
Abstract
Recent evidence strongly supports the idea that common general anesthetics (GAs) such as isoflurane (Iso) and nitrous oxide (N2O; laughing gas), as well as sedative drugs such as midazolam are neurotoxic for the developing mammalian brain having deleterious effects on neural circuits involved in cognition, learning and memory. However, to date, very little is known about epigenetic mechanisms involved in GA-induced plasticity of synaptic transmission in the hippocampus, the main memory-processing region in the brain. Here, we used patch-clamp recordings of miniature inhibitory post-synaptic currents (mIPSCs) from hippocampal neurons in slice cultures exposed to the clinically relevant GA combination. We found that in vitro exposure to a combination of midazolam, 0.75% Iso, and 70% N2O for 6 h leads to lasting increase in frequency of mIPSCs, while amplitudes and kinetics of the events were spared. Importantly, co-application of entinostat (MS-275), a selective inhibitor of class I histone deacetylases (HDAC), completely reversed GA-induced synaptic plasticity. Furthermore, when given in vivo to P7 pups exposed to GA with midazolam, Iso and N2O for 6 h, MS-275 reversed GA-induced histone-3 hypoacetylation as shown by an increase in Ac-H3 protein expression in the hippocampus. We conclude that exposure to a combination of Iso with N2O and midazolam causes plasticity of mIPSCs in hippocampal neurons by epigenetic mechanisms that target presynaptic sites. We hypothesize that GA-induced epigenetic alterations in inhibitory synaptic transmission in the hippocampus may contribute to altered neuronal excitability and consequently abnormal learning and memory later in life.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most currently used GA agents have either N-methyl-D-aspartate (NMDA) receptor-blocking or/and γ-aminobutyric acid A (GABAA) receptor-enhancing properties, which are thought to be essential for their clinically useful properties such as sedation and hypnosis [1]. Unfortunately, it has been well documented that increased activation of GABAA receptors and/or blockade of NMDA receptors can trigger widespread neurodegeneration in many regions of developing rodent and non-human primate brains including the hippocampus [2,3,4,5,6]. Although human studies addressing the issue of safety of clinical anesthesia in the developing brain are still at an early stage, at least some concerns have been raised in regard to learning disability later in life following early exposure to GAs [7].
Plasticity of both excitatory and inhibitory synaptic transmission in hippocampus and hippocampal-cortical circuitry is strongly implicated in cognitive functions, learning, and memory [8,9,10]. We have previously published that in vivo exposure to common GAs during critical brain development preferentially alters inhibitory synaptic transmission in subiculum, a main output structure of hippocampus [11]. In another study, we showed that exposure of rat pups at the age of P7 to a clinically relevant anesthetic cocktail consisting of 0.75% isoflurane (Iso), 70% N2O, and 9 mg/kg of midazolam triggers lasting plasticity of synaptic (both inhibitory and excitatory) and intrinsic ion channels such as T-type calcium channels (T-channels) in thalamic inhibitory interneurons [12]. Collectively, these studies signify importance of lasting alterations of synaptic transmission in different brain regions following exposure to GAs during critical brain development period. However, molecular mechanisms of synaptic plasticity induced by an early exposure to GAs are not well studied. We have recently suggested using a global HDAC inhibitor, sodium butyrate (NaB), a strong and important role of epigenetic mechanisms in neurotoxicity and lasting functional impairment in rodent brain exposed to GA at critical age of P7 [13]. This signifies that future research is warranted to investigate epigenetic mechanisms by which common GAs may affect neuronal communication in the immature brain and to develop possible therapeutic strategies that could unable safer pediatric anesthesia practice. Here, we used in vitro patch-clamp recordings in cultured hippocampal slices and in vivo experiments with a selective class I HDAC inhibitor [N-(2-aminophenyl)-4-[N-(pyridine-3-ylmethoxy-carbonyl)aminomethyl]benzamide)] (entinostat, MS-275). Our main goal for this study was to further investigate possible epigenetic mechanisms involved in lasting alterations of inhibitory synaptic transmission after exposure of immature brain to GA, as assessed by properties of mIPSCs mediated by GABAA receptors.
