPA VT if PIB-positive. In the frontal lobe, this difference was also seen in the PDn group (p = 0.009). Further, PIB-positive PD-MCIs showed significantly higher FEPPA VT than PIB-positive HCs in the frontal (p = 0.028) and temporal lobes (p = 0.002), striatum (p = 0.017), and DLPFC (p = 0.046), as well as significantly higher VT than PIB-positive PDns in the striatum (p = 0.023) and temporal lobe (p = 0.009). In the temporal lobe (p = 0.043) and DLPFC (p = 0.024), PIB-negative HCs had significantly higher FEPPA VT than PIB-negative PD-MCIs. When applying multiple-test correction (p < 0.007) for the number of assessed ROIs, interaction effects between PIB positivity and groups in the frontal (p = 0.006) and temporal lobes (p = 0.001) remained significant.

[18F]-FEPPA total distribution volume (VT) in regions of interest with partial volume error correction (pvec). PIB+, PIB positive; PIB−, PIB negative
The Influence of Disease Duration on Microglial Activation
A total of 15 PD patients were in the short disease duration group and 14 PD patients were in the long disease duration group. Factorial ANOVA analyses revealed significant main effects of PIB positivity (F(1, 24) = 7.7, p = 0.010) and disease duration (F(1, 24) = 4.3, p = 0.048) in the precuneus (Fig. 2). In this region, the short disease duration group had significantly higher [18F]-FEPPA VT if PIB-positive. Similar findings were also observed in the temporal lobe (F(1, 24) = 6.9, p = 0.015). In the frontal lobe, there was a significant main effect of PIB positivity (F(1, 24) = 20.4, p < 0.001) and disease duration (F(1, 24) = 6.0, p = 0.022), with an interaction effect (F(1, 24) = 4.7, p = 0.040). In this region, patients had significantly higher [18F]-FEPPA VT if PIB-positive, with PIB-positive patients in the short disease duration group having significantly higher FEPPA VT than PIB-positive patients in the long disease duration group (p = 0.022) (Fig. 3).

[18F]-FEPPA total distribution volume (VT) in regions of interest with partial volume error correction (pvec). PIB+, PIB positive; PIB−, PIB negative
Associations Between β-Amyloid Deposition, Microglial Activation, and Clinical Characteristics
Partial correlation analyses in the PD-MCI group revealed a significant, positive association between [18F]-FEPPA VT and [11C]-PIB DVR in the ventrolateral prefrontal cortex (VLPFC) (r = 0.669, p = 0.034), medial prefrontal cortex (MPFC) (r = 0.668, p = 0.030), DLPFC (r = 0.648, p = 0.031), and striatum (r = 0.777, p = 0.005), suggesting higher β-amyloid deposition was associated with increased microglial activation (Fig. 4 and Fig. 5). Partial correlation analyses remained significant in the striatum after correcting for multiple comparisons. There were no significant correlations between [18F]-FEPPA VT and [11C]-PIB DVR in the PD normal and HC group (p > 0.05). In the patient group, there was a negative correlation between the MoCA sub-category “attention” and [11C]-PIB DVR in the frontal (r = − 0.373, p = 0.046) and temporal lobes (r = − 0.481, p = 0.008), implying β-amyloid deposition in these regions was associated with poor attention performance. Further, there was a positive correlation between disease duration and [11C]-PIB DVR in the MPFC (r = 0.501, p = 0.006), temporal lobe (r = 0.408, p = 0.028), frontal lobe (r = 0.369, p = 0.049), and precuneus (r = 0.507, p = 0.005), suggesting β-amyloid deposition increased with duration of the disease. Positive correlations between disease duration and [11C]-PIB DVR in the MPFC and precuneus remained significant after applying Bonferroni’s correction.

