
Abstract
Mounting evidence suggests that aberrations in immune-inflammatory pathways contribute to the pathophysiology of major depressive disorder (MDD), and individuals with MDD may have elevated levels of predominantly pro-inflammatory cytokines and C-reactive protein. In addition, previous meta-analyses suggest that antidepressant drug treatment may decrease peripheral levels of interleukin-1 beta (IL-1β) and IL-6. Recently, several new studies examining the effect of antidepressants on these cytokines have been published, and so we performed an updated meta-analysis of studies that measured peripheral levels of cytokines and chemokines during antidepressant treatment in patients with MDD. The PubMed/MEDLINE, EMBASE, and PsycInfo databases were searched from inception through March 9, 2017. Forty-five studies met inclusion criteria (N = 1517). Peripheral levels of IL-6, tumor necrosis factor-alpha (TNF-α), IL-1β, IL-10, IL-2, IL-4, interferon-γ, IL-8, the C-C motif ligand 2 chemokine (CCL-2), CCL-3, IL-1 receptor antagonist, IL-13, IL-17, IL-5, IL-7, and the soluble IL-2 receptor were measured in at least three datasets and thus were meta-analyzed. Antidepressant treatment significantly decreased peripheral levels of IL-6 (Hedges g = −0.454, P <0.001), TNF-α (g = −0.202, P = 0.015), IL-10 (g = −0.566, P = 0.012), and CCL-2 (g = −1.502, P = 0.006). These findings indicate that antidepressants decrease several markers of peripheral inflammation. However, this meta-analysis did not provide evidence that reductions in peripheral inflammation are associated with antidepressant treatment response although few studies provided separate data for treatment responders and non-responders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Accumulating evidence indicates that activation of immune-inflammatory pathways may contribute to the development of major depressive disorder (MDD) in at least a subset of patients [1, 2]. In particular, activation of cell-mediated immunity (CMI) may play a significant role in the biology of MDD [3]. Cytokines and chemokines are key regulators of immune function, with different roles (for example, some of these mediators are predominantly pro-inflammatory, whereas others are mainly anti-inflammatory) [4, 5]. A recent meta-analysis of 82 studies found elevated peripheral levels of interleukin-6 (IL-6), tumor necrosis factor (TNF)-alpha, IL-10, the soluble IL-2 receptor, C-C chemokine ligand 2 (CCL-2), IL-13, IL-18, IL-12, the IL-1 receptor antagonist, and the soluble TNF receptor 2 in patients with MDD compared to healthy controls [6].
Most antidepressants are thought to primarily act by increasing or otherwise modulating monoamine function [7]. However, emerging evidence suggests that immune mechanisms may contribute to the therapeutic benefits of some of these drugs [8]. For example, a study found that selective serotonin reuptake inhibitors (SSRIs) but not venlafaxine inhibited lipopolysaccharide (LPS)-stimulated microglia in vitro [9]. A previous meta-analysis provided evidence that antidepressant drugs may decrease IL-1β levels in patients with MDD [10], whereas another meta-analysis indicated that antidepressants decrease IL-6 levels [11]. Heterogeneity across studies was high in these estimates. Since these meta-analyses were published, new studies have appeared in the literature [12, 13]. In addition, a few studies suggest that peripheral immune activation may predict treatment non-response [12, 14].
This present systematic review and meta-analysis aims to reassess available evidence of the effects of antidepressants on peripheral levels of cytokines and chemokines in individuals with MDD. In addition, we sought to explore potential sources of heterogeneity across studies and to investigate whether antidepressant-related changes in cytokine/chemokine levels differed between treatment responders and non-responders.
Methods
This study comprised a within-group meta-analysis of studies that compared peripheral levels of cytokines and chemokines in participants with MDD at baseline and after treatment with an approved antidepressant. We complied with the Preferred Reported Items for Systematic Reviews and Meta-analysis (PRISMA) statement [15]. The literature search, title/abstract screening, final decision on eligibility after full-text-review, and data extraction were independently performed by two investigators (THF and NQA). Disagreements were resolved through consensus. If a consensus could not be achieved, the decision was made independently by a third investigator (CAK).