Materials and Methods
Anesthesia Delivery In Vivo
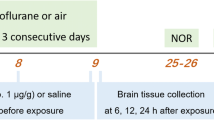
At post-natal day 7 (P7), both male and female Sprague-Dawley rats were randomly assigned to experimental group and then exposed to 6 h of clinically relevant triple cocktail of anesthesia with combination of midazolam 9 mg/kg intraperitoneally (i.p.), 70% N2O, 25% oxygen, plus 0.75% Iso. Typically, sham controls were littermates exposed to 6 h of mock anesthesia consisting of separation from their mother in an air-filled chamber and i.p. injections of dimethyl sulfoxide (DMSO), a vehicle used to dissolve midazolam (Fig. 1A). An agent-specific vaporizer was used to deliver a set percentage of Iso with a mixture of O2 and N2O gases into a temperature-controlled chamber preset to maintain 33–34 °C. The composition of the gas chamber was analyzed using real-time feedback (Datex Capnomac Ultima) for N2O, Iso, CO2, and O2 percentages. For control (sham) animals, 0.1% DMSO was used to substitute midazolam. Tissue collections for western blot studies were done at 24-h post-exposure to GA. The experiments were approved by the Institutional Animal Use and Care Committee of the University of Colorado Anschutz Medical Campus, Aurora, CO and by the Animal Use and Care Committee of the University of Virginia, Charlottesville, VA. All experiments were done in accordance with the Public Health Service’s Policy on Humane Care and Use of Laboratory Animals. Efforts were made to minimize the number of animals used.

Scheme depicts timeline of events in our experimental procedures
Anesthesia Delivery In Vitro
Modulation of hippocampal synaptic transmission was assessed with electrophysiological patch-clamp recordings by focusing on synaptic GABAA-mediated currents. We used hippocampal slices from P7 rat pups and cultured for up to 4 weeks following the procedure described elsewhere [13, 14]. Figure 1B summarizes our experiments where experimental slice cultures were exposed to our routine GA protocol (25μg/ml midazolam, 70% nitrous oxide, and 0.75% isoflurane for 6 h). Sham cultures were exposed to 1% DMSO for 6 h. GABA-dependent miniature inhibitory post-synaptic currents (mIPSCs) were recorded in pyramidal neurons of the CA1 hippocampal region 1–10 days post-treatment in the presence of 1μM tetrodotoxin (TTX) (to block action potentials) and 10μM NBQX and 50μM d-APV (to block AMPA and NMDA currents, respectively).
Western Blot Studies
For protein quantification, we dissected the hippocampus, including the subiculum, immediately after the brains were removed from the individual pups using a dissecting scope (×10 magnification). Tissue was collected on ice and was snap-frozen immediately in liquid nitrogen. The protein concentration of the lysates was determined with the Total Protein Kit using the Bradford method (Cayman Chemical, Ann Arbor, MI). Ten to twenty-five micrograms of total protein was heat-denaturized, separated by SDS-PAGE through 4–20% Tris-glycine polyacrylamide gradient gels (Bio Rad, Hercules, CA). Proteins were transferred to PVDF membrane (Millipore, Billerica, MA), blocked at room temperature for 1 h in 3% bovine serum albumin (BSA), and followed by incubation at 4 °C overnight with primary antibodies such as rabbit polyclonal anti-acetyl-histone H3 (1:3000, 06-599, Millipore, Billerica, MA) or anti-GAPDH (1:16,500 Millipore, Billerica, MA) antibodies as loading controls.
Membranes were incubated for 1 h at room temperature with peroxidase conjugated (HRP) secondary antibodies goat anti-rabbit IgG (1:10,000, Santa Cruz, Dallas, TX). Immunoreactivity was detected by enhanced chemiluminescence substrate (Super Signal west Femto; Thermo Scientific, UT). Images were captured using GBOX (Chemi XR 5, Syngene, MD) and gels were analyzed densitometrically with the computerized image analysis program ImageQuant 5.0 (GE Healthcare, Life Sciences, Piscataway, NJ).
