
Abstract
As mitochondrial dysfunction is evident in neurodegenerative disorders that are accompanied by pain, we generated inducible mutant mice with disruption of mitochondrial respiratory chain complex IV, by COX10 deletion limited to sensory afferent neurons through the use of an Advillin Cre-reporter. COX10 deletion results in a selective energy-deficiency phenotype with minimal production of reactive oxygen species. Mutant mice showed reduced activity of mitochondrial respiratory chain complex IV in many sensory neurons, increased ADP/ATP ratios in dorsal root ganglia and dorsal spinal cord synaptoneurosomes, as well as impaired mitochondrial membrane potential, in these synaptoneurosome preparations. These changes were accompanied by marked pain hypersensitivity in mechanical and thermal (hot and cold) tests without altered motor function. To address the underlying basis, we measured Ca2+ fluorescence responses of dorsal spinal cord synaptoneurosomes to activation of the GluK1 (kainate) receptor, which we showed to be widely expressed in small but not large nociceptive afferents, and is minimally expressed elsewhere in the spinal cord. Synaptoneurosomes from mutant mice showed greatly increased responses to GluK1 agonist. To explore whether altered nucleotide levels may play a part in this hypersensitivity, we pharmacologically interrogated potential roles of AMP-kinase and ADP-sensitive purinergic receptors. The ADP-sensitive P2Y1 receptor was clearly implicated. Its expression in small nociceptive afferents was increased in mutants, whose in vivo pain hypersensitivity, in mechanical, thermal and cold tests, was reversed by a selective P2Y1 antagonist. Energy depletion and ADP elevation in sensory afferents, due to mitochondrial respiratory chain complex IV deficiency, appear sufficient to induce pain hypersensitivity, by ADP activation of P2Y1 receptors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondrial dysfunction is associated with a wide range of neuropathic pain states [1], including peripheral neuropathies induced by anti-cancer [2, 3] and anti-retroviral therapy [4], diabetic hyperglycaemia [5, 6] and traumatic nerve injury [7]. Neurodegenerative disorders displaying pain phenotypes, such as Charcot-Marie-Tooth disease and multiple sclerosis [8,9,10,11], also display mitochondrial defects [12,13,14,15].
A number of processes ensuing from mitochondrial dysfunction have been implicated in leading to pain hypersensitivity. These include neurotoxicity from the production of reactive oxygen species (ROS), disturbances in mitochondrial fission/fusion dynamics and frank depletion of ATP levels [2, 16,17,18,19,20,21,22,23,24]. The reduced efficiency of Na+/K+ ATPase resulting from ATP depletion has been suggested to cause increased excitability and neuropathy-associated ectopic firing in primary sensory afferents [25]. In animal models of chemotherapy-induced peripheral neuropathy (CIPN), where mitochondrial dysfunction is associated with hyperalgesia, beneficial effects of antioxidants and a selective inhibitor of mitochondrial fission have been reported [19, 26,27,28,29,30]. In peripheral neuropathy patients, while currently available antioxidants display partial reversal of hypersensitivity, this is relatively modest in magnitude [19], suggesting that mechanisms additional to ROS toxicity play a significant part in driving pain hypersensitivity.
Genetically modified mouse models of disrupted mitochondrial function in sensory neurons could potentially help to elucidate downstream pathogenic mechanisms including those responsible for causing pain hypersensitivity [31]. Interestingly, inducible disruption of mitochondrial respiratory chain complex IV function in the nervous system (targeting the COX10 gene) produced a delayed onset of neuropathology associated with energy deficit in the absence of ROS production [32]. Using a similar approach here, but regionally targeting the mutation to primary sensory neurons by crossing to an Advillin gene Cre-reporter [33], we have been able to elucidate the functional outcome of selective mitochondrial energy depletion in sensory afferents. We report robust pain hypersensitivity, implicate activation of the ADP-sensitive P2Y1 receptor in its generation and demonstrate effective analgesia in this model with a highly selective P2Y1 antagonist.
Methods
Animals
All animal breeding, maintenance and experimental procedures were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986 and were approved by the University of Edinburgh’s Local Ethical Review Board. Animals were housed under a 12-h light-dark cycle and given access to food and water ad libitum.
C57/Bl6J double transgenic mice were produced using an inducible Cre-LoxP recombination system. Mutants expressed floxed COX10fl/fl, encoding a farnesyl transferase, essential for the function of mitochondrial respiratory chain complex IV [34] and tamoxifen-inducible Cre-recombinase [35] under the promoter for Advillin (Adv), a peripheral sensory neuron-specific marker expressed in over 90% of dorsal root ganglion (DRG) cells [33]. This produced DRG-specific mitochondrial mutants (COX10fl/flAdv-CreERT2+/−), which were compared with COX10-floxed, but Cre-lacking controls (COX10fl/flAdv-CreERT2−/−) that had been similarly treated with tamoxifen. COX10 deletion in the double transgenics was achieved by intraperitoneal injection of tamoxifen (Sigma). Tamoxifen was dissolved in 10:1 sunflower oil/clinical grade ethanol at 10 mg/ml and injected intraperitoneally at a dose of 0.18 mg/g body weight per day from P28 for 5 consecutive days. Experiments were carried out 6.5 weeks later using mice of either sex. Tissue-specific gene deletion was previously confirmed using Southern blots [32] and in situ hybridisation [36]. Disruption of complex IV by COX10 deletion using a constitutive Cre promoter has been reported to represent a specific energy failure phenotype with minimal ROS production for up to 4 months [32].
Sensory/Motor Testing In Vivo
Six and a half weeks following induction with tamoxifen, mutant and control mice were weighed and general motor function/co-ordination was tested by Rotarod (UgoBasile, Varese), by measuring the duration of maintained running at 24 rpm for 4 cycles of 2 min with 5 min intervals of rest in between the running cycles. In addition, hindlimb muscle strength was assessed by Grip Strength Meter (BioGS3, BioSeb) as maximum maintained force in g. Mechanical and thermal nociceptive sensitivity were also assessed using standard behavioural tests, during which, the investigator was blinded to genotype. Mechanical nociception was assessed as paw withdrawal threshold (in g) from force-calibrated von Frey nylon filaments (Stoetling, Illinois). Nociceptive heat sensitivity was measured as paw withdrawal latency (in s) using Hargreaves’ infrared apparatus (Linton Instrumentation), set to a maximum temperature of 52 °C and a cut-off time of 20 s. Testing was always separated by at least 5 min to avoid sensitisation of responses. Nociceptive cold sensitivity was measured as cold-plate standing duration (in s) using a simple thermal choice chamber with a metal plate kept at the temperature of iced water (~ 4 °C) versus a polystyrene platform at room temperature. In experiments assessing drug effects, time-course data were obtained for the von Frey and Hargreaves’ reflex paw withdrawal tests, but in the cold avoidance test, readings were taken just before and 30 min after drug administration to avoid any effect of learning due to repeated testing.
Assays of Mitochondrial Function
To quantify any changes in ATP and ADP levels in sensory afferent neuronal cell bodies of mutant mice compared to controls, DRG were rapidly removed and snap frozen on dry ice before weighing and homogenisation in ice cold 1 M perchloric acid. After micro-titration to pH 7.0 with 4 M KOH, samples were centrifuged to obtain neutralised, de-proteinised extracts. Aliquots were added to 96-well assay plates and assayed independently for ATP and ADP using fluorometric enzyme-linked assay kits (Abcam, ab83355 and ab83359), according to the manufacturer’s protocols. Internal standard curves allowed quantification, and after volume correction, values were expressed as nmol/mg DRG wet weight. To assess any alterations in relative ADP:ATP levels in dorsal spinal cord (where sensory afferent terminals are located), dorsal, as well as ventral, spinal cord synaptoneurosomes (see below) were assayed using an enzyme-linked bioluminescence kit (Abcam, ab65313) [18], which displays very high sensitivity measurement of the nucleotides, but in terms of relative, rather than absolute concentrations.
Mitochondrial respiratory chain functionality was assessed by sequential histochemical evaluation of cytochrome c oxidase (COX; complex IV) and succinate dehydrogenase (SDH; complex II) activity [14]. In brief, COX media (100 μM cytochrome c, 4 mM diaminobenzidine tetrahydrochloride and 20 μg ml−1 catalase in 0.1 M phosphate buffer pH 7.0) was applied to snap-frozen cryosections of whole spinal cord with DRG for 40 min, washed in PBS before SDH medium (130 mM sodium succinate, 200 mM phenazine methosulphate, 1 mM sodium azide and 1.5 mM nitroblue tetrazolium in 0.1 M phosphate buffer pH 7.0) was applied for a further 20 min at 37 °C.
To assess whether AdvCOX10 mutants showed elevated levels of ROS compared to controls, dorsal and ventral spinal cord synaptoneurosomes (see below) were loaded with the broad-spectrum fluorometric ROS reporter, 5-(and 6)-chloromethyl-2′,7′-dichlorohydrofluorescein diacetate (CM-H2DCFDA; Molecular Probes), 2 μM, 30 min, 37 °C [37]. Fluorescence was read at excitation 495 nm, emission 525 nm, and compared in each case to a maximal positive control provided by the ROS donor tert-butyl hydroperoxide (t-BuOOH; Sigma), 5 mM, 30 min, 37 °C [17, 21].
Assessment of relative mitochondrial membrane potential was carried out using the ratiometric fluorescent probe JC-1 (Molecular Probes). JC-1 is a cationic carbocyanine that accumulates in mitochondria according to their membrane potential and at higher concentration transitions from the green-fluorescing monomer to a red-fluorescing aggregate [38, 39]. Synaptoneurosomes were prepared from dorsal or ventral spinal cord of mutant or control mice (see below), aliquoted into 48-well plates and pre-incubated at 37 °C before addition of 1.5 μM JC-1 for 30 min before fluorescent measurement at excitation, 488 nm, emission 525 and 595 nm. In some control experiments, samples were incubated with the mitochondrial respiratory chain uncoupler, carbonyl cyanide-p-trifluomethoxyphenylhydrazone (FCCP; 1 μM) for 45 min to depolarise the mitochondrial membrane potential, and in all cases, this resulted in around 75% reduction in the red/green emission ratio.