Search Strategy
A systematic search was conducted in the PubMed/MEDLINE, EMBASE, and PsycInfo databases from inception up until March 9, 2017. The detailed search strings used in this review are presented in the supplementary online material that accompanies the online version of this article. This search strategy was augmented by tracking the citation lists of included articles in Google Scholar [16].
Study Selection
We included original peer-reviewed articles published in any language. Eligible studies had to measure peripheral cytokine or chemokine levels in adult subjects (age ≥18 years old) who met either DSM [17] or ICD [18] criteria for MDD. The following exclusion criteria were adopted: (1) studies in which participants had medical and/or psychiatric comorbidities (except current smoking), (2) studies which included pregnant women or women in the postpartum period, (3) case reports or case series (N < 10), (4) studies that assayed the immune variables in specimens/tissues other than blood (e.g., CSF), (5) studies in animals or assessing cytokine/chemokine production in vitro, and (6) studies which included other interventions (e.g., exercise) unless data for patients treated with antidepressants were separately provided. The authors of meeting abstracts which met inclusion criteria were contacted by e-mail to provide data for analysis (no additional data were provided).
Data Extraction
For each immune mediator, we extracted means, variance estimates (standard deviation (SD), standard error of the mean (SEM), or 95% confidence interval (CI)) and sample sizes of each study. In studies that provided median ± IQR or median ± range, we estimated the mean ± SD following a standard procedure [19]. For purposes of data extraction, we considered cytokine/chemokine levels at baseline and at the time during treatment when the largest number of participants was included in analysis (follow-up time ≥4 weeks). We also extracted the following data whenever available: (1) first author, (2) publication year, (4) gender distribution (% females), (5) mean age and body mass index (BMI), (6) mean illness duration (years), (7) treatment status (drug-free during assessment and/or treatment-naïve), (8) measurement of depressive symptoms at baseline and endpoint, (9) response rates (defined as the percentage of participants who achieved a 50% reduction in baseline depression scores at endpoint), (9) follow-up time (weeks), (10) studies in which a single antidepressant class was used vs. those in which agents from more than one antidepressant class were used, (11) frequency of melancholic and atypical depression in the sample, and (12) current smokers (%). For studies that included healthy controls (HCs), we also extracted the following data in these participants: (1) sample size and (2) chemokine/cytokine levels.
Methodological Quality Assessment of Included Studies
Seven parameters were used to estimate the methodological quality of included studies: (1) enrolled at least 40 participants with MDD at baseline (1 = Yes; 0 = No), (2) attrition rate ≤20% (1 = Yes; 0 = No), (3) provided treatment response rates (1 = Yes; 0 = No), (4) contrasted cytokine/chemokine levels between responders versus non-responders (1 = Yes; 0 = No), (5) a washout period was conducted prior to trial initiation or otherwise participants were treatment-naïve (1 = Yes; 0 = No), (6) time of sample collection was reported (e.g. morning vs. evening) (1 = Yes; 0 = No), and (7) the manufacturer of the test was reported (and test parameters could be verified at the proper website) or other test parameters were provided (i.e., detection limit and coefficient of variation was reported).
Statistical Analysis
Because studies used different measurement methods, we estimated a standardized mean difference (Hedges’s g) and 95% confidence intervals (CIs) for each immune mediator, which provides an unbiased effect size (ES) adjusted for small sample sizes [20]. We assessed the heterogeneity across studies using the Cochran Q test, a weighted sum of the squares of the deviations of individual study ES estimates from the overall estimate. In addition, heterogeneity across studies was quantified with the I 2 statistic, which in brief indicates the percentage of total variation across several studies due to heterogeneity, and it is considered large when ≥50% [21]. We anticipated a high degree of heterogeneity. Therefore, we pooled ES using a random-effects model according to the DerSimonian and Laird, using the inverse variance method to estimate heterogeneity [22]. Meta-analyses were carried only for mediators with at least three individual datasets. Random-effects modeling assumes a genuine diversity across studies and incorporates a between-study variance into the calculations [20]. An ES of 0.2 was considered low, 0.5 moderate, and 0.8 large [23].
We computed composite measures to provide an indication of the profiles of peripheral immune activation involved in MDD (i.e., T Helper (TH1), TH2, regulatory T cells (TRegs), and macrophage polarized M1 phenotype responses). To this end, we averaged the ES estimates from each study which contributed with a mediator included in the a priori defined biosignatures. The rationale for the estimation of each aggregate measure is provided in the Supplementary online material that accompanies the online version of this article (Supplementary Table S1 describes the mediators and number of studies that contributed to each composite ES estimate, available online).