Recording Procedures
The standard extracellular solution for recording of mIPSCs consisted in mM, of 2 CaCl2, 130 NaCl, 1 MgCl2, 10 glucose, 26 NaHCO3, 1.25 NaH2PO4, 2 KCl, and 0.001 TTX. For recordings of mIPSCs, we used an internal solution containing, in mM, 130 KCl, 4 NaCl, 0.5 CaCl2, 5 EGTA, 10 HEPES, 2 MgATP2, and 0.5 Tris-GTP. To eliminate glutamatergic excitatory currents, all recordings of mIPSCs were done in the presence of 10μM 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) and 50μM (2R)-amino-5-phosphonovaleric acid (d-APV); (2R)-amino-5 phosphonopentanoate (AP5). All experiments were done at room temperature (20–24 °C). Whole-cell recordings were obtained from hippocampal neurons visualized with an infrared (IR) DIC camera (Hammamatsu, C2400) on a Zeiss 2 FS Axioscope (Carl Zeiss, Jena) with a ×40 lens.
Electrodes were fabricated from thin-walled microcapillary glass with a final resistance of 3–6 MΩ. Membrane currents were recorded with an Axoclamp 200B amplifier (Molecular Devices, Foster City, CA). Voltage commands and digitization of membrane currents were done with Clampex 8.2 of the pClamp software package (Molecular Devices) running on an IBM-compatible computer. Neurons typically were held at − 70 mV. Currents were filtered at 5–10 kHz.
Analysis of Current
All data were analyzed using MiniAnalysis software (Synaptasoft). The limits for mIPSCs were set in most of recordings at three times the root mean square of baseline noise. In our analysis of kinetics of spontaneous synaptic currents, we included only isolated (i.e. non-overlapping) events. All mIPSCs were analyzed with respect to peak amplitude and 10–90% rise time and fastest events (rise times ≤ 3 ms) where chosen for further analysis of decay kinetics assessed by measuring half-width of isolated events.
Cumulative Distribution Functions
Plots describing the cumulative distribution functions (CDFs) of mIPSC properties (i.e. amplitude, inter-event interval (IEI) and half-width) were derived empirically using custom scripts written in Matlab (Mathworks, Natick, MA). In brief, the CDF describes the probability that an event amplitude (or IEI, half-width) will be found that is less than or equal to that event. CDFs were derived for individual neurons, as well as for data pooled from all neurons.
Statistical Analysis
Statistical analysis was done with two-tailed Student’s t test, one-way ANOVA, and Mann-Whitney rank sum test where indicated, with statistical significance determined at p < 0.05.
Drugs and Chemicals
Isoflurane was obtained from Abbott (Abbott Park, IL). All other salts and chemicals were obtained from Sigma Chemical (St. Louis, MO). All drugs were prepared as stock solutions and were diluted freshly to the appropriate concentrations at the time of experiments. All stocks were prepared in sterile water except for NBQX and MS-275, which were prepared in DMSO. The maximum final concentration of DMSO in any one experiment was 1% and was also used in sham (control) exposures of hippocampal slice cultures.
Results
To further assess whether agents that modulate H3 acetylation can reverse GA-induced H3 hypoacetylation, we tested in our experiments a selective class I HDAC inhibitor, MS-275. Bar graphs from western blots of hippocampal tissues on Fig. 2A depict average effect of GA exposure (black bar) when compared to sham group (green bar) with a decrease in Ac-H3 protein expression of about 50% (***, p < 0.001, n = 6). MS-275, at 10 mg/kg [15] given 60 min before triple cocktail, caused significant up-regulation of H3 acetylation (about 40%) compared to GA alone (**, p < 0.01, Fig. 2A, red bar). The magnitude of the effect was sufficient to significantly improve H3 acetylation status as compared with GA animals (**, p < 0.01). At this does, MS-275 + GA group still remained significantly decreased compared to sham controls (Fig. 2A, red bar; *, p < 0.05). MS-275 had no effect on the baseline H3 acetylation compared to sham animals (blue bar, p = 0.41). Panels on Fig. 2B depict raw data from all western blot experiments used to generate graphs on Fig. 2A (n = 6 animals per group).