Synaptoneurosome Preparation and Measurement of Ca2+ Fluorescence Responses
A key measure of the significance of biochemical changes in nociceptive primary sensory neurons is the impact on neurotransmission at synapses between their central terminals and postsynaptic neurons in spinal dorsal horn. To investigate this in AdvCOX10 mutant mice, compared to controls, we prepared synaptoneurosomes from dorsal spinal cord. Synaptoneurosomes are re-sealed pre-synaptic and closely apposed post-synaptic elements, freshly prepared from CNS tissue [40, 41]. Protocols that optimise metabolic and ionic integrity in synaptoneurosomes have been recently developed in our laboratory so dynamic Ca2+ fluorescence responses to receptor stimuli can be measured [42, 43]. Thus, we were able to assay neurotransmission at the first central synapses of nociceptive afferents, using as a stimulus, an excitatory receptor abundantly expressed on presynaptic terminals of nociceptive afferents (the GluK1, kainate receptor) and as a read-out, Ca2+ fluorescence, which reflects mainly the signal from the postsynaptic compartment [42, 43].
Lumbar spinal cord segments (L4–6) were rapidly removed from animals after cull and immediately homogenised in ice-cold, highly oxygenated medium using a hand-held Teflon-glass homogeniser. The medium was divalent ion-free Krebs-Henseleit buffer, additionally containing HEPES (10 mM, pH 7.4), sodium pyruvate (0.5 mM), glutathione (5 μM), creatine phosphate (2 mM), magnesium chloride (5 mM), sodium kynurenate (0.3 mM) and type III protease inhibitor cocktail (Calbiochem, 1:1000). The homogenate was rapidly syringe-filtered through a 30-μm nylon mesh filter and then through a 5-μm pore mixed cellulose fibre matrix filter (Millipore) before centrifugation at 1500 g for 15 min at 4 °C. The pellet was resuspended in ice-cold highly oxygenated medium as above but lacking magnesium ions and kynurenate. Electron microscopic analysis of this subcellular fraction revealed the abundant presence of small dual-compartment re-sealed profiles consistent with the previously described characteristics of synaptoneurosomes [40]. Aliquots of synaptoneurosome suspension (150 μl) were dispensed into 48-well plates, and Calcium-5 (a sensitive, no-wash Ca2+ fluorophore, formulated with an extracellular fluorescence quenching agent; Molecular Devices) was added in 50-μl resuspension buffer containing 8 mM CaCl2, before incubation for 45 min at 37 °C under 95% O2/5% CO2. For the current series of experiments, Concanavalin A (ConA; 300 μg/ml) was added 10 min prior to assay, to minimise GluK1 receptor desensitisation [44,45,46]. Plates were transfered to the plate reader (Varioskan Flash, ThermoScientific; thermostatted at 28 °C). Pharmacological agents were then added (modulatory antagonists, then agonists, before GluK1 activator) immediately before recording at 12 × 30 s intervals or 6 × 1 min intervals. Intracellular Ca2+ fluorescence was read at excitation 488 nm, emission 518 nm. Ionomycin (10 μM) and basal measurements were included in every plate to calibrate the dynamic range of the assay. The following pharmacological agents were used: the selective GluK1 agonist, 5-iodowillardiine (5-IW; Tocris); the selective GluK1 antagonist, ACET (Tocris); the selective GluA agonist, R,S-AMPA (Abcam); the inhibitor of synaptic vesicle exocytosis, tetanus toxin (Sigma-Aldrich); the inhibitors of glutamate receptor desensitisation, Concanavalin A (ConA, Sigma-Aldrich) and cyclothiazide (Tocris); the selective P2Y1 agonist, MRS 2365 (Tocris); the selective P2Y1, P2Y12 and P2Y13 antagonists, MRS 2500, AZD 1283 and MRS 2211, respectively (Tocris); the selective AMP-Kinase (AMPK) activators, AICAR and A697662 (Abcam); the AMPK inhibitor, dorsomorphin (Abcam); and the Ca2+ ionophore, ionomycin (Abcam). The concentrations used for each agent were selected on the basis of publications showing substantial efficacy in cellular assays or at generally 10–20-fold the reported in vitro EC50/IC50 concentrations.
Immunofluorescence Histochemistry
DRGs from spinal segments L4–6 were dissected, rapidly embedded in OCT cryo-sectioning medium (Thermo Scientific) and frozen on dry ice. Twenty-micrometre sections were cut by cryostat and mounted onto poly-L-lysine coated slides (Thermo Scientific). Sections were washed in Tris-buffered saline (TBS) pH 7.60 and then incubated for 1 h at room temperature in blocking buffer (10% normal donkey serum, 4% fish skin gelatin, 0.2% Triton X-100 in TBS) prior to overnight incubation at 4 °C with a combination of primary antibodies in buffer (4% normal donkey serum, 4% fish skin gelatin and 0.2% Triton X-100 in TBS). The primary antibodies used were rabbit polyclonal anti-GluK1 (Alomone, AGC-008, 1:500), chicken polyclonal anti-peripherin (Abcam, ab39374, 1:7500), guinea pig polyclonal anti-TRPV1 (Abcam, ab10295, 1:3000), rabbit polyclonal anti-P2Y1 (Alomone, APR-021, 1:650). Sections were washed in TBS and incubated for 1 h at room temperature with a combination of secondary antibodies or Alexa Fluor-488 conjugated isolectin IB4 (Invitrogen; I21411; 1:3000) in buffer (4% normal donkey serum, 4% fish skin gelatin in TBS). Extensively cross-adsorbed secondary antibodies were donkey anti-guinea pig CF-405 (Sigma-Aldrich; SAB4600231; 1:600), donkey anti-rabbit Alexa Fluor-568 (Life Technologies; A10042; 1:600) and donkey anti-chicken CF-488 (Sigma-Aldrich; SAB4600031; 1:600). Sections were washed three further times in TBS and then mounted in ProLong® Gold Antifade (Life Technologies) before visualisation through a × 20 objective using a Nikon A1R confocal microscope. Standard primary antibody omission or blocking peptide controls, wherever possible, confirmed that non-specific staining was minimal.
Western Blotting
Samples were solubilised in × 2 Laemmli buffer: 50 mM Tris, pH 7.4 containing 5% mercaptoethanol and 2% sodium dodecyl sulphate, SDS, Sigma). The samples were thoroughly mixed and heated at 80 °C for 10 min and then either used immediately or stored at − 20 °C. Proteins were separated by SDS-PAGE (SDS-polyacrylamide gel electrophoresis) using the NuPage XCell SureLock™ Minicell gel electrophoresis system (Invitrogen). Samples (5 μl) were mixed with 1-μl loading solution (0.04% w/v Bromophenol Blue in glycerol) and added to the wells of 4–12% Bis-Tris NuPage gels. Pre-stained standard molecular weight proteins (Spectra Multicolor Broad Range Protein Ladder; Thermo-Pierce) were run alongside. Samples were electrophoresed using NuPage MOPS running buffer (Invitrogen) under a 200 V potential difference. Proteins were transferred to polyvinylidene difluoride membrane (Immobilon-PSQ, Millipore) at 30 V in transfer buffer (5% NuPage transfer buffer, 10% methanol) for 90 min. Membranes were incubated for 60 min in blocking buffer (5% bovine serum albumin (BSA) in 0.1 M Tris-buffered saline (TBS), pH 7.60, with 0.1% Tween-20) to reduce non-specific binding. Membranes were then washed and incubated overnight at 4 °C in 2% BSA in 0.1 M TBS with 0.1% Tween-20, containing rabbit monoclonal anti-phospho-AMPKα (Thr172) (Cell Signalling Technology, #2535, 1:500). Membranes were washed and incubated for 50 min at room temperature with peroxidase-conjugated goat anti-rabbit secondary antibody (Chemicon, 1:40,000). Bands were detected by peroxidase-linked enhanced chemiluminescence and X-ray film (ECL reagent; Immobilon, Millipore, and Hyperfilm; GE Healthcare). After washing, membranes were re-probed with mouse monoclonal anti-GAPDH (Millipore, #MAB 374, 1:5000) for 30 min at room temperature. Membranes were washed and incubated with peroxidase-conjugated goat anti-mouse secondary antibody (Chemicon, 1:40,000) for 30 min at room temperature. Bands were detected by ECL reagent and X-ray film, as before. Films were scanned and band intensities were quantified by densitometry using Image J.
Non-linear Curve-Fitting and Statistical Analysis
All data analysis was carried out using GraphPad Prism. Non-linear curve-fitting used a sigmoidal dose-response (variable slope) model. Data in two-group format were analysed statistically by Student’s t test. Comparisons between more than two groups were made by one-way ANOVA with Tukey’s or Dunnett’s test or by two-way ANOVA with Bonferroni’s test.
Results
Assessments of Mitochondrial Function Confirm an Energy Failure Phenotype in Sensory Afferents of AdvCOX10 Mutants
We used a range of distinct approaches to establish that disruption of mitochondrial energy production was occurring in neuronal sensory pathways of the AdvCOX10 mutant mice at 6.5 weeks after the start of tamoxifen induction. In flash-frozen DRG, we used micro-scaled, enzyme-linked fluorometric assays to quantify ATP and ADP content. Figure 1a shows that ATP concentrations were significantly reduced and ADP concentrations were significantly increased in AdvCOX10 mutant compared to control mice. Correspondingly, the ADP/ATP ratio was more than two-fold greater in AdvCOX10 mutants than in controls.