Effect of disease duration on microglial activation. PIB+, PIB positive; PIB−, PIB negative
Results with PVE-Corrected DVR
The Influence of β-Amyloid Deposition on Microglial Activation
The influence of β-amyloid deposition on microglial activation remained significant after applying PVE-corrected DVR in the temporal lobe, VLPFC, and striatum. There was a significant interaction between PIB positivity and patient groups in the temporal lobe (p = 0.009) and striatum (p = 0.004) (Fig. 6). In these regions, the PD-MCI group had significantly higher [18F]-FEPPA VT if PIB-positive. In the striatum, PIB-positive PD-MCIs showed significantly higher FEPPA VT than PIB-positive HCs (p = 0.025) and PIB-positive PDns (p = 0.002). In the temporal lobe, PIB-positive PD-MCIs showed also significantly higher FEPPA VT than PIB-positive PDns (p = 0.010). There was a significant main effect of PIB positivity in the VLPFC (F(1, 32) = 9.1, p = 0.005) (Fig. 6). In this region, PD patients and controls showed significantly higher [18F]-FEPPA VT when PIB-positive.

Positive correlations between [18F]-FEPPA and [11C]-PIB in regions of interest shown in PD-MCI while controlling for the TSPO genotype. VLPFC, ventrolateral prefrontal cortex; MPFC, medio-prefrontal cortex
The Influence of Disease Duration on Microglial Activation
There was no significant effect between disease duration and PIB positivity.
Associations Between β-Amyloid Deposition, Microglial Activation, and Clinical Characteristics
Partial correlation analyses in the PD-MCI group revealed a significant, positive association between [18F]-FEPPA VT and [11C]-PIB DVR in the in the striatum (r = 0.708, p = 0.015), suggesting higher β-amyloid deposition was associated with increased microglial activation (Fig. 7).

Positive correlations between [18F]-FEPPA and [11C]-PIB in regions of interest shown in PD-MCI while controlling for the TSPO genotype. DLPFC, dorsolateral prefrontal cortex
In the patient group, there was a negative correlation between the MoCA sub-category “attention” and [11C]-PIB DVR in the temporal lobe (r = − 0.386, p = 0.038), implying β-amyloid deposition in these regions was associated with poor attention performance. Further, there was a positive correlation between disease duration and [11C]-PIB DVR in the inferior parietal lobe (r = 0.418, p = 0.024), temporal lobe (r = 0.381, p = 0.042), and precuneus (r = 0.619, p < 0.001), suggesting β-amyloid deposition increased with duration of the disease (Figs. 6 and 7).

[18F]-FEPPA total distribution volume (VT) in regions of interest with partial volume error corrected distribution volume ratios (DVRs). PIB+, PIB positive; PIB−, PIB negative

Positive correlations between [18F]-FEPPA and [11C]-PIB in the striatum shown in PD-MCIs while controlling for the TSPO genotype and using partial volume error corrected distribution volume ratios (DVRs)
Discussion
This is the first dual-tracer PET study exploring the relationship between microglial activation and β-amyloid accumulation in PD with normal and impaired cognition using a second-generation TSPO radiotracer. For the first time, our results revealed significant interactions between β-amyloid and neuroinflammation, particularly in patients with cognitive deficits. As previously mentioned, β-amyloid is a characteristic feature of AD [6]; however, it is also present to a lesser extent in PD [7]. It has been proposed that β-amyloid, in combination with neuroinflammatory mechanisms and α-syn pathology, may play a role in the cognitive decline in PD [24]. In this regard, it is well-known that α-syn and β-amyloid deposition can induce microglial activation and neuroinflammation [24]. Our current findings are in line with this hypothesis, since we observed that the presence of amyloid was associated with higher microglial activation in various cortical and subcortical regions. This effect was most prominent in PD-MCIs, whereas an interaction between “PIB-positivity” and neuroinflammation was less consistent in PD with normal cognition. Further, in the PD-MCI group, we observed a positive correlation between [18F]-FEPPA and [11C]-PIB binding in striatal and prefrontal regions (i.e., DLPFC, VLPFC, and MPFC), thus suggesting this association may influence cortical areas involved in executive functions and decision-making processes. In fact, β-amyloid has been shown to correlate with cognitive performance [8, 25], and we found that β-amyloid deposition in the frontal and temporal lobes was also associated with poor attention performance in PD patients.
It has been proposed that β-amyloid may contribute to chronic microglial activation resulting in a sustained production of pro-inflammatory mediators [12], which ultimately may lead to further neurodegeneration and disease progression. This observation is consistent with previous reports showing a relationship between first-generation biomarkers of microglia ([11C]-PK11195) and [11C]-PIB binding in AD and PD patients with dementia [26].