Selective class I HDAC inhibitor MS-275 reverses GA-induced histone-3 hypoacetylation in the immature hippocampus. MS-275 at 10 mg/kg was given 60 min before tissue collection which was done 24 h after administration of GA with 9 mg/kg i.p. of midazolam, 0.75% Iso, and 70% N20. GAPDH glyceraldehyde phosphate dehydrogenase. a GA-induced H3 hypoacetylation, as evidenced by about 50% decrease levels of Ac-H3 protein expression, was almost significantly reversed (about 20% decrease) with MS-275 treatment when compared to sham controls. Note that MS-275 given alone did not significantly affect levels of Ac-H3. (*, p < 0.05; **, p < 0.01; ***, p < 0.001). b Original western blot gels from all experiments used to generate graphs with average data presented on panel a of this figure (n = 6 pups per data point in all cohorts)
Modulation of hippocampal synaptic transmission is an important form of synaptic plasticity thought to underlie the storage and processing of memories in the hippocampus [8]. To begin to understand the link between GA-induced epigenetic modulations and functional alteration of synaptic transmission, we performed electrophysiological patch-clamp recordings of synaptic GABAA-mediated currents using hippocampal slices from P7 rat pups that were cultured for up to 4 weeks [13, 14]. Slices were exposed to our routine GA protocol in vitro (25μg/ml midazolam, 70% nitrous oxide, and 0.75% isoflurane for 6 h). GABA-dependent mIPSCs were recorded in pyramidal neurons of the CA1 hippocampal region 1–10 days post-treatment. Representative traces from these experiments are depicted in Fig. 3A in the following order from the top: sham group (green trace), GA group (black trace), MS-275 (blue trace), and GA + MS-275 (red trace). Quantification of similar recordings from multiple cells is depicted in Fig. 3B and showed about 40% increase in the frequency of mIPSCs in GA-treated slices (black bar in middle panel) compared with sham controls (green bar in middle panel *, p < 0.05). Importantly, co-treatment with 10μM MS-275 completely reversed GA-induced up-regulation in frequency of mIPSCs (Fig. 3B, blue bar in middle panel). Furthermore, when MS-275 was given alone it had no significant effect on frequency of mIPSCs when compared to sham controls (Fig. 3B, red bar in middle panel). Similarly, there was no significant difference among the groups in mIPSCs amplitudes (bottom panel of Fig. 3B) or their decay time kinetics estimated by measuring half-widths (top panel of Fig. 3B). Total number of cells per group is indicated in parentheses on the X axis of Fig. 3B.