Studies with statistically non-significant (i.e., negative) results are less likely to be published than studies with significant results [24, 25]. To assess publication bias, we inspected a funnel plot graph for asymmetry, and calculated the Egger’s regression test for funnel plot asymmetry [26]. Evidence of small-study effects (indicative of publication bias) was considered when the P value of the Egger’s test was <0.1 and the ES of the largest study was more conservative or changed direction when compared with the overall ES estimate (funnel plots of ES estimates in which evidence of publication bias was observed are presented in Supplementary Figs. S12–S14, available online) [25]. The trim-and-fill procedure was used to estimate the ES adjusting to publication bias [27], while the fail-safe N (i.e., the file drawer) statistic was used to determine how many additional studies would be necessary to turn significant ES non-significant [28].
We explored potential sources of heterogeneity across studies in each mediator, using either subgroup (if there were at least three studies in each subgroup) or random-effects meta-regression analyses (if there were at least five studies with available moderator data). We grouped studies in which response rates were above the median value for a specific immune mediator and contrasted them with studies in which response rates were below the median value. Fewer datasets often provide underpowered and unreliable estimates [29]. The following variables were considered in meta-regression analyses: sample size, mean age, mean BMI, gender distribution (% females), percent of current smokers, response rates (%), changes in depressive symptoms from baseline (normalized to the threshold for severe depression for each rating scale), mean illness duration in years, and treatment response rate (%). Studies were weighted in such a way that investigations with more precise parameters (indicated by sample size and 95% CIs) had more influence in meta-regression analyses [29]. For statistically significant ES estimates, we performed sensitivity analyses in which we excluded each study from analyses to verify whether a single study turned results non-significant or otherwise changed the direction of the ES. In addition, a cumulative meta-analysis was performed for immune mediators with significant ES estimates and at least 10 datasets. These analyses address the influence of new studies on prior pooled results. For these analyses, individual data sets were sorted in chronological order. The earliest available study was included in the analysis first. At each subsequent step of the cumulative meta-analysis, one more study was included in the analysis, and the summary ES and 95% CI were recalculated. The “Proteus phenomenon” refers to the situation in which the first published studies are often the most biased toward inflated effect sizes (i.e., the winner’s curse); subsequent replication studies tend to be less biased toward the extreme, often finding evidence of smaller effects or even contradicting the findings of initial studies. Thus, cumulative meta-analyses allow the appreciation of these phenomena.
All analyses were conducted in the Stata MP software version 14.0 (Stata-Corp, College Station, TX, USA) using the metan package. Statistical significance was considered at an alpha level of 0.05.
Results
Study Selection
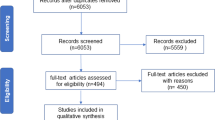
Following removal of duplicates, the title/abstracts of 5521 unique references were screened for eligibility. A total of 5102 references were excluded, while 419 full texts were retrieved and screened for eligibility. Of those articles, 374 were excluded (see Supplementary Table S2 that accompanies to online version of this article for reasons for exclusion). Finally, 45 original studies met inclusion criteria. Figure 1 provides the PRISMA flowchart for study selection.

PRISMA flowchart of study selection
Characteristics and Methodological Quality of Included Studies
A total of 45 studies were included (N = 1517). The mean follow-up was 7.6 weeks (SD = 3.3; range = 4–20). Twenty-one studies (46.7%) included only SSRIs, while 5 (11.1%) investigations included only SNRIs and 19 (42.2%) included “miscellaneous” antidepressants. Thirteen (28.9%) contrasted cytokine/chemokine levels between treatment responders and non-responders. Characteristics of included studies are provided in Supplementary Table S3 (available online).
Quality scores of the included studies ranged from 2 to 7 (median 4). The scores of each study are presented in supplementary Table S3 (available online).