However, the exact role of microglial activation in PD is still not clear, as earlier investigations have been inconclusive. While some studies detected increased neuroinflammation in PD patients compared to HCs [27], others have not [28]. Interestingly, for the first time, our dual-tracer PET study revealed that PIB-positive PD patients with shorter disease duration had significantly higher [18F]-FEPPA uptake than patients with a longer disease duration, thus possibly suggesting a neuroprotective/reparative function [29] of microglia activation during early stages of disease. This is consistent with previous findings in AD studies [30] where microglial activation appeared more evident at the prodromal and possibly at the preclinical stage of AD. Thus, one possible interpretation is that increased microglial activation in earlier stages might play a beneficial and neuroprotective role in the clinical progression of the disease. However, chronic overactivation and concomitant pathologies, such as abnormal protein accumulation, may cause microglia to exhibit their neurotoxic phenotype [29] with further neurodegeneration and amyloid accumulation, as our patients showed a positive association between disease duration and amyloid deposition in various frontal and temporal regions.
Consistent with other studies, no correlations were detected between [18F]-FEPPA uptake and antiparkinsonian medication, disease severity, and duration [27].
Our study has a few limitations. Overall, exploration and quantification of TSPO expression remains a challenge, considering the rs6971 polymorphism and the need for calculating the plasma free fraction (fp) values for TSPO tracers.
Another important limitation is to consider the lack of specificity of TSPO binding for activated microglia, since TSPO is also expressed on astrocytes [31]. Further, we cannot differentiate between the two phenotypes of microglia (i.e., M1 and M2), which exhibit pro-inflammatory and anti-inflammatory properties [29].
Conclusion
To our knowledge, this is the first dual-tracer study to explore the relationship between microglial activation and β-amyloid accumulation in patients with PD using a second-generation TSPO radiotracer. Given the methodological limitations of previous investigations, we quantified activated microglia by using a novel radioligand exhibiting optimal chemical and pharmacokinetic properties, as well as a full kinetic model with an arterial input function. We demonstrate, for the first time, significant interactions between beta-amyloid and activated microglia affecting cognitive performance in PD. Cognitive complications are increasingly recognized as important contributors to long-term disability in PD; however, exact pathophysiological mechanisms are still unknown. The use of in vivo imaging biomarkers could allow early detection of functional hallmarks, enabling therapeutic interventions to prevent further neurodegeneration and disease progression.
Current findings suggest that elevated microglial activation early in the disease onset may be neuroprotective and failing to appropriately adapt to amyloid accumulation might be an underlying mechanism for further neurodegeneration. However, increased microglial activation at earlier stages of the disease could also drive other pathogenic mechanisms that become more independent of inflammation later with disease progression. Studies in preclinical PD cohorts, such as patients with idiopathic REM sleep behavior disorder, observed increased microglia activation in the substantia nigra [32] and occipital lobe [33]. These findings suggest that early dysregulated microglial activation may contribute to the development of PD pathology. The concept of an early inflammatory response in the development of PD has important implications for preventive and therapeutic strategies for PD as recent findings revealed a significant reduction of TSPO binding in PD patients after being treated with an inhibitor of microglial activity [34].
Further studies will be needed to identify the exact role of microglial activation in the development and progression of PD and if amyloid deposits cause neuroinflammation and further neurodegeneration or if increased microglia activation develops as a protective response.