MS-275, applied 60 min before anesthesia exposure, completely reversed increased frequency of mIPSCs in hippocampal slice cultures exposed to the combination of 25μg/ml of midazolam, 0.75% Iso, and 70% N2O. a Sample of original mIPSC traces from sham (green), GA-treated (black), MS-275-treated (blue), and GA + MS-275-treated hippocampal neurons. b Graphs representing the effects of GA, MS-275, and their combination on half-width, frequency, and amplitude of mIPSCs. Note that the application of MS-275 (10 μM) reversed the GA-induced increase in mIPSC frequency. Number of neurons in each group is indicated in parenthesis bellow the X axis of the bottom panel of this figure. c Plots describing the cumulative distribution functions (CDFs) of average mIPSC events in four treatment groups demonstrate very little change in the half-width and amplitude (upper and lower panels), as opposed to frequency (middle panel) of events. Number of events in each cohort is as follows: sham (green lines) 3910; GA (black lines) 4227; MS-275 (blue lines) 3233; MS-275 + GA (red lines) 2247
Cumulative distribution fractions (CDFs) of mIPSCs amplitudes, decay times, and frequencies were also measured from IEIs from all events from the recordings in the sham group (green lines), GA group (black lines), MS-275 group (blue lines), and GA + MS-275 groups (red lines) and shown in Fig. 3C. CDFs were first derived for individual neurons and then for data pooled from all neurons. The graphs of CDFs of half-widths of mIPSCs (top panel in Fig. 3C) and amplitudes of events (bottom panel in Fig. 3C) show very little change after GA treatment. In contrast, middle panel in Fig. 3C indicates that GA treatment (solid black line) caused CDF curve for IEIs to shift to the left when compared to sham (green solid line) indicating decreased IEIs. CDF curve for MS-275 treatment alone (blue line) largely overlapped with sham controls. Furthermore, treatment of slices with combination of GA + MS-275 (red solid line) induced a slight shift of CDF to the right when compared to sham controls. Overall, decreased IEIs with GA treatment were consistent with increased frequency of mIPSCs as depicted on Fig. 3B (middle panel).
Discussion
Here, we found that 6-h-long GA treatment increased frequency of mIPSCs that was completely reversed by treatment of slices with MS-275, while GA minimally affected decay kinetics and amplitude of events in the same cells. It is generally accepted in studies of mIPSCs that presynaptic modulators alter the frequency, whereas post-synaptic modulators alter the amplitude and/or decay of post-synaptic events. Hence, our data suggest that GA alters presynaptic component(s) of inhibitory neurotransmission and that this alteration is influenced by the state of histone-3 acetylation. Since GABAA currents are excitatory in the developing hippocampal neurons, we conclude that GA may induce lasting and perhaps excessive depolarization of hippocampal neurons by presynaptic mechanism(s).
It has been well established that even single exposure to anesthesia causes substantial neurodegenerative changes in the brain [2,3,4,5,6]. Interestingly, Chalon and colleagues reported in 1981 long-term learning difficulties in the first generation of mice exposed in utero to general anesthetics such as halothane and enflurane, as well as second generation offspring never exposed to GA [16]. This suggests that even single exposure to GA during critical period of brain development causes changes that become embedded in the genetic information, which in turn results in the impairment of proper and timely neuronal development. However, although a crucial role for epigenetic changes in long-term memory formation has been reasonably well documented, the importance of epigenetic changes in GA-induced cognitive impairments has only recently been reported by our group [13]. In that study, we described using both in vivo and in vitro approaches that exposure of P7 rat pups to GA with triple anesthetic cocktail consisting of midazolam, Iso, and N2O caused epigenetic modulation manifested as hystone-3 hypoacetylation. Furthermore, we found that reversal of histone hypoacetylation with a broad HDAC inhibitor NaB blocked GA-induced morphological and functional impairment of neuronal development, such as alterations in inhibitory synaptic transmission [13]. Here, we continue our studies using patch-clamp recordings of mIPSCs from hippocampal neurons in slice cultures exposed to the same clinically relevant GA combination as used in our previous studies. We conclude that exposure to a combination of Iso with N2O and midazolam causes plasticity of mIPSCs in hippocampal neurons by Ac-H3-mediated epigenetic mechanisms that target presynaptic sites. We further hypothesize that GA-induced epigenetic alterations in inhibitory synaptic transmission in the hippocampus may contribute to altered neuronal excitability and consequently abnormal learning and memory processing later in life.