Cellular changes in AdvCOX10 mutant mice. a ATP/ADP concentrations in flash-frozen lumbar DRGs from AdvCOX10 mutant mice, compared to controls, were measured using fluorometric enzyme assay kits in 96-well plates. Values are means ± SEM from 8 control and 7 mutant mice. The left panel shows absolute concentrations in nmol/mg DRG wet weight, while the right panel shows ADP/ATP ratio. ATP concentrations were significantly reduced in mutant compared to control, ADP concentrations were significantly raised, and the ADP/ATP ratio was significantly greater (***p < 0.001, **p < 0.01 and ***p < 0.001, respectively, t tests). b Deficits in functional activity of mitochondrial respiratory chain complex IV, with intact SDH activity, were revealed by blue staining using a sequential histochemical assay for COX and SDH (COX/SDH). Cells or neuropil with intact complex IV are labelled brown, whereas complex IV-deficient cells with blue staining, lack the brown stain. Numerous primary sensory neurons in DRG of AdvCOX10 mutants showed prominent blue staining, while this was never observed in cells or the dense neuropil of spinal cord ventral horn. No blue staining was detected in either DRG or ventral horn of control animals. Typical images are shown for control and AdvCOX10 mutants. Scale bar = 100 μm. c Relative ADP/ATP concentration ratios were measured in dorsal and ventral spinal cord synaptoneurosomes from control and AdvCOX10 mutant mice, using a high sensitivity (ratiometric) enzyme-linked bioluminescence assay. The relative ADP/ATP ratio was significantly greater in dorsal horn of AdvCOX10 mutants compared to control animals (*p < 0.05) and compared to ventral horn of mutants (†p < 0.05); analysis by one-way ANOVA with Tukey’s test, n = 7 in each case. d The relative mitochondrial membrane potential in synaptoneurosomes from dorsal and ventral spinal cord (segments L4–6) of AdvCOX10 mutants and control mice was measured using the fluorometric probe JC-1. JC-1 monomer emits at 525 nm, but accumulates in mitochondria according to membrane potential, which results in aggregates that emit at 595 nm (red). The red/green fluorescence ratio reflecting mitochondrial membrane potential was significantly reduced in dorsal, but not ventral spinal cord of AdvCOX10 mutant mice compared to controls (*p < 0.05, t test). Values are means ± SEM, n = 5 in each case. e CM-H2DCFA fluorescence readings of dorsal and ventral spinal cord synaptoneurosomes from control and AdvCOX10 mutant mice, reflecting relative levels of ROS. Values were normalised to those in the presence of a positive control, the ROS donor, t-BuOOH (5 mM, 30 min). In all cases, values were less than 15% of the positive control, with no significant differences between dorsal and ventral cord of either mutant or control animals, by one-way ANOVA with Tukey’s test, n = 4 in each case
To measure ADP/ATP ratios in the region of primary afferent neuron termination, in spinal dorsal horn, we used an enzyme-linked, bioluminescent ratiometric assay in synaptoneurosomes from dorsal and ventral spinal cord of AdvCOX10 mutants and control mice (Fig. 1c). ADP/ATP ratios were significantly increased in dorsal (but not ventral) spinal cord synaptoneurosomes from AdvCOX10 mutant mice compared to controls.
In addition, we used a sequential histochemical staining approach to identify DRG cells showing cytochrome c oxidase deficiency (lacking complex IV activity with intact complex II). Figure 1b shows that numerous complex IV-deficient DRG cell bodies lacking the brown staining and identified by the greatly increased complex II activity, with blue staining, were evident in AdvCOX10 mutants, but not in control mice. In contrast, spinal cord ventral horn neurons showed no blue staining that would have been indicative of complex IV deficiency.
We further evaluated mitochondrial membrane potential in dorsal/ventral spinal cord synaptoneurosomes using the ratiometric fluorescent dye, JC-1 (Fig. 1d). In dorsal spinal cord, the JC-1 595:525 nm fluorescence ratio (a measure of effective mitochondrial membrane potential) was significantly reduced in AdvCOX10 mutants compared to controls. No such change was apparent in ventral spinal cord samples, consistent with the expectation that the Adv promoter would target the COX10 deletion to neurons of peripheral origin, that is sensory afferents terminating in dorsal rather than ventral spinal cord. In support of the idea that JC-1 was effectively reporting mitochondrial membrane potential here, the mitochondrial depolarising agent and ATP-synthesis uncoupler (FCCP, 1 μM, 45 min, 37 °C) significantly reduced 595:525 fluorescence ratios in all cases (p < 0.01, n = 5, t test). The mean percentage inhibition values due to FCCP were 74.3% and 73.5% in control dorsal and ventral spinal cord samples, with corresponding values of 74.5% and 57.4% in AdvCOX10 mutants.
To assess whether elevated ROS levels (a possible contributor to hypersensitivity) were present in AdvCOX10 mutants compared to controls, we prepared synaptoneuromes from dorsal and ventral spinal cords and loaded these with the broad-spectrum fluorometric ROS reporter, CM-H2DCFA. In comparison with a robust positive control signal from the potent ROS donor, t-BuOOH, we found no evidence of elevated ROS levels in AdvCOX10 mutant mice relative to controls (Fig. 1e). Fluorescence values from the ROS reporter were less than 15% of those in the presence of t-BuOOH in all cases, with no significant difference between dorsal spinal cord of AdvCOX10 mutants or control mice, and similar results in ventral spinal cord.
All these observations are consistent with the idea that disruption of COX10 produces predominantly an energy failure phenotype that is not associated with excess ROS production [32].
AdvCOX10 Mutants Display Marked Hypersensitivity in Mechanical and Thermal (Hot and Cold) Pain Behaviours
To assess any functional sensory consequences of Adv-directed COX10 deletion, we used von Frey filament testing of mechanical paw withdrawal threshold (Fig. 2a), Hargreaves’ apparatus testing of thermal paw withdrawal threshold to noxious heat (Fig. 2b) and noxious cold plate standing duration (Fig. 2c). In each test, there was greater than 2-fold hypersensitivity in AdvCOX10 mutants compared to controls (statistically significant in each case). These findings indicate that disrupted mitochondrial energy production in sensory neurons leads to marked pain hypersensitivity. Figure 2d shows that the general health (weight gain) and motor function in AdvCOX10 mutants were not significantly different compared to controls at 6.5 weeks after completing tamoxifen treatment. This confirms the sensory modality-specific hypersensitivity in the mutants, while general health and motor function showed no discernible deficit.

Changes in nociceptive behavioural responses and general physiological characteristics of AdvCOX10 mutant mice. a The threshold for paw withdrawal responses to mechanical stimulation with von Frey filaments was significantly reduced in mutants compared to control mice. Values are means ± SEM, n = 17 for control and 13 for mutant, ***p < 0.001 by t test. b The latency for paw withdrawal responses from a noxious heat stimulus (Hargreaves’ test) was significantly reduced in mutant mice. Values are means ± SEM, n = 5 in each case, **p < 0.01 by t test. In vivo noxious cold avoidance behaviour in terms of standing duration on a 4 °C cold plate (before escaping to an insulated, room temperature platform) was significantly reduced in mutant mice. Values are means ± SEM, n = 6 for control and 7 for mutant, *p < 0.05 by t test. d There were no significant differences between control mice and AdvCOX10 mutants for weight, Rotarod running duration or grip strength. Values are means ± SEM, n in parentheses; t test p values were 0.57, 0.34 and 0.47, respectively
GluK1 Receptor Expression in Nociceptive Afferents
Excitatory GluK1 receptors and their mRNA are reported to be widely expressed in small nociceptive afferents and minimally expressed elsewhere in spinal cord [44, 45, 47,48,49,50]. A major role for GluK1 in particular in DRG is emphasised by the abrogation of DRG neuron excitatory responses to kainate receptor agonists in GluK1−/− mice [51, 52]. We therefore investigated whether pharmacologically targeting GluK1 would be an appropriate way to monitor responsiveness to a broad spectrum of nociceptive afferents. Dual-label immunofluorescence evaluation of GluK1 expression in DRG confirmed its selective distribution, with more than 90% of GluK1-positive cells co-staining for the unmyelinated C-fibre marker, peripherin (Fig. 3a). Both presumed peptidergic C-fibres, immunoreactive for TRPV1 [53] and presumed non-peptidergic C-fibres, labelled with isolectin IB4 [54], showed GluK1-immunoreactivity in around half of the population (Fig. 3b). There were no significant differences between control and AdvCOX10 mutant mice in the percentages of peripherin+, TRPV1+ or IB4+ DRG cells expressing GluK1.

GluK1 is widely expressed in DRG nociceptive afferents and at their central terminals in dorsal spinal cord. a Example images of dual label immunofluorescence identification of DRG cells expressing GluK1, peripherin (a marker of small nociceptive C-fibre afferents), and their co-localisation in control and AdvCOX10 mutant mice. Scale bar = 50 μm. b Quantification of results from experiments outlined in a. A substantial proportion of nociceptors labelled by peripherin, by TRPV1 (to preferentially label the peptidergic sub-class), and by IB4 (to preferentially label the non-peptidergic subclass) were each shown to co-express GluK1. No significant changes in co-expression between control and AdvCOX10 mutant mice were identified. c Concentration-dependent Ca2+ fluorescence responses of dorsal spinal cord synaptoneurosomes from control mice to the selective GluK1 agonist, 5-iodowillardiine (5-IW) and their attenuation in the presence of the selective GluK1 antagonist, ACET. Values are means ± SEM, n = 4 in each case. The EC50 for 5-IW was significantly (5.3-fold) increased in the presence of 100 nM ACET, from a mean of 242 nM to a mean of 1.29 μM (p < 0.0001, extra sum of squares F test). d Ca2+ fluorescence responses of synaptoneurosomes to a near-maximal 5-IW stimulus (2 μM) were significantly greater in mutant compared to control animals, consistent with the central sensitisation known to occur in chronic pain states (*p < 0.05). 5-IW responses were significantly lower in ventral than dorsal spinal cord in both control (†p < 0.05, n = 6) and mutant mice (†††p < 0.001, n = 6). Statistical analyses were by one-way ANOVA with Tukey’s test
Ca2+ Fluorescence Responses Elicited by GluK1 Receptor Activation in Dorsal Spinal Cord Synaptoneurosomes
In ex vivo dorsal spinal cord synaptoneurosomes from control mice, we showed that the selective GluK1 agonist, 5-iodowillardiine (5-IW; [46, 55,56,57]), produced a concentration-dependent increase in Ca2+ fluorescence with a mean EC50 value [95% confidence interval] of 242 [205/285] nM (Fig. 3c). The selective GluK1 antagonist ACET [58], at a concentration of 100 nM, produced a rightward shift in the 5-IW concentration-response curve with a 5.3-fold increase in mean EC50 value to 1.29 [0.88/1.87] μM (p < 0.0001 by extra sum of squares F test; Fig. 3c). Responses to 2 μM 5-IW were markedly reduced by 45-min pre-incubation with the inhibitor of exocytosis, tetanus toxin [59], reaching a maximum effect of 85.8 ± 4.9% inhibition (mean ± SEM, n = 4) at a concentration of 20 nM (p < 0.01 by one-way ANOVA with Dunnett’s test), consistent with a largely presynaptic site of action for 5-IW. In contrast, responses to activation of GluA receptors, which are widely distributed postsynaptically, using 20 μM AMPA (in the presence of 10 μM cyclothiazide to attenuate desensitisation [60, 61], were minimally affected (21.7 ± 8.7% inhibition at 20 nM tetanus toxin; p > 0.05). Figure 3d shows not only that Ca2+ fluorescence responses to 2 μM 5-IW were significantly greater in dorsal compared to ventral spinal cord in both control and AdvCOX10 mutant mice (p < 0.05 and p < 0.001, by one-way ANOVA with Tukey’s test), but also that dorsal responses were significantly greater in AdvCOX10 mutants compared to those in control mice (p < 0.05). This is entirely consistent with the idea that mitochondrial deficiency due to the AdvCOX10 mutation has brought about hypersensitivity at the first central synapses of nociceptors in dorsal spinal cord.