References
Chaudhuri KR, Healy DG, Schapira AHV, National Institute for Clinical Excellence (2006) Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 5:235–245 http://linkinghub.elsevier.com/retrieve/pii/S1474442206703738. Accessed November 4, 2018
Petrou M, Dwamena BA, Foerster BR, MacEachern MP, Bohnen NI, Müller MLTM, Albin RL, Frey KA (2015) Amyloid deposition in Parkinson’s disease and cognitive impairment: a systematic review. Mov Disord 30:928–935
Imamura K, Hishikawa N, Sawada M et al (2003) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol 106:518–526 http://springerlink.bibliotecabuap.elogim.com/10.1007/s00401-003-0766-2. Accessed November 4, 2018
Suridjan I, Pollock BG, Verhoeff NPLG, Voineskos AN, Chow T, Rusjan PM, Lobaugh NJ, Houle S et al (2015) In-vivo imaging of grey and white matter neuroinflammation in Alzheimer’s disease: a positron emission tomography study with a novel radioligand, “18 F”-FEPPA. Mol Psychiatry 20:1579–1587
Edison P, Ahmed I, Fan Z et al (2013) Microglia, amyloid, and glucose metabolism in Parkinson’s disease with and without dementia. Neuropsychopharmacology 38:938–949 http://www.nature.com/articles/npp2012255. Accessed November 4, 2018
Masters CL, Simms G, Weinman NA et al (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82:4245–4249 http://www.ncbi.nlm.nih.gov/pubmed/3159021. Accessed November 4, 2018
Mastaglia FL, Johnsen RD, Byrnes ML et al (2003) Prevalence of amyloid-beta deposition in the cerebral cortex in Parkinson’s disease. Mov Disord 18:81–86 http://doi.wiley.com/10.1002/mds.10295. Accessed November 4, 2018
Petrou M, Bohnen NI, Muller MLTM et al (2012) Abeta-amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology 79:1161–1167 http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3525303&tool=pmcentrez&rendertype=abstract
Mielke MM, Wiste HJ, Weigand SD, Knopman DS, Lowe VJ, Roberts RO, Geda YE, Swenson-Dravis DM et al (2012) Indicators of amyloid burden in a population-based study of cognitively normal elderly. Neurology. 79:1570–1577
Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N, Villemagne VL, O'Keefe G et al (2008) Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 79:1331–1338
Gomperts SN, Locascio JJ, Rentz D et al (2013) Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology 80:85–91 7p. Accessed at: http://search.ebscohost.com/login.aspx?direct=true&db=ccm&AN=108089057&site=ehost-live
Prokop S, Miller KR, Heppner FL (2013) Microglia actions in Alzheimer’s disease. Acta Neuropathol 126:461–477 http://www.ncbi.nlm.nih.gov/pubmed/24224195. Accessed November 4, 2018
Litvan I, Goldman JG, Tröster AI et al (2012) Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov Disord 27:349–356 http://doi.wiley.com/10.1002/mds.24893. Accessed October 1, 2018
Wilson AA, Garcia A, Parkes J et al (2008) Radiosynthesis and initial evaluation of [18F]-FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl Med Biol 35:305–314 http://linkinghub.elsevier.com/retrieve/pii/S0969805108000073. Accessed November 4, 2018
Mathis CA, Bacskai BJ, Kajdasz ST et al (2002) A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett 12:295–298 http://www.ncbi.nlm.nih.gov/pubmed/11814781. Accessed November 4, 2018
Rusjan PM, Wilson AA, Bloomfield PM, Vitcu I, Meyer JH, Houle S, Mizrahi R (2011) Quantitation of translocator protein binding in human brain with the novel radioligand [18F]-FEPPA and positron emission tomography. J Cereb Blood Flow Metab 31:1807–1816 http://journals.sagepub.com/doi/10.1038/jcbfm.2011.55. Accessed October 1, 2018
Rusjan P, Mamo D, Ginovart N, Hussey D, Vitcu I, Yasuno F, Tetsuya S, Houle S et al (2006) An automated method for the extraction of regional data from PET images. Psychiatry Res - Neuroimaging 147:79–89
Bencherif B, Stumpf MJ, Links JM et al (2004) Application of MRI-based partial-volume correction to the analysis of PET images of mu-opioid receptors using statistical parametric mapping. J Nucl Med 45:402–408 http://www.ncbi.nlm.nih.gov/pubmed/15001679. Accessed November 4, 2018
Lopresti BJ, Klunk WE, Mathis CA et al (2005) Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med 46:1959–1972 http://www.ncbi.nlm.nih.gov/pubmed/16330558. Accessed October 1, 2018
Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, Madison C, Ayakta N, Ghosh PM, la Joie R, Arthur-Bentil SK et al (2015) Existing Pittsburgh Compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain. 138:2020–2033
Villemagne VL, Pike KE, Chételat G et al (2011) Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol 69:181–192 http://doi.wiley.com/10.1002/ana.22248. Accessed October 1, 2018