We have previously reported that treatment with the same GA cocktail of immature neurons causes alterations in inhibitory synaptic transmission mediated by GABAA receptors in different preparations. For example, we used an acute slice preparation of subiculum from adolescent rats and reported that GA treatment at age of P7 in vivo caused lasting decrease of the amplitudes of evoked inhibitory post-synaptic currents (eIPSCs) and speeded decay time constant of eIPSCs with alterations of paired-pulse ratio suggesting that both presynaptic and post-synaptic mechanisms contribute [11]. In contrast, 24-h-long in vitro exposure of Iso, N2O, and midazolam in CA1 region of hippocampal slice cultures caused slowing of kinetics of mIPSCs suggesting involvement of predominantly post-synaptic mechanisms [11]. Interestingly, shorter applications (6 h) of the same GA cocktail in the same preparation used in current study resulted in increased frequency of mIPSCs without alterations of the amplitudes and current kinetics, which in turn implicates presynaptic mechanisms of modulation. Hence, lasting effects of GA on inhibitory synaptic transmission may differ in various preparations (subiculum vs. CA1), mode of application (in vivo vs. in vitro), and with different durations (6 vs. 24 h) of GA exposure.
Epigenetic mechanisms were also implicated in alterations of inhibitory synaptic transmission that have been reported after exposure to other neurotoxic drugs. For example, cocaine administration induced changes in GABAA receptor subunit expression and increased frequency of mIPSCs in nucleus accumbens neurons, an effect which was completely reversed with administration of MS-275 [17]. In a more recent study, Subburaju et al. [18] showed that HDAC1 isoform of class I HDACs modulates the expression of GAD65 and 67, the key genes involved in the synthesis of GABA, in stratum oriens of hippocampal CA3/2 region. Moreover, it appears that this modulation happens primarily within GABAergic interneurons, which could also explain the effects of MS-275 on GA-induced increase in frequency of mIPSCs observed in our experiments.
In addition, it was shown that MS-275 is beneficial in different animal models of depression, as well as fear, anxiety, and trauma-related psychiatric disorders [19, 20]. Regarding the role of histone acetylation in memory formation, it has been shown that during certain behavioral tasks, acetylation occurs preferentially in learning- and memory-associated genes (e.g., cAMP-response-element-binding protein—Creb1) within areas such as hippocampus, amygdala, and prefrontal cortex [21]. More specifically, within class I of HDACs, it appears that hippocampal HDAC1 isoform regulates the extinction of contextual fear memories [22], whereas HDAC2-knockout mice exhibit enhanced memory performance and facilitated synaptic plasticity [23]. Finally, the selective deletion of hippocampal HDAC3 enhanced object-location memory [24]. Bearing in mind that MS-275, at the concentration used in our study, most likely blocked HDAC1–3 isoforms [25], it is expected that this compound may have significant effects on synaptic plasticity and memory processing in the hippocampus.
We conclude that further comprehensive preclinical and clinical studies are needed to establish a possible link and precise mechanisms of anesthetic-induced plasticity of inhibitory synaptic transmission in hippocampus, epigenetics, and disturbances in learning and memory that was observed in young animals and children after exposure to GA at the critical period of brain development. This and future studies may provide a rationale for new strategies to prevent abnormalities and/or to normalize synaptic function and neuronal excitability of the hippocampus and hippocampal-cortical circuitry after exposure to GA. The ultimate clinical value lies in being able to devise more targeted and individualized preventive interventions that are based on newly acquired understanding of how anesthesia modulates histones and target genes in the developing neurons of the most sensitive individuals and thus to provide safer pediatric anesthesia.