Interleukin-6
Interleukin-6 (IL-6) was measured in 24 studies (N = 722). Antidepressant treatment significantly reduced IL-6 levels with a moderate ES (Hedge’s g = −0.454) (Table 1; Fig. 2a). Heterogeneity was large (I 2 = 84.7%). There was no evidence of publication bias. In meta-regression analysis, mean difference in depressive symptom scores was a significant moderator (the higher the difference in depressive symptom scores, the lower the difference in IL-6 levels between endpoint and baseline) (Supplementary Table S4, available online). Heterogeneity was higher in SSRI studies compared to studies using “miscellaneous” antidepressants (Supplementary Table S5, available online). Finally, IL-6 levels decreased in studies which sampled this cytokine from plasma, but not in those in which IL-6 was sampled from serum (Table S5, available online). In sensitivity analysis, the exclusion of any single study one-at-a-time did not alter the direction or statistical significance of the ES (Fig. S15). In cumulative meta-analysis, this ES estimate has been consistent since 2005 (Fig. S19, available online).

Forest plots of studies which measured changes in peripheral a IL-6, b TNF-α, c IL-1β, d IL-10, or e CCL-2 in individuals with MDD after antidepressant therapy. Effect size estimates are presented as Hedges’s g with 95% confidence intervals (CIs). Square sizes are proportional to the ES of each study. References are presented in the Supplementary online material
Tumor Necrosis Factor-Alpha
Levels of tumor necrosis factor-alpha (TNF-α) were significantly lower after antidepressant treatment (23 studies; N = 797). The ES estimate was small (Hedge’s g = −0.202; P = 0.015) (Table 1; Fig. 2b). There was evidence of publication bias, but the ES remained small and significant after adjustment with the trim-and-fill procedure. Heterogeneity was large (I 2 = 80.0%). Mean baseline depressive symptoms were associated with lower differences in TNF-α between endpoint and baseline in meta-regression analysis (Table S4, available online). Subgroup analyses indicated that heterogeneity was lower in studies which used either SNRIs or “miscellaneous” antidepressants compared to SSRI studies (Table S5, available online). Levels of TNF-α significantly decreased in studies in which its levels were assayed with ELISA but not in those studies that used other types of assay (Table S5, available online), and levels decreased only in studies where treatment time was longer than the median (Table S5, available online). In addition, sensitivity analysis revealed that the individual exclusion of two studies one-at-a-time rendered this ES non-significant (Fig. S16, available online). Finally, the cumulative meta-analysis indicates that this ES estimate has not been consistent over time (Fig. S20, available online).
Interleukin-1 Beta
Levels of interleukin-1 beta (IL-1β) were not significantly reduced after antidepressant drug treatment (Hedge’s g = −0.255; P = 0.176; 15 studies; N = 331; Fig. 2C). There was no evidence of publication bias. Heterogeneity was large (I 2 = 92.0%) (Table 1). In meta-regression analyses, the longer the mean follow-up time, the larger the difference in IL-1β between endpoint and baseline (Table S4, available online). Subgroup analyses indicated that heterogeneity was significantly lower in studies using either “miscellaneous” antidepressants or SNRIs compared to studies using SSRIs (Table S5, available online). In addition, heterogeneity was lower in studies in which IL-1β was sampled from plasma compared to studies in which this cytokine derived from serum (Table S5, available online).
Interleukin-10
Levels of IL-10 were measured in 10 studies (N = 331), and were significantly reduced after antidepressant drug treatment (Hedge’s g = −0.566) (Fig. 2d). However, there was evidence of small-study effects, but adjustment for publication bias with the trim-and-fill procedure did not change the ES (Table 1). In meta-regression analysis, the longer the mean follow-up time of the study, the lower the difference in IL-10 levels between endpoint and baseline (Table S4, available online). In subgroup analyses, levels of IL-10 significantly decreased with low heterogeneity in studies using miscellaneous antidepressants but not in studies using SSRIs, which had high heterogeneity. In addition, IL-10 decreased with low heterogeneity in studies which sampled this cytokine from plasma but not in studies which sampled this cytokine from serum, which had high heterogeneity. IL-10 levels were found to be reduced in studies that used other assay types but not in those that used ELISA (Table S5, available online). Levels were also decreased only when treatment duration was above the median. Sensitivity analysis revealed one possible outlier [30] (Fig. S17, available online), while the ES appears stable since 2009 (Fig. S21, available online).