Lahiri DK, Nurnberger JI (1991) A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res 19:5444 http://www.ncbi.nlm.nih.gov/pubmed/1681511. Accessed November 4, 2018
Mizrahi R, Rusjan PM, Kennedy J, Pollock B, Mulsant B, Suridjan I, de Luca V, Wilson AA et al (2012) Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [18F]-FEPPA. J Cereb Blood Flow Metab 32:968–972
Alvarez-Erviti L, Couch Y, Richardson J et al (2011) Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res 69:337–342 http://linkinghub.elsevier.com/retrieve/pii/S0168010211000101. Accessed November 4, 2018
Akhtar RS, Xie SX, Chen YJ, Rick J, Gross RG, Nasrallah IM, van Deerlin VM, Trojanowski JQ et al (2017) Regional brain amyloid-β accumulation associates with domain-specific cognitive performance in Parkinson disease without dementia. PLoS One 12:1–18. https://doi.org/10.1371/journal.pone.0177924
Fan Z, Aman Y, Ahmed I et al (2015) Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement 11:608–21.e7 https://linkinghub.elsevier.com/retrieve/pii/S1552526014025011. Accessed November 4, 2018
Gerhard A, Pavese N, Hotton G et al (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21:404–412 http://linkinghub.elsevier.com/retrieve/pii/S0969996105002263. Accessed November 4, 2018
Kobylecki C, Counsell SJ, Cabanel N et al (2013) Diffusion-weighted imaging and its relationship to microglial activation in parkinsonian syndromes. Parkinsonism Relat Disord 19:527–532 https://linkinghub.elsevier.com/retrieve/pii/S1353802013000527. Accessed November 4, 2018
Cai Z, Hussain MD, Yan LJ (2014) Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci 124:307–321
Hamelin L, Lagarde J, Dorothée G, Leroy C, Labit M, Comley RA, de Souza LC, Corne H et al (2016) Early and protective microglial activation in Alzheimer’s disease: a prospective study using18F-DPA-714 PET imaging. Brain. 139:1252–1264
Lavisse S, Guillermier M, Hérard A-S et al (2012) Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci 32:10809–10818 http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.1487-12.2012. Accessed November 4, 2018
Stokholm MG, Iranzo A, Østergaard K et al (2017) Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. Lancet Neurol 16:789–796 https://linkinghub.elsevier.com/retrieve/pii/S1474442217301734. Accessed November 4, 2018
Stokholm MG, Iranzo A, Østergaard K et al (2018) Extrastriatal monoaminergic dysfunction and enhanced microglial activation in idiopathic rapid eye movement sleep behaviour disorder. Neurobiol Dis 115:9–16 https://linkinghub.elsevier.com/retrieve/pii/S0969996118300494. Accessed November 4, 2018
Jucaite A, Svenningsson P, Rinne JO, Cselényi Z, Varnäs K, Johnström P, Amini N, Kirjavainen A et al (2015) Effect of the myeloperoxidase inhibitor AZD3241 on microglia: a PET study in Parkinson’s disease. Brain 138:2687–2700 https://academic.oup.com/brain/article-lookup/doi/10.1093/brain/awv184. Accessed November 4, 2018
Funding
This study was supported by the National Parkinson Foundation and the Canadian Institutes of Health Research (MOP-136778). Antonio Strafella is supported also by the Canada Research Chair program from the Canadian Institutes of Health Research. Dr. Strafella has received honoraria from GE Healthcare Canada.
Author information
Authors and Affiliations
Contributions
Christine Ghadery: acquisition of data, analysis and interpretation of data, and drafting of the manuscript. Yuko Koshimori: acquisition of data and critical revision of the manuscript for intellectual content. Leigh Christopher: acquisition of data and critical revision of the manuscript for intellectual content. Jinhee Kim: analysis and interpretation of data and critical revision of the manuscript for intellectual content. Pablo Rusjan: methodological PET supervision of data analyses and critical revision of the manuscript for intellectual content. Anthony E. Lang: critical revision of the manuscript for intellectual content. Sylvain Houle: critical revision of the manuscript for intellectual content. Antonio P. Strafella: study concept and design, critical revision of the manuscript for intellectual content, acquisition of funding, and overall study supervision.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declared that they have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ghadery, C., Koshimori, Y., Christopher, L. et al. The Interaction Between Neuroinflammation and β-Amyloid in Cognitive Decline in Parkinson’s Disease. Mol Neurobiol 57, 492–501 (2020). https://doi.org/10.1007/s12035-019-01714-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-01714-6