Investigation of Potential Roles for ADP or AMP in the Nociceptive Hypersensitivity Seen in AdvCOX10 Mutants
As the disruption of mitochondrial respiratory chain complex IV by mutations including COX10 is reported to produce predominantly an energy failure phenotype [32], we postulated that lower energy forms of adenosine phosphate (ADP or AMP, rather than ATP) might play a role in the pain hypersensitivity phenotype. Figure 1 shows explicitly that the ADP/ATP concentration ratio was increased in AdvCOX10 mutant DRG and dorsal spinal cord, so we tested whether this might represent an active signal that contributes to the development of hypersensitivity.
Various purinergic receptors are expressed in sensory afferents including several (P2Y1, P2Y12 and P2Y13), which are selectively activated by ADP [62, 63], so could potentially respond to the increased ADP/ATP ratio seen in DRG of AdvCOX10 mutants. Of these candidates, P2Y1 signals primarily through Gq/11, Ca2+ and PKC, so is anticipated to cause increased excitability, whereas P2Y12 and P2Y13 are primarily Gi linked, so are likely to produce broadly inhibitory outcomes [62]. Although the literature contains some conflicting reports, the consensus view is that P2Y1 is mainly expressed in small DRG cells, negative for the marker of larger myelinated neurons, NF-200, with both IB4-positive (non-peptidergic) and CGRP-positive/capsaicin-responsive (peptidergic) C-nociceptors being represented [64,65,66,67]. Figure 4 compares the effects of selective P2Y1, P2Y12 and P2Y13 agents [68, 69] on the 5-IW-induced Ca2+ fluorescence responses of dorsal spinal cord synaptoneurosomes from control and AdvCOX10 mutant mice. The increase in 5-IW responses seen in mutants compared to controls was significantly reversed by blocking P2Y1, but not P2Y12 or P2Y13 (using MRS 2500, AZD 1283 and MRS 2211, respectively). Addition of a P2Y1 agonist (MRS 2365) increased 5-IW-induced Ca2+ fluorescence responses in controls, but not to beyond a level that was apparent in AdvCOX10 mutants. These results clearly implicate P2Y1 (but not P2Y12 or P2Y13) in the hypersensitivity of 5-IW responses seen in AdvCOX10 mutants.

Hypersensitivity of dorsal spinal cord synaptoneurosomes to GluK1 agonist in AdvCOX10 mutant mice is associated with P2Y1 activation. Ca2+ fluorescence responses of dorsal spinal cord synaptoneurosomes to 2 μM 5-IW, together with modulatory effects of P2Y agents were compared between AdvCOX10 mutant mice and controls. Values are means ± SEM, n = 6–14. These included the P2Y1 antagonist, MRS 2500 (100 nM), the P2Y12 antagonist, AZD 1283 (350 nM), the P2Y13 antagonist, MRS 2211 (20 μM) and the P2Y1 agonist, MRS 2365 (50 nM). Statistically significant differences between 5-IW responses of AdvCOX10 mutants and control mice were assessed by two-way ANOVA with Bonferroni’s test (*p < 0.05, **p < 0.01, ***p < 0.001). Statistically significant differences between the drug-free 5-IW responses of AdvCOX10 mutants or controls and their responses in the presence of P2Y agents were assessed by one-way ANOVA with Dunnett’s test (††p < 0.01)
Considering that relative levels of AMP might also be increased in the AdvCOX10 mutant energy failure phenotype, we investigated any potential role of AMP-activated protein kinase (AMPK) in the nociceptive hypersensitivity observed. Activation of AMPK has, however, been reported to exert antinociceptive effects in models of ongoing peripheral neuropathic pain due to trauma, chemotherapeutic agents or hyperglycaemia, as well as pain due to inflammation, surgical tissue injury or metastatic bone cancer [70,71,72,73,74,75,76]. Figure 5a shows that Western blots in dorsal spinal cord synaptoneurosomes revealed no increase in phospho-AMPKα (Thr172)/GAPDH ratios in AdvCOX10 mutants compared to control mice. Figure 5b further shows that the AMPK activators AICAR and A697662 actually reduced the 5-IW-induced Ca2+ fluorescence responses of dorsal spinal cord synaptoneurosomes. These antinociceptive effects were similar in extent in both control and AdvCOX10 mutant mice and clearly were not contributing to the hypersensitivity. The AMPK inhibitor, dorsomorphin, had no discernible effect. These observations are at odds with the idea of AMPK activation contributing to the nociceptive hypersensitivity seen in AdvCOX10 mutants.

Evaluation of any potential involvement of AMPK in the hypersensitivity observed in AdvCOX10 mutant mice. Using dorsal spinal cord synaptoneurosomes from control and AdvCOX10 mutant mice, neither Western blots for phospho-AMPKα (Thr172), the activated form, nor the effects of AMPK-modulating pharmacological agents on 5-IW-induced Ca2+ fluorescence responses, provided support for the idea that AMPK activation might contribute to hypersensitivity in the mutants. a Typical example images from control and AdvCOX10 samples probed with specific antibodies for phospho-AMPKα (Thr172), and the ubiquitous housekeeping enzyme GAPDH, as loading control. Single bands were detected close to the expected running positions of 62 kDa and 36 kDa, respectively, with no discernible differences in phospho-AMPKα (Thr172) staining between control and AdvCOX10 mutants. The bar chart shows that phospho-AMPKα (Thr172)/GAPDH densitometric ratios from Image J analysis of scanned films were very similar between control and AdvCOX10 mutants (n = 4 in each case, not significant by t test, p = 0.89). b The effects of AMPK activators, and an inhibitor, on 5-IW-induced Ca2+ fluorescence responses of synaptoneurosomes. While 5-IW responses were greater in AdvCOX10 mutants, they were similarly attenuated by the AMPK activators, AICAR and A697662, and unaffected by the inhibitor, dorsomorphin; inconsistent with any role of AMPK activation in hypersensitivity. Values are shown as mean ± SEM, n in parentheses. The statistical significance of differences between corresponding control and mutant values was assessed by two-way ANOVA with Bonferroni’s test (*p < 0.05, **p < 0.01). Differences between drug-free controls and corresponding drug-treated values were evaluated by one-way ANOVA with Dunnett’s test (†p < 0.05, ††p < 0.01)
P2Y1 Is Widely Expressed in Nociceptive Afferents and This Is Further Increased in AdvCOX10 Mutants Compared to Controls
As P2Y1 activation had been specifically implicated in the hypersensitivity at first central synapses of nociceptive inputs seen in AdvCOX10 mutants, we investigated its expression in DRG of both control and AdvCOX10 mutant mice. Figure 6 shows that a substantial proportion of peripherin-immunoreactive small nociceptive afferents were also positive for P2Y1 and that this proportion was significantly increased in AdvCOX10 mutants compared to control mice. The increase was apparent in both TRPV1-positive (largely peptidergic) and IB4-positive (non-peptidergic) subpopulations of nociceptive afferents. Clearly, not only the increased relative concentration of ADP compared to ATP, but also the increased expression of P2Y1 in small afferent neurons, is likely to contribute to the nociceptive hypersensitivity in AdvCOX10 mutants.