C-C Motif Ligand 2 Chemokine
Levels of the C-C motif ligand 2 chemokine (CCL-2) were examined in five studies (N = 163). Antidepressant drug treatment significantly reduced CCL-2 levels with a large ES estimate (Hedge’s g = −1.502) (Fig. 2e). There was no evidence of small-study effects (Table 1), and the heterogeneity was large (I 2 = 96.0%). Meta-regression analyses did not identify any moderator (Table S4, available online). Sensitivity analysis revealed that the exclusion of a single study one-at-a-time turned this ES non-significant (Fig. S18, available online).
Other Immune Variables
Eleven additional immune variables (interferon gamma-IFN-γ, IL-4, IL-2, IL-8, CCL-3, IL-1 receptor antagonist, IL-13, IL-17, IL-5, IL-7, and the soluble IL-2 receptor) were investigated in at least three studies, and were meta-analyzed. Overall, levels of these cytokine/chemokines were not significantly altered after antidepressant drug treatment (Table 1). The forest plots for these estimates are provided in the supplementary online material that accompanies the online version of this article (Figs. S1 to S11, available online). Heterogeneity for these estimates was large (I 2 between 64.2 and 95.5%), with the exception of the soluble IL-2 receptor where heterogeneity was low.
Composite Scores
Composite measures of cytokine/chemokine profiles suggestive of the activation of different immune cells were calculated. We found evidence that antidepressant drug treatment may lead to a significant reduction in cytokines/chemokines predominantly secreted by M1 macrophages (Hedge’s g = −0.35; P < 0.001), whereas cytokines/chemokines predominantly secreted by TH1, TH2, and TRegs were not significantly altered (Fig. 3).

Effect size estimates in Hedges’s g (with 95% CI) of composite measures indicative of cytokine/chemokine profiles related to the activation of T Helper (TH) 1, TH2, regulatory T cells (TReg), and M1 polarized macrophages. The number of datasets (k) included in each estimate is also depicted
Treatment Response
Based on data provided by individual studies, we were able to contrast ES for changes in TNF-α and IL-6 levels between antidepressant treatment respondents vs. non-responders. These ES estimates were non-significant (TNF-α for responders: g = −0.346, k = 8, P = 0.115; TNF-α for non-responders: g = 0.049, k = 7, P = 0.590; IL-6 for responders: g = −0.222, k = 4, P = 0.480; IL-6 for non-responders: g = −0.010, k = 4, P = 0.964). IL-1β levels were not altered in treatment responders (g = 0.617, k = 3, P = 0.407), while peripheral levels of this cytokine for treatment non-responders from at least three independent datasets were not available. In addition, we could contrast baseline levels of TNF-α and IL-8 between responders and non-responders. There was no difference in the baseline levels of both cytokines (TNF-α: Hedges’s g = 0.248, k = 7, P = 0.353; IL-8: Hedges’s g = −0.082; k = 3; P = 0.595) in responders versus non-responders.
Discussion
This meta-analysis suggests that the pharmacological treatment of MDD is accompanied by a significant decrease in levels of IL-6, TNF-α, IL-10, and CCL-2. Previous meta-analyses have found that antidepressant drug treatment may reduce TNF-α and IL-6 levels in individuals with MDD [10, 11]. These previous studies provided effect size estimates for TNF-α, IL-1β, IL-6, and IL-10. In addition, similarly to the current meta-analysis, a high degree of heterogeneity was observed [10, 11]. Furthermore, two recent meta-analysis found different results regarding changes in peripheral level immune mediators after antidepressant treatment [31, 32]. One of this meta-analysis verified that antidepressant drug treatment decreased levels of IL-6, IL-10, and IL-12, and increased levels of IL-1β and IL-4 [32]. However, this meta-analysis included relatively few studies, and estimated ES through fixed-effects modeling, which can provide unreliable results when heterogeneity across studies is high [32, 21]. The largest previous meta-analysis synthesized data from 35 original studies [31]. Nevertheless, this effort included participants with bipolar depression, while non-pharmacological treatments for depression were also considered [31]. Due to the largest amount of data available, we were able to estimate effect sizes for 16 immune mediators. In addition, we could explore more potential sources of heterogeneity than has previously been possible.