References
Franks NP (2008) General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci 9(5):370–386
Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF (2003) Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci 23(3):876–882
Zou X, Liu F, Zhang X, Patterson TA, Callicott R, Liu S, Hanig JP, Paule MG et al (2011) Inhalation anesthetic-induced neuronal damage in the developing rhesus monkey. Neurotoxicol Teratol 33(5):592–597
Zou X, Patterson TA, Divine RL, Sadovova N, Zhang X, Hanig JP, Paule MG, Slikker W Jr et al (2009) Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int J Dev Neurosci 27(7):727–731
Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, Dissen GA, Creeley CE et al (2010) Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology 112(4):834–841
Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Martin LD, Dissen GA, Creeley CE et al (2012) Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology 116(2):372–384
Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, Gleich SJ, Schroeder DR et al (2009) Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology 110(4):796–804
Mody I (2005) Aspect of the homeostatic plasticity of GABAA receptor-mediated inhibition. J Phyisol 562(1):37–46
Tokuda K, O’Deli KA, Izumi Y, Zorumski CF (2010) Midazolam inhibits hippocampal long-term potentiation and learning through dual central and peripheral benzodiazepine receptor activation and neurosteroidogenesis. J Neurosci 30(5):16788–16795
Zorumski CF, Izumi Y (2012) NMDA receptors and metaplasticity: mechanisms and possible roles in neuropsychiatric disorders. Neurosci Biobehav Rev 36(3):989–1000
Sanchez V, Feinstein SD, Lunardi N, Joksovic PM, Boscolo A, Todorovic SM, Jevtovic-Todorovic V (2011) Long-term impairment of mitochondrial morphogenesis and synaptic transmission in developing rat brain. Anesthesiology 115(5):992–1002
DiGruccio MR, Joksimovic S, Joksovic PM, Lunardi N, Salajegheh R, Jevtovic-Todorovic V, Beenhakker MP, Goodkin HP et al (2014) Hyperexcitability of rat thalamocortical networks after exposure to general anesthesia during brain development. J Neurosci 35(4):1481–1492
Dalla Massara L, Osuru HP, Oklopcic A, Milanovic D, Joksimovic SM, Caputo V, DiGruccio MR, Ori C et al (2016) General anesthesia causes epigenetic histone modulation of c-Fos and brain-derived neurotrophic factor, target genes important for neuronal development in the immature rat hippocampus. Anesthesiology 124(6):1311–1327
McCormack SG, Stornetta RL, Zhu JJ (2006) Synaptic AMPA receptor exchange maintains bidirectional plasticity. Neuron 50:75–88
Whittle N, Schmuckermair C, Gunduz Cinar O, Hauschild M, Ferraguti F, Holmes A, Singewald N (2013) Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in a genetic mouse model. Neuropharmacology 64:414–423
Chalon J, Tang CK, Ramanathan S, Eisner M, Katz R, Turndorf (1981) Exposure to halothane and enflurane affects learning function of murine progeny. Anesth Analg 60:794–797
Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, Chaudhury D, Damez-werno DM et al (2013) Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat Neurosci 16(4):434–440
Subburaju S, Coleman AJ, Cunningham MG, Ruzicka WB, Benes FM (2016) Epigenetic regulation of glutamic acid decarboxylase 67 in a hippocampal circuit. Cereb Cortex
Covington HE, Maze I, LaPlant QC, Vialou VF, Yoshinori ON, Berton O, Fass DM, Renthal W et al (2009) Antidepressant actions of HDAC inhibitors. J Neurosci 29(37):11451–11460
Singewald N, Schumuckermair C, Whittle N, Holmes RKJ (2015) Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther 149:150–190
Graff J, Tsai LH (2013) The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol 53:311–330
Bahari-Javan S, Maddalena A, Kerimoglu C, Wittnam J, Held T et al (2012) HDAC1 regulates fear extinction in mice. J Neurosci 32:5062–5073
Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N et al (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459:55–60
McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA et al (2011) HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci 31:764–774
Khan N, Jeffers M, Kumar S, Hackett C, Boldog F et al (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 409(2):581–589
Acknowledgments
We thank Drs. G. Wang and J.J. Zhu for preparation of hippocampal slice cultures.
Funding Information
This study was supported in part by NIH grants HD-044517 (VJT), HD-044517S (VJT), GM 118197 (VJT), GM-102525 (SMT), March of Dimes (VJT), Harold Carron Endowment (VJT), and funds from the Department of Anesthesiology at University of Virginia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Joksimovic, S.M., Osuru, H.P., Oklopcic, A. et al. Histone Deacetylase Inhibitor Entinostat (MS-275) Restores Anesthesia-induced Alteration of Inhibitory Synaptic Transmission in the Developing Rat Hippocampus. Mol Neurobiol 55, 222–228 (2018). https://doi.org/10.1007/s12035-017-0735-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0735-8