P2Y1 is expressed in DRG nociceptive afferents and its expression is increased in AdvCOX10 mutants compared to control mice. a Example images of dual label immunofluorescence identification of DRG cells expressing P2Y1, peripherin and their co-localisation in control and AdvCOX10 mutant mice. Scale bar = 50 μm. b Example images of P2Y1 co-localisation with TRPV1 and IB4 staining in small DRG cells of AdvCOX10 mutant mice. Scale bar = 50 μm. c Quantification of results from experiments outlined in a, b. Substantial proportions of nociceptors labelled by peripherin, TRPV1 and IB4 were each shown to co-express P2Y1. In each case, the level of P2Y1 expression was increased in AdvCOX10 mutants compared to control mice (***p < 0.001, ***p < 0.001 and *p < 0.05, respectively, by t test)
P2Y1 Antagonist Treatment Reverses the Pain-Associated Hypersensitivity of AdvCOX10 Mutants In Vivo and in Ex Vivo Synaptoneurosomes
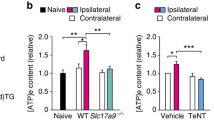
To assess whether P2Y1 activation plays a critical role in the pain hypersensitivity in AdvCOX10 mutants, we treated animals in vivo with the highly selective P2Y1 antagonist, MRS 2500 (intraperitoneal). Figure 7a shows that reflex paw withdrawal thresholds to von Frey filament stimulation were much lower (i.e., hypersensitive) in AdvCOX10 mutants compared to control mice and that this hypersensitivity was clearly reversed by MRS 2500 over 15–60 min following administration. MRS 2500 had no discernible effect in controls. Similar results were observed in the Hargreaves’ thermal test (Fig. 7b), where MRS 2500 caused significant reversal of hypersensitivity in the AdvCOX10 mutants from 15 to 45 min following administration. Increased noxious cold avoidance behaviour in the AdvCOX10 mutants was also significantly reversed 30 min after MRS 2500 administration (Fig. 7c). Assessments were not made at multiple time points in this test, in order to avoid learning-associated behavioural adaptation. Figure 7d shows 5-IW-induced Ca2+ fluorescence responses of dorsal spinal cord synaptoneurosomes taken from animals that had been treated in vivo with MRS 2500, 45 min prior to tissue collection (or untreated). The clear increase in 5-IW responses in untreated AdvCOX10 mutants compared to untreated control mice was significantly attenuated (by around 50%) in animals treated with MRS 2500. Thus, the hypersensitivity of mutants (both in terms of reflex pain behaviour in vivo and exaggerated responsiveness of ex vivo synaptoneurosomes) was significantly attenuated by in vivo P2Y1 blockade.

Reversal by P2Y1 antagonist of pain-associated hypersensitivity in AdvCOX10 mutants both in vivo and in ex vivo tissue preparations. a, b Time-courses of in vivo reflex paw withdrawal responses to von Frey (mechanical) and Hargreaves’ (thermal) tests, respectively, in both control animals and in COX10 mutants and the effects of intraperitoneal injection of the selective P2Y1 antagonist, MRS 2500 (2 mg/kg in sterile saline). Mutants displayed hypersensitivity compared to controls (*, **, ***p < 0.05, 0.01 and 0.001, respectively, by repeated measures two-way ANOVA with Bonferroni’s test. In mutants, but not control mice, MRS 2500 caused marked reversal of hypersensitive paw withdrawal responses towards normal values. Values are means ± SEM, n = 4–5 in each case. († and ††p < 0.05 and p < 0.01, respectively, by repeated measures one-way ANOVA with Dunnett’s test. No changes were seen in responses from control animals. c In vivo noxious cold avoidance behaviour in terms of standing duration on a 4 °C cold plate compared to an insulated, room temperature, platform for control and AdvCOX10 mutant mice, before and 30 min after intraperitoneal injection of 2 mg/kg MRS 2500. **p < 0.01; a significant reduction in cold plate standing duration for mutant compared to control mice and †p < 0.05; a significant reversal of this hypersensitivity due to MRS 2500, by one-way ANOVA with Tukey’s test. Values are means ± SEM, n = 4 in each case. d Ca2+ fluorescence responses to 5-IW (2 μM) of lumbar dorsal spinal cord synaptoneurosomes from control or AdvCOX10 mutant mice that had been treated in vivo with MRS 2500 (2 mg/kg ip, 45 min before tissue collection). The 5-IW response was significantly increased in untreated mutant animal samples compared to control animals (**p < 0.01), whereas this increment was significantly attenuated in preparations from mutant animals treated in vivo with MRS 2500 (†p < 0.05). Values are means ± SEM, n = 12 for untreated controls and mutants and n = 5 for MRS 2500-treated controls and mutants. Statistical significance was assessed by one-way ANOVA with Tukey’s test
Discussion
Deficits in mitochondrial function have been reported in a variety of models of neuropathic pain. The current results, studying the targeted mutation of a key component of the mitochondrial respiratory chain complex IV, underline that mitochondrial deficits, have a causal role in bringing about pain hypersensitivity and identify aspects of the mechanism(s) responsible. In models of neuropathic pain hypersensitivity due to the administration of chemotherapeutic agents (platinum compounds, paclitaxel or bortezomib), deficits in ATP production have been widely described [30, 77, 78]. Similar inhibition of mitochondrial respiratory chain function is observed in various models of diabetic neuropathy [79]. Mitochondrial dysfunction can induce cellular oxidative stress; generating ROS and leading to cellular oxidative damage, including in DRG neurons [80, 81]. Both local and systemic treatment with antioxidants, or even toxins to inhibit ROS-producing mitochondrial respiratory chain complexes, is reported to attenuate CIPN-associated mechanical hyperalgesia [20, 27, 82, 83].
However, the mitochondrial gene deletion model used here (which brings about a selective energy-deficient phenotype rather than one of excess ROS generation) indicates that energy deficiency alone is sufficient to cause pain hypersensitivity. Direct measurement of ATP/ADP content (in DRG and dorsal spinal cord) and histochemical assessment of SDH enzyme activity in DRG, as well as monitoring of relative mitochondrial membrane potential using JC-1 indicator in dorsal spinal cord all matched the anticipated energy-deficient phenotype, while experiments with a broad spectrum ROS-reporter indicated only low levels of ROS that were no greater in AdvCOX10 mutants than controls. The Advillin-promoter targeting of the deletion to peripheral neurons, caused, as expected, selective biochemical deficits in DRG and dorsal, but not ventral, spinal cord. These were matched by selective changes in sensory function with no detectable impact on motor function or general well-being of the animals. Significant hypersensitivity was found in mechanical, heat and cold-induced responses, suggesting a wide impact across classes of nociceptive afferents.
To investigate the underlying basis in an ex vivo synaptic preparation, readily amenable to pharmacological interrogation, we measured Ca2+ fluorescence responses of dorsal spinal cord synaptoneurosomes that were elicited by a selective GluK1 agonist, 5-IW. The selective GluK1 antagonist, ACET, caused an apparently parallel right-shift in the 5-IW concentration response curve, in accordance with its reported competitive mode of blockade. Responses were also strongly inhibited by the presynaptically acting inhibitor of synaptic release, tetanus neurotoxin. This is consistent with the reported distribution of GluK1 receptors (selectively in small nociceptive afferents and at their corresponding presynaptic terminals in the synaptoneurosome preparation). This also matches evidence that neuronal mitochondria are particularly enriched at presynaptic terminals, where synaptic vesicle trafficking places high demands on ATP synthesis, so presynaptically initiated responses may be particularly vulnerable to mitochondrial dysfunction here [84, 85]. We provided quantitative evidence that GluK1 is expressed in both non-peptidergic (IB4+) and likely peptidergic (TRPV1+) nociceptors. This matches previous findings, which described GluK1 in both IB4+ and some TRPV1+ cells [48] and suggests that GluK1 activation might provide a fair reflection of consensus nociceptive input. Correspondingly, both GluK1 antagonists and genetic ablation of GluK1 reduce reflex pain behaviour in a number of models of neuropathic or inflammatory pain [86,87,88]. GluK1-mediated Ca2+ fluorescence responses were significantly greater in AdvCOX10 mutant dorsal spinal cord than in controls.
As the ADP/ATP ratio was increased in the DRG and dorsal spinal cord of mutants, we investigated the potential contribution of purinergic receptors that might respond to this change and are known to be expressed in nociceptive pathways. Of the wide range of P2X (ionotropic) and P2Y (metabotropic) purinergic receptors that respond to adenine nucleotides, a number have been specifically implicated in pain transmission [62, 89,90,91]. While many of these are activated by ATP, a few, notably P2Y1, P2Y12 and P2Y13 are preferentially activated by ADP, so could be of particular relevance to the pain hypersensitivity in AdvCOX10 mutant mice. P2Y12 and P2Y13 signal predominantly through Gi and are generally thought to lead to inhibitory consequences, such as inhibition of Ca2+ channels and facilitation of K+ channels [62]. In contrast, P2Y1 is coupled to Gq and will lead to increased Ca2+ mobilisation and activation of protein kinase C, both excitatory processes that in presynaptic terminals would be expected to lead to increased transmitter release and central sensitisation in pain pathways [62, 92]. Further, P2Y1 is not only activated by ADP, but inhibited by ATP [63], so will act as an exquisitely sensitive detector of any increase in the ADP/ATP ratio. We showed here that P2Y1, but not P2Y12 and P2Y13 antagonists, reversed the increment in GluK1 agonist responses seen in AdvCOX10 mutant mice. Furthermore, in synaptoneurosomes from control animals, a selective P2Y1 agonist replicated the increment in GluK1 agonist responses seen in AdvCOX10 mutants. P2Y1 receptors seem likely to be responsible for the hypersensitivity seen in AdvCOX10 mutants.
As AMP levels could also be increased in an energy-deficient situation due to mitochondrial respiratory chain disruption, and thereby activate AMPK, we looked for any evidence of AMPK activation in AdvCOX10 mutants compared to controls by immunoblotting for phospho-AMPKα (Thr172) in dorsal spinal cord synaptoneurosomes and found no support for this idea. Indeed, reports of neuropathic pain hypersensitivity due to hyperglycaemia, nerve injury or chemotherapeutic toxicity describe reductions, rather than increases in AMPK activity [72, 73, 93]. We further evaluated whether activators or inhibitors of AMPK modified the hypersensitivity in GluK1 responses of dorsal spinal cord synaptoneurosomes seen in AdvCOX10 mutant mice. The AMPK activators, AICAR and A697662, actually reduced GluK1 agonist-induced Ca2+ fluorescence responses in both control and AdvCOX10 mutant animals. These observations are consistent with the antinociceptive effects of AMPK activators widely reported in neuropathic, inflammatory, bone cancer and acute pain models [70,71,72,73,74,75,76]. Our findings do not support the idea of AMPK activation contributing to the hypersensitivity seen in AdvCOX10 mutant mice.