Meta-analytic evidence suggests that IL-6, TNF-α, IL-10, the soluble IL-2 receptor, CCL-2, IL-12, IL-13, IL-18, the IL-1 receptor antagonist, the soluble TNF receptor 2, and C-reactive protein levels are elevated in individuals with MDD compared to healthy controls [33, 6, 34]. We found that antidepressants significantly decreased peripheral levels of IL-6, IL-10, TNF-α, and CCL-2. Notwithstanding the fact that approved antidepressants are thought to primarily act via monoaminergic mechanisms, a compelling body of evidence indicates that an activation of neurotrophic mechanisms in the brain may significantly contribute to the therapeutic effects of antidepressants [35]. Interestingly, preclinical evidence indicates that peripheral inflammation may influence hippocampal plasticity via microglial activation [36]. In addition, IL-6 and TNF-α may reduce hippocampal synaptic plasticity [37]. Thus, our findings are consistent with the view that antidepressants may diminish peripheral inflammation and its impact on the brain [8] although our analysis indicate that these effects may not be consistently associated with treatment response. Previous evidence indicates that peripheral inflammation may be observed in a subset but not in all individuals with MDD [1]. In addition, a previous study suggests that individuals with MDD and higher peripheral inflammation may respond to the TNF-α antagonist infliximab, whereas in patients with MDD and lower levels of peripheral inflammation the effects of infliximab did not significantly differ from placebo [38]. Therefore, it is possible that the observed effects of standard antidepressants observed herein (i.e., an overall decrease in predominantly inflammatory cytokines and chemokines) may not be the main mechanism contributing to the therapeutic benefits of these drugs [39]. However, no antidepressant study included in this meta-analysis a priori stratified participants with lower versus higher levels of pro-inflammatory cytokines at baseline. In addition, both baseline as well as differences in levels of immune mediators were not been consistently provided across included studies as a function of treatment response.
A previous meta-analysis found that levels of TNF-α decreased in treatment responders but not in treatment non-responders [31]. In addition, it was suggested that baseline inflammation may predict antidepressant treatment response. However, this meta-analysis included several treatments other than antidepressant drugs, as well as participants with bipolar depression, and also included studies in which part of the sample had significant baseline comorbidities [31]. The current meta-analysis avoids these potential confounders and includes more studies and participants. We found that although antidepressants may decrease TNF-α levels, these results should be interpreted cautiously due to the high heterogeneity across studies and the fact that sensitivity analysis indicated that some individual studies might have biased the overall ES estimate. In addition, we found no evidence that changes in peripheral levels of TNF-α significantly differed as a function of antidepressant treatment response. Furthermore, baseline TNF-α did not differ when antidepressant treatment responders were compared to non-responders. Finally, although antidepressants significantly decreased IL-6 levels, this effect did not significantly differ as a function of treatment response.
It has been postulated that the trafficking and redistribution of pro-inflammatory monocytes to the brain may interact and activate microglial cells in ways that contribute to the pathophysiology of MDD [40]. Interestingly, evidence indicates that TNF-α, IL-6, and CCL-2 may be predominantly secreted by M1 polarized macrophages albeit not selectively [41]. In addition, an emerging body of preclinical investigation suggests that SSRIs may decrease the secretion of inflammatory mediators by lipopolysaccharide-stimulated microglial cells [42, 9]. Thus, our results are in agreement with these experimental data although we found no conclusive evidence to demonstrate a differential impact of different classes of antidepressants upon peripheral immune activation.
We found evidence that antidepressants may decrease peripheral levels of the chemokine CCL-2. This chemokine is predominantly pro-inflammatory and has been implicated in the chemotaxis of peripheral monocytes to the brain [43]. The inhibition of the traffic of peripheral monocytes to the brain may constitute a promising novel therapeutic mechanism for MDD [44]. However, these results should be cautiously interpreted due to the limited number of available studies and by the fact that this ES did not survive sensitivity analysis.
IL-10 is predominantly secreted by regulatory T cells (TRegs) and exerts mainly an anti-inflammatory effect [45]. It has been suggested that a “compensatory (anti)inflammatory reflex system” (CIRS) may operate in MDD [46]. This system has been hypothesized to play a counter-regulatory (i.e., homeostatic) role in the context of peripheral inflammation. We found that antidepressants may reduce IL-10 levels in individuals with MDD. Thus, it is possible in theory that this effect may occur secondarily to an overall reduction of peripheral inflammation promoted by antidepressants.