To further assess the likely role of P2Y1 receptors that the pharmacology had implied, we measured expression of P2Y1 in afferents of both likely peptidergic and non-peptidergic classes. P2Y1 is thought to be expressed in small sensory afferents, but not glia [65]. It has been reported that many small nociceptors (of both peptidergic and non-peptidergic classes) are immunoreactive for P2Y1 [67], although a preferential expression in the non-peptidergic class has been highlighted [66]. We found substantial P2Y1 expression in both non-peptidergic (IB4+) and largely peptidergic (TRPV1+) nociceptors. Although TRPV1+ nociceptors have been described to largely map to the peptidergic subpopulation [53], other reports indicate that TRPV1 may be more widespread [94]. There is evidence that P2Y1 expression in nociceptors is up-regulated during neuropathic and inflammatory pain states [95,96,97], potentially enhancing the degree of influence of P2Y1-selective pharmacological agents.
We then assessed the impact of P2Y1 blockade in vivo on mechanical and thermal reflex pain behaviours, noxious cold avoidance behaviour and the GluK1 responsiveness in ex vivo tissue from AdvCOX10 mutant and control mice. A highly selective P2Y1 antagonist, MRS 2500, at a dose of 2 mg/kg ip, shown to effectively block peripheral P2Y1 receptors [98], caused striking analgesia in AdvCOX10 mutants and correspondingly, MRS 2500 diminished the hypersensitivity in ex vivo synaptoneurosomes from the mutant mice. P2Y1 has been suggested to play a role in mechanical and thermal sensation [66, 99], in the hypersensitivity seen in inflammatory, neuropathic and bone-metastasis-induced pain models and in regulating new post-inflammation thermal responsiveness [64, 95, 100,101,102,103]. Similar roles for P2Y1 have been described in chronic pain-associated sensitisation of visceral and muscle afferent inputs [97, 104, 105]. These findings in multiple types of pain state (including that due to mitochondrial deficiency here) support the idea that P2Y1 could represent a widely applicable analgesic target worthy of translational development.
Conclusions
Overall, these findings indicate that the energy-deficient phenotype in primary sensory afferents found in AdvCOX10 mutants is entirely sufficient to produce pain hypersensitivity. Although the tamoxifen-induced deletion was not related here to a specific pain model, there is abundant evidence that functional deficiency of the mitochondrial respiratory chain complex IV is a common observation in neuropathic pain models, so the current findings are likely to be widely applicable in neuropathic pain of various origins [106, 107]. Blockade of P2Y1, which appears to be selectively activated in response to an increased ADP/ATP ratio, could be a useful new strategy for the clinical treatment of neuropathic pain, although any unintended side effects of P2Y1 blockade would need to be considered. Potential applications could include neuropathic pain due to nerve damage, toxicity from cancer and retroviral therapeutics or diabetic hyperglycaemia, and a range of neurodegenerative disorders, each of which has been shown to involve mitochondrial deficits.
References
Bennett GJ, Doyle T, Salvemini D (2014) Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol 10(6):326–336. https://doi.org/10.1038/nrneurol.2014.77
Flatters SJ, Bennett GJ (2006) Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: Evidence for mitochondrial dysfunction. Pain 122(3):245–257. https://doi.org/10.1016/j.pain.2006.01.037
Canta A, Pozzi E, Carozzi VA (2015) Mitochondrial dysfunction in chemotherapy-induced peripheral neuropathy (CIPN). Toxics 3(2):198–223. https://doi.org/10.3390/toxics3020198
Dalakas MC, Semino-Mora C, Leon-Monzon M (2001) Mitochondrial alterations with mitochondrial DNA depletion in the nerves of AIDS patients with peripheral neuropathy induced by 2′3'-dideoxycytidine (ddC). Lab Investig 81(11):1537–1544
Chowdhury SK, Zherebitskaya E, Smith DR, Akude E, Chattopadhyay S, Jolivalt CG, Calcutt NA, Fernyhough P (2010) Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 59(4):1082–1091. https://doi.org/10.2337/db09-1299
Hinder LM, Vincent AM, Burant CF, Pennathur S, Feldman EL (2012) Bioenergetics in diabetic neuropathy: What we need to know. J Peripher Nerv Syst 17(Suppl 2):10–14. https://doi.org/10.1111/j.1529-8027.2012.00389.x
Lim TK, Rone MB, Lee S, Antel JP, Zhang J (2015) Mitochondrial and bioenergetic dysfunction in trauma-induced painful peripheral neuropathy. Mol Pain 11:58. https://doi.org/10.1186/s12990-015-0057-7
Carter GT, Jensen MP, Galer BS, Kraft GH, Crabtree LD, Beardsley RM, Abresch RT, Bird TD (1998) Neuropathic pain in Charcot-Marie-tooth disease. Arch Phys Med Rehabil 79(12):1560–1564
Foley PL, Vesterinen HM, Laird BJ, Sena ES, Colvin LA, Chandran S, MacLeod MR, Fallon MT (2013) Prevalence and natural history of pain in adults with multiple sclerosis: Systematic review and meta-analysis. Pain 154(5):632–642. https://doi.org/10.1016/j.pain.2012.12.002
Shy ME, Patzko A (2011) Axonal Charcot-Marie-tooth disease. Curr Opin Neurol 24(5):475–483. https://doi.org/10.1097/WCO.0b013e32834aa331
Truini A, Barbanti P, Pozzilli C, Cruccu G (2013) A mechanism-based classification of pain in multiple sclerosis. J Neurol 260(2):351–367. https://doi.org/10.1007/s00415-012-6579-2
Baloh RH, Schmidt RE, Pestronk A, Milbrandt J (2007) Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-tooth disease from mitofusin 2 mutations. J Neurosci 27(2):422–430. https://doi.org/10.1523/JNEUROSCI.4798-06.2007
Kalman B, Laitinen K, Komoly S (2007) The involvement of mitochondria in the pathogenesis of multiple sclerosis. J Neuroimmunol 188(1–2):1–12. https://doi.org/10.1016/j.jneuroim.2007.03.020
Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM et al (2011) Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 69(3):481–492. https://doi.org/10.1002/ana.22109
Pareyson D, Saveri P, Sagnelli A, Piscosquito G (2015) Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci Lett 596:66–77. https://doi.org/10.1016/j.neulet.2015.04.001
Duggett NA, Griffiths LA, McKenna OE, de Santis V, Yongsanguanchai N, Mokori EB, Flatters SJ (2016) Oxidative stress in the development, maintenance and resolution of paclitaxel-induced painful neuropathy. Neuroscience 333:13–26. https://doi.org/10.1016/j.neuroscience.2016.06.050
Kim HY, Lee I, Chun SW, Kim HK (2015) Reactive oxygen species donors increase the responsiveness of dorsal horn neurons and induce mechanical hyperalgesia in rats. Neural Plast 2015:293423. https://doi.org/10.1155/2015/293423
Duggett NA, Griffiths LA, Flatters SJL (2017) Paclitaxel-induced painful neuropathy is associated with changes in mitochondrial bioenergetics, glycolysis, and an energy deficit in dorsal root ganglia neurons. Pain 158(8):1499–1508. https://doi.org/10.1097/j.pain.0000000000000939
Ferrari LF, Chum A, Bogen O, Reichling DB, Levine JD (2011) Role of Drp1, a key mitochondrial fission protein, in neuropathic pain. J Neurosci 31(31):11404–11410. https://doi.org/10.1523/JNEUROSCI.2223-11.2011
Joseph EK, Levine JD (2006) Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain 121(1–2):105–114. https://doi.org/10.1016/j.pain.2005.12.010
Lee KY, Chung K, Chung JM (2010) Involvement of reactive oxygen species in long-term potentiation in the spinal cord dorsal horn. J Neurophysiol 103(1):382–391. https://doi.org/10.1152/jn.90906.2008
Sui BD, Xu TQ, Liu JW, Wei W, Zheng CX, Guo BL, Wang YY, Yang YL (2013) Understanding the role of mitochondria in the pathogenesis of chronic pain. Postgrad Med J 89(1058):709–714. https://doi.org/10.1136/postgradmedj-2012-131068
Xiao WH, Zheng H, Zheng FY, Nuydens R, Meert TF, Bennett GJ (2011) Mitochondrial abnormality in sensory, but not motor, axons in paclitaxel-evoked painful peripheral neuropathy in the rat. Neuroscience 199:461–469. https://doi.org/10.1016/j.neuroscience.2011.10.010
Xiao WH, Bennett GJ (2012) Effects of mitochondrial poisons on the neuropathic pain produced by the chemotherapeutic agents, paclitaxel and oxaliplatin. Pain 153(3):704–709. https://doi.org/10.1016/j.pain.2011.12.011
Lim TK, Shi XQ, Johnson JM, Rone MB, Antel JP, David S, Zhang J (2015) Peripheral nerve injury induces persistent vascular dysfunction and endoneurial hypoxia, contributing to the genesis of neuropathic pain. J Neurosci 35(8):3346–3359. https://doi.org/10.1523/JNEUROSCI.4040-14.2015
Flatters SJ, Xiao WH, Bennett GJ (2006) Acetyl-L-carnitine prevents and reduces paclitaxel-induced painful peripheral neuropathy. Neurosci Lett 397(3):219–223. https://doi.org/10.1016/j.neulet.2005.12.013
Joseph EK, Chen X, Bogen O, Levine JD (2008) Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J Pain 9(5):463–472. https://doi.org/10.1016/j.jpain.2008.01.335
Joseph EK, Levine JD (2010) Multiple PKCepsilon-dependent mechanisms mediating mechanical hyperalgesia. Pain 150(1):17–21. https://doi.org/10.1016/j.pain.2010.02.011
Toyama S, Shimoyama N, Ishida Y, Koyasu T, Szeto HH, Shimoyama M (2014) Characterization of acute and chronic neuropathies induced by oxaliplatin in mice and differential effects of a novel mitochondria-targeted antioxidant on the neuropathies. Anesthesiology 120(2):459–473. https://doi.org/10.1097/01.anes.0000435634.34709.65