Strengths and Limitations
The main strength of this meta-analysis was the inclusion of the largest amount of data currently available, and the proper exploration of potential sources of heterogeneity. However, some potential sources of heterogeneity could not be explored due the lack of data reported across studies. For example, body mass index it appears may influence both antidepressant treatment response [47] and peripheral inflammation [48]. In meta-regression analysis, mean BMI did not significantly moderate changes in peripheral IL-6, TNF-α, IL-1β, IFN-γ, and IL-10 levels after antidepressant treatment. However, included studies did not provide data to reliably control for this potential moderator for other cytokines/chemokines included in our analysis. Furthermore, our meta-regression analyses suggest that antidepressant-related differences in TNF-α levels were not moderated by percent of current smokers. Nevertheless, these results should be cautiously interpreted because relatively few studies provided data on these potential moderators, and an accumulating body of evidence suggests that obesity and smoking may contribute to peripheral inflammation in patients with MDD [49,50,51,52]. Second, the methodological quality of included studies in this meta-analysis significantly varied. Third, we could not contrast differences in cytokine/chemokine levels after antidepressant treatment in individuals with melancholic and atypical depression (due to lack of data) although evidence suggests that peripheral immune activation may differ in these depression subtypes [53]. Finally, differences in the standardization of assays across different laboratories as well as technical challenges to assay certain mediators (e.g., IL-2 and IFN-γ) may have contributed to the heterogeneity of findings [54].
Conclusion
In summary, this meta-analysis showed that, overall, antidepressants decreased peripheral levels of IL-6, TNF-α, IL-10, and CCL-2. This meta-analysis suggests that antidepressants may decrease peripheral inflammation. However, this effect did not appear to consistently differ between responders and non-responders. In addition, baseline TNF-α levels did not predict antidepressant treatment response. Future studies should contrast peripheral cytokine/chemokine levels between responders and non-responders. In addition, an individual patient meta-analysis in which participants are stratified according to the degree of baseline inflammation could represent a next step to investigate the hypothesis that antidepressants may be more efficacious for patients with lower peripheral inflammation, whereas anti-inflammatory agents may be a promising strategy for those patients with higher immune activation [1, 39]. Finally, other antidepressant treatment modalities with proven efficacy like electroconvulsive therapy (ECT) may also impact peripheral immune activation notwithstanding evidence remains limited [55, 56]. Therefore, future studies may investigate whether cytokines/chemokines may serve as peripheral biomarkers of treatment response considering the new framework of precision psychiatry [57].
References
Miller AH, Raison CL (2016) The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16(1):22–34. doi:10.1038/nri.2015.5
Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9(1):46–56. doi:10.1038/nrn2297
Leonard B, Maes M (2012) Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev 36(2):764–785. doi:10.1016/j.neubiorev.2011.12.005
Raison CL, Capuron L, Miller AH (2006) Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol 27(1):24–31. doi:10.1016/j.it.2005.11.006
Stuart MJ, Baune BT (2014) Chemokines and chemokine receptors in mood disorders, schizophrenia, and cognitive impairment: a systematic review of biomarker studies. Neurosci Biobehav Rev 42:93–115. doi:10.1016/j.neubiorev.2014.02.001
Kohler CA, Freitas TH, Maes M, de Andrade NQ, Liu CS, Fernandes BS, Stubbs B, Solmi M, Veronese N, Herrmann N, Raison CL, Miller BJ, Lanctot KL, Carvalho AF (2017) Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. doi:10.1111/acps.12698
Li X, Frye MA, Shelton RC (2012) Review of pharmacological treatment in mood disorders and future directions for drug development. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 37(1):77–101. doi:10.1038/npp.2011.198
Leonard BE (2014) Impact of inflammation on neurotransmitter changes in major depression: an insight into the action of antidepressants. Prog Neuro-Psychopharmacol Biol Psychiatry 48:261–267. doi:10.1016/j.pnpbp.2013.10.018
Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR (2012) A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behav Immun 26(3):469–479. doi:10.1016/j.bbi.2011.12.011
Hannestad J, DellaGioia N, Bloch M (2011) The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 36(12):2452–2459. doi:10.1038/npp.2011.132
Hiles SA, Baker AL, de Malmanche T, Attia J (2012) Interleukin-6, C-reactive protein and interleukin-10 after antidepressant treatment in people with depression: a meta-analysis. Psychol Med 42(10):2015–2026. doi:10.1017/s0033291712000128
Carvalho LA, Torre JP, Papadopoulos AS, Poon L, Juruena MF, Markopoulou K, Cleare AJ, Pariante CM (2013) Lack of clinical therapeutic benefit of antidepressants is associated overall activation of the inflammatory system. J Affect Disord 148(1):136–140. doi:10.1016/j.jad.2012.10.036
Fornaro M, Rocchi G, Escelsior A, Contini P, Martino M (2013) Might different cytokine trends in depressed patients receiving duloxetine indicate differential biological backgrounds. J Affect Disord 145(3):300–307. doi:10.1016/j.jad.2012.08.007
Cattaneo A, Ferrari C, Uher R, Bocchio-Chiavetto L, Riva MA, Pariante CM (2016) Absolute measurements of macrophage migration inhibitory factor and interleukin-1-beta mRNA levels accurately predict treatment response in depressed patients. The international journal of neuropsychopharmacology / official scientific journal of the Collegium Internationale Neuropsychopharmacologicum (CINP). doi:10.1093/ijnp/pyw045
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, Clarke M, Devereaux PJ et al (2009) The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ (Clinical research ed) 339:b2700. doi:10.1136/bmj.b2700
Bakkalbasi N, Bauer K, Glover J, Wang L (2006) Three options for citation tracking: Google Scholar, Scopus and Web of Science. Biomedical digital libraries 3:7. doi:10.1186/1742-5581-3-7
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn (DSM-5), 5th edn. American Psychiatric Association
World Health Organization (1993) The ICD-10 classification of mental and behavioural disorders: diagnostic criteria for research. World Health Organization, Geneva
Hozo SP, Djulbegovic B, Hozo I (2005) Estimating the mean and variance from the median, range, and the size of a sample. BMC Med Res Methodol 5:13. doi:10.1186/1471-2288-5-13
Lau J, Ioannidis JP, Schmid CH (1997) Quantitative synthesis in systematic reviews. Ann Intern Med 127(9):820–826
Patsopoulos NA, Evangelou E, Ioannidis JP (2009) Heterogeneous views on heterogeneity. Int J Epidemiol 38(6):1740–1742. doi:10.1093/ije/dyn235
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7(3):177–188
Cohen J (1992) A power primer. Psychol Bull 112(1):155–159
Carvalho AF, Kohler CA, Fernandes BS, Quevedo J, Miskowiak KW, Brunoni AR, Machado-Vieira R, Maes M et al (2016) Bias in emerging biomarkers for bipolar disorder. Psychol Med 46(11):2287–2297. doi:10.1017/s0033291716000957
Carvalho AF, Kohler CA, Brunoni AR, Miskowiak KW, Herrmann N, Lanctot KL, Hyphantis TN, Quevedo J et al (2016) Bias in peripheral depression biomarkers. Psychother Psychosom 85(2):81–90. doi:10.1159/000441457
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ (Clinical research ed) 315(7109):629–634
Duval S, Tweedie R (2000) Trim and fill: a simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics 56(2):455–463
Rosenthal R (1979) The file drawer problem and tolerance for null results. Psychol Bull 86(3):638
Thompson SG, Higgins JP (2002) How should meta-regression analyses be undertaken and interpreted? Stat Med 21(11):1559–1573. doi:10.1002/sim.1187
Hernandez ME, Mendieta D, Martinez-Fong D, Loria F, Moreno J, Estrada I, Bojalil R, Pavon L (2008) Variations in circulating cytokine levels during 52 week course of treatment with SSRI for major depressive disorder. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology 18(12):917–924. doi:10.1016/j.euroneuro.2008.08.001
Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ (2015) Inflammation and clinical response to treatment in depression: a meta-analysis. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology 25(10):1532–1543. doi:10.1016/j.euroneuro.2015.06.007
Goldsmith DR, Rapaport MH, Miller BJ (2016) A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry 21(12):1696–1709. doi:10.1038/mp.2016.3
Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimaki M (2015) Cumulative meta-analysis of interleukins 6 and 1beta, tumour necrosis factor alpha and C-reactive protein in patients with major depressive disorder. Brain Behav Immun 49:206–215. doi:10.1016/j.bbi.