Zheng H, Xiao WH, Bennett GJ (2011) Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp Neurol 232(2):154–161. https://doi.org/10.1016/j.expneurol.2011.08.016
Torraco A, Diaz F, Vempati UD, Moraes CT (2009) Mouse models of oxidative phosphorylation defects: Powerful tools to study the pathobiology of mitochondrial diseases. Biochim Biophys Acta 1793(1):171–180. https://doi.org/10.1016/j.bbamcr.2008.06.003
Diaz F, Garcia S, Padgett KR, Moraes CT (2012) A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum Mol Genet 21(23):5066–5077. https://doi.org/10.1093/hmg/dds350
Hasegawa H, Abbott S, Han BX, Qi Y, Wang F (2007) Analyzing somatosensory axon projections with the sensory neuron-specific Advillin gene. J Neurosci 27(52):14404–14414. https://doi.org/10.1523/JNEUROSCI.4908-07.2007
Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM et al (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485(7399):517–521. https://doi.org/10.1038/nature11007
Feil R, Wagner J, Metzger D, Chambon P (1997) Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun 237(3):752–757. https://doi.org/10.1006/bbrc.1997.7124
Booker SA, Campbell GR, Mysiak KS, Brophy PJ, Kind PC, Mahad DJ, Wyllie DJ (2017) Loss of protohaem IX farnesyltransferase in mature dentate granule cells impairs short-term facilitation at mossy fibre to CA3 pyramidal cell synapses. J Physiol 595(6):2147–2160. https://doi.org/10.1113/JP273581
Wojtala A, Bonora M, Malinska D, Pinton P, Duszynski J, Wieckowski MR (2014) Methods to monitor ROS production by fluorescence microscopy and fluorometry. Methods Enzymol 542:243–262. https://doi.org/10.1016/B978-0-12-416618-9.00013-3
Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA (2011) Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 50(2):98–115. https://doi.org/10.2144/000113610
Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A (1997) JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: Implications for studies on mitochondrial functionality during apoptosis. FEBS Lett 411(1):77–82
Hollingsworth EB, McNeal ET, Burton JL, Williams RJ, Daly JW, Creveling CR (1985) Biochemical characterization of a filtered synaptoneurosome preparation from Guinea pig cerebral cortex: Cyclic adenosine 3′:5′-monophosphate-generating systems, receptors, and enzymes. J Neurosci 5(8):2240–2253
Villasana LE, Klann E, Tejada-Simon MV (2006) Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods 158(1):30–36. https://doi.org/10.1016/j.jneumeth.2006.05.008
Sun L, Gooding HL, Brunton PJ, Russell JA, Mitchell R, Fleetwood-Walker S (2013) Phospholipase D-mediated hypersensitivity at central synapses is associated with abnormal behaviours and pain sensitivity in rats exposed to prenatal stress. Int J Biochem Cell Biol 45(11):2706–2712. https://doi.org/10.1016/j.biocel.2013.07.017
Vinuela-Fernandez I, Sun L, Jerina H, Curtis J, Allchorne A, Gooding H, Rosie R, Holland P et al (2014) The TRPM8 channel forms a complex with the 5-HT(1B) receptor and phospholipase D that amplifies its reversal of pain hypersensitivity. Neuropharmacology 79:136–151. https://doi.org/10.1016/j.neuropharm.2013.11.006
Huettner JE (1990) Glutamate receptor channels in rat DRG neurons: Activation by kainate and quisqualate and blockade of desensitization by con a. Neuron 5(3):255–266
Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A (1993) Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin a. Neuron 11(6):1069–1082
Wong LA, Mayer ML, Jane DE, Watkins JC (1994) Willardiines differentiate agonist binding sites for kainate- versus AMPA-preferring glutamate receptors in DRG and hippocampal neurons. J Neurosci 14(6):3881–3897
Furuyama T, Kiyama H, Sato K, Park HT, Maeno H, Takagi H, Tohyama M (1993) Region-specific expression of subunits of ionotropic glutamate receptors (AMPA-type, KA-type and NMDA receptors) in the rat spinal cord with special reference to nociception. Brain Res Mol Brain Res 18(1–2):141–151
Lee CJ, Kong H, Manzini MC, Albuquerque C, Chao MV, MacDermott AB (2001) Kainate receptors expressed by a subpopulation of developing nociceptors rapidly switch from high to low Ca2+ permeability. J Neurosci 21(13):4572–4581
Sato K, Kiyama H, Park HT, Tohyama M (1993) AMPA, KA and NMDA receptors are expressed in the rat DRG neurones. Neuroreport 4(11):1263–1265
Tolle TR, Berthele A, Zieglgansberger W, Seeburg PH, Wisden W (1993) The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and in periaqueductal gray. J Neurosci 13(12):5009–5028
Kerchner GA, Wilding TJ, Huettner JE, Zhuo M (2002) Kainate receptor subunits underlying presynaptic regulation of transmitter release in the dorsal horn. J Neurosci 22(18):8010–8017
Mulle C, Sailer A, Swanson GT, Brana C, O'Gorman S, Bettler B, Heinemann SF (2000) Subunit composition of kainate receptors in hippocampal interneurons. Neuron 28(2):475–484
Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ (2009) Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A 106(22):9075–9080. https://doi.org/10.1073/pnas.0901507106
Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ (2001) A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 106(5):619–632
Alt A, Weiss B, Ogden AM, Knauss JL, Oler J, Ho K, Large TH, Bleakman D (2004) Pharmacological characterization of glutamatergic agonists and antagonists at recombinant human homomeric and heteromeric kainate receptors in vitro. Neuropharmacology 46(6):793–806. https://doi.org/10.1016/j.neuropharm.2003.11.026
Patneau DK, Mayer ML, Jane DE, Watkins JC (1992) Activation and desensitization of AMPA/kainate receptors by novel derivatives of willardiine. J Neurosci 12(2):595–606
Swanson GT, Green T, Heinemann SF (1998) Kainate receptors exhibit differential sensitivities to (S)-5-iodowillardiine. Mol Pharmacol 53(5):942–949
Dolman NP, More JC, Alt A, Knauss JL, Pentikainen OT, Glasser CR, Bleakman D, Mayer ML et al (2007) Synthesis and pharmacological characterization of N3-substituted willardiine derivatives: Role of the substituent at the 5-position of the uracil ring in the development of highly potent and selective GLUK5 kainate receptor antagonists. J Med Chem 50(7):1558–1570. https://doi.org/10.1021/jm061041u
Verderio C, Coco S, Bacci A, Rossetto O, De Camilli P, Montecucco C, Matteoli M (1999) Tetanus toxin blocks the exocytosis of synaptic vesicles clustered at synapses but not of synaptic vesicles in isolated axons. J Neurosci 19(16):6723–6732
Kessler M, Rogers G, Arai A (2000) The norbornenyl moiety of cyclothiazide determines the preference for flip-flop variants of AMPA receptor subunits. Neurosci Lett 287(2):161–165
Voitenko N, Gerber G, Youn D, Randic M (2004) Peripheral inflamation-induced increase of AMPA-mediated currents and Ca2+ transients in the presence of cyclothiazide in the rat substantia gelatinosa neurons. Cell Calcium 35(5):461–469. https://doi.org/10.1016/j.ceca.2003.11.002
Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M et al (2006) International Union of Pharmacology LVIII: Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58(3):281–341. https://doi.org/10.1124/pr.58.3.3
Leon C, Hechler B, Vial C, Leray C, Cazenave JP, Gachet C (1997) The P2Y1 receptor is an ADP receptor antagonized by ATP and expressed in platelets and megakaryoblastic cells. FEBS Lett 403(1):26–30
Jankowski MP, Rau KK, Soneji DJ, Ekmann KM, Anderson CE, Molliver DC, Koerber HR (2012) Purinergic receptor P2Y1 regulates polymodal C-fiber thermal thresholds and sensory neuron phenotypic switching during peripheral inflammation. Pain 153(2):410–419. https://doi.org/10.1016/j.pain.2011.10.042
Kobayashi K, Yamanaka H, Noguchi K (2013) Expression of ATP receptors in the rat dorsal root ganglion and spinal cord. Anat Sci Int 88(1):10–16. https://doi.org/10.1007/s12565-012-0163-9
Molliver DC, Rau KK, McIlwrath SL, Jankowski MP, Koerber HR (2011) The ADP receptor P2Y1 is necessary for normal thermal sensitivity in cutaneous polymodal nociceptors. Mol Pain 7:13. https://doi.org/10.1186/1744-8069-7-13
Ruan HZ, Burnstock G (2003) Localisation of P2Y1 and P2Y4 receptors in dorsal root, nodose and trigeminal ganglia of the rat. Histochem Cell Biol 120(5):415–426. https://doi.org/10.1007/s00418-003-0579-3
Jacobson KA, Ivanov AA, de Castro S, Harden TK, Ko H (2009) Development of selective agonists and antagonists of P2Y receptors. Purinergic Signal 5(1):75–89. https://doi.org/10.1007/s11302-008-9106-2
von Kugelgen I (2006) Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther 110(3):415–432. https://doi.org/10.1016/j.pharmthera.2005.08.014
Burton MD, Tillu DV, Mazhar K, Mejia GL, Asiedu MN, Inyang K, Hughes T, Lian B et al (2017) Pharmacological activation of AMPK inhibits incision-evoked mechanical hypersensitivity and the development of hyperalgesic priming in mice. Neuroscience 359:119–129. https://doi.org/10.1016/j.neuroscience.2017.07.020
Hasanvand A, Amini-Khoei H, Hadian MR, Abdollahi A, Tavangar SM, Dehpour AR, Semiei E, Mehr SE (2016) Anti-inflammatory effect of AMPK signaling pathway in rat model of diabetic neuropathy. Inflammopharmacology 24(5):207–219. https://doi.org/10.1007/s10787-016-0275-2
Ling YZ, Li ZY, Ou-Yang HD, Ma C, Wu SL, Wei JY, Ding HH, Zhang XL et al (2017) The inhibition of spinal synaptic plasticity mediated by activation of AMP-activated protein kinase signaling alleviates the acute pain induced by oxaliplatin. Exp Neurol 288:85–93. https://doi.org/10.1016/j.expneurol.2016.11.009
Maixner DW, Yan X, Gao M, Yadav R, Weng HR (2015) Adenosine monophosphate-activated protein kinase regulates interleukin-1beta expression and glial glutamate transporter function in rodents with neuropathic pain. Anesthesiology 122(6):1401–1413. https://doi.org/10.1097/ALN.0000000000000619
Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J et al (2011) Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain 7:70. https://doi.org/10.1186/1744-8069-7-70
Russe OQ, Moser CV, Kynast KL, King TS, Stephan H, Geisslinger G, Niederberger E (2013) Activation of the AMP-activated protein kinase reduces inflammatory nociception. J Pain 14(11):1330–1340. https://doi.org/10.1016/j.jpain.2013.05.012
Song H, Han Y, Pan C, Deng X, Dai W, Hu L, Jiang C, Yang Y et al (2015) Activation of adenosine monophosphate-activated protein kinase suppresses Neuroinflammation and ameliorates bone Cancer pain: Involvement of inhibition on mitogen-activated protein kinase. Anesthesiology 123(5):1170–1185. https://doi.org/10.1097/ALN.0000000000000856
Garrido N, Perez-Martos A, Faro M, Lou-Bonafonte JM, Fernandez-Silva P, Lopez-Perez MJ, Montoya J, Enriquez JA (2008) Cisplatin-mediated impairment of mitochondrial DNA metabolism inversely correlates with glutathione levels. Biochem J 414(1):93–102. https://doi.org/10.1042/BJ20071615
Zheng H, Xiao WH, Bennett GJ (2012) Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Exp Neurol 238(2):225–234. https://doi.org/10.1016/j.expneurol.2012.08.023
Fernyhough P (2015) Mitochondrial dysfunction in diabetic neuropathy: A series of unfortunate metabolic events. Curr Diab Rep 15(11):89. https://doi.org/10.1007/s11892-015-0671-9
Carozzi VA, Canta A, Chiorazzi A (2015) Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci Lett 596:90–107. https://doi.org/10.1016/j.neulet.2014.10.014
Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, Doetsch PW (2013) Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One 8(11):e81162. https://doi.org/10.1371/journal.pone.0081162
Fidanboylu M, Griffiths LA, Flatters SJ (2011) Global inhibition of reactive oxygen species (ROS) inhibits paclitaxel-induced painful peripheral neuropathy. PLoS One 6(9):e25212. https://doi.org/10.1371/journal.pone.0025212
Griffiths LA, Flatters SJ (2015) Pharmacological modulation of the mitochondrial Electron transport chain in paclitaxel-induced painful peripheral neuropathy. J Pain 16(10):981–994. https://doi.org/10.1016/j.jpain.2015.06.008
Hollenbeck PJ, Saxton WM (2005) The axonal transport of mitochondria. J Cell Sci 118(Pt 23):5411–5419. https://doi.org/10.1242/jcs.02745
Rangaraju V, Calloway N, Ryan TA (2014) Activity-driven local ATP synthesis is required for synaptic function. Cell 156(4):825–835. https://doi.org/10.1016/j.cell.2013.12.042
Bhangoo SK, Swanson GT (2013) Kainate receptor signaling in pain pathways. Mol Pharmacol 83(2):307–315. https://doi.org/10.1124/mol.112.081398
Ruscheweyh R, Sandkuhler J (2002) Role of kainate receptors in nociception. Brain Res Brain Res Rev 40(1–3):215–222
Wu LJ, Ko SW, Zhuo M (2007) Kainate receptors and pain: From dorsal root ganglion to the anterior cingulate cortex. Curr Pharm Des 13(15):1597–1605
Gerevich Z, Illes P (2004) P2Y receptors and pain transmission. Purinergic Signal 1(1):3–10. https://doi.org/10.1007/s11302-004-4740-9
Burnstock G (2006) Purinergic P2 receptors as targets for novel analgesics. Pharmacol Ther 110(3):433–454. https://doi.org/10.1016/j.pharmthera.2005.08.013
Burnstock G (2016) Purinergic mechanisms and pain. Adv Pharmacol 75:91–137. https://doi.org/10.1016/bs.apha.2015.09.001
Latremoliere A, Woolf CJ (2009) Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J Pain 10(9):895–926. https://doi.org/10.1016/j.jpain.2009.06.012
Roy Chowdhury SK, Smith DR, Saleh A, Schapansky J, Marquez A, Gomes S, Akude E, Morrow D et al (2012) Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 135(Pt 6):1751–1766. https://doi.org/10.1093/brain/aws097
Hwang SJ, Oh JM, Valtschanoff JG (2005) Expression of the vanilloid receptor TRPV1 in rat dorsal root ganglion neurons supports different roles of the receptor in visceral and cutaneous afferents. Brain Res 1047(2):261–266. https://doi.org/10.1016/j.brainres.2005.04.036
Barragan-Iglesias P, Pineda-Farias JB, Bravo-Hernandez M, Cervantes-Duran C, Price TJ, Murbartian J, Granados-Soto V (2016) Predominant role of spinal P2Y1 receptors in the development of neuropathic pain in rats. Brain Res 1636:43–51. https://doi.org/10.1016/j.brainres.2016.01.042
Lu P, Hudgins RC, Liu X, Ford ZK, Hofmann MC, Queme LF, Jankowski MP (2017) Upregulation of P2Y1 in neonatal nociceptors regulates heat and mechanical sensitization during cutaneous inflammation. Mol Pain 13:1744806917730255. https://doi.org/10.1177/1744806917730255
Wu J, Cheng Y, Zhang R, Liu D, Luo YM, Chen KL, Ren S, Zhang J (2017) P2Y1R is involved in visceral hypersensitivity in rats with experimental irritable bowel syndrome. World J Gastroenterol 23(34):6339–6349. https://doi.org/10.3748/wjg.v23.i34.6339
Hechler B, Nonne C, Roh EJ, Cattaneo M, Cazenave JP, Lanza F, Jacobson KA, Gachet C (2006) MRS2500 [2-iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate], a potent, selective, and stable antagonist of the platelet P2Y1 receptor with strong antithrombotic activity in mice. J Pharmacol Exp Ther 316(2):556–563. https://doi.org/10.1124/jpet.105.094037
Nakamura F, Strittmatter SM (1996) P2Y1 purinergic receptors in sensory neurons: Contribution to touch-induced impulse generation. Proc Natl Acad Sci U S A 93(19):10465–10470
Malin SA, Molliver DC (2010) Gi- and Gq-coupled ADP (P2Y) receptors act in opposition to modulate nociceptive signaling and inflammatory pain behavior. Mol Pain 6:21. https://doi.org/10.1186/1744-8069-6-21
Chen J, Wang L, Zhang Y, Yang J (2012) P2Y1 purinoceptor inhibition reduces extracellular signal-regulated protein kinase 1/2 phosphorylation in spinal cord and dorsal root ganglia: Implications for cancer-induced bone pain. Acta Biochim Biophys Sin Shanghai 44(4):367–372. https://doi.org/10.1093/abbs/gms007
Kwon SG, Roh DH, Yoon SY, Moon JY, Choi SR, Choi HS, Kang SY, Han HJ et al (2014) Blockade of peripheral P2Y1 receptors prevents the induction of thermal hyperalgesia via modulation of TRPV1 expression in carrageenan-induced inflammatory pain rats: Involvement of p38 MAPK phosphorylation in DRGs. Neuropharmacology 79:368–379. https://doi.org/10.1016/j.neuropharm.2013.12.005
Barragan-Iglesias P, Mendoza-Garces L, Pineda-Farias JB, Solano-Olivares V, Rodriguez-Silverio J, Flores-Murrieta FJ, Granados-Soto V, Rocha-Gonzalez HI (2015) Participation of peripheral P2Y1, P2Y6 and P2Y11 receptors in formalin-induced inflammatory pain in rats. Pharmacol Biochem Behav 128:23–32. https://doi.org/10.1016/j.pbb.2014.11.001
Hockley JR, Tranter MM, McGuire C, Boundouki G, Cibert-Goton V, Thaha MA, Blackshaw LA, Michael GJ et al (2016) P2Y receptors sensitize mouse and human colonic nociceptors. J Neurosci 36(8):2364–2376. https://doi.org/10.1523/JNEUROSCI.3369-15.2016
Queme LF, Ross JL, Lu P, Hudgins RC, Jankowski MP (2016) Dual modulation of nociception and cardiovascular reflexes during peripheral ischemia through P2Y1 receptor-dependent sensitization of muscle afferents. J Neurosci 36(1):19–30. https://doi.org/10.1523/JNEUROSCI.2856-15.2016
Lehmann HC, Chen W, Borzan J, Mankowski JL, Hoke A (2011) Mitochondrial dysfunction in distal axons contributes to human immunodeficiency virus sensory neuropathy. Ann Neurol 69(1):100–110. https://doi.org/10.1002/ana.22150
Lax NZ, Whittaker RG, Hepplewhite PD, Reeve AK, Blakely EL, Jaros E, Ince PG, Taylor RW et al (2012) Sensory neuronopathy in patients harbouring recessive polymerase gamma mutations. Brain 135(Pt 1):62–71. https://doi.org/10.1093/brain/awr326
Acknowledgments
We are grateful to Professors Carlos Moraes and John Wood FRS, for generously providing COX10flox/flox and Adv-CreERT2−/− mutants, respectively. Thanks to Anisha Kubasik-Thayil from the IMPACT Confocal Imaging facility at the University of Edinburgh for expert imaging assistance.
Funding
This work was supported by a Progressive Multiple Sclerosis Alliance Challenge Award to DJM (PA0051).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Don Mahad and Sue M. Fleetwood-Walker are communicating authors
Rights and permissions
About this article

Cite this article
Mitchell, R., Campbell, G., Mikolajczak, M. et al. A Targeted Mutation Disrupting Mitochondrial Complex IV Function in Primary Afferent Neurons Leads to Pain Hypersensitivity Through P2Y1 Receptor Activation. Mol Neurobiol 56, 5917–5933 (2019). https://doi.org/10.1007/s12035-018-1455-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1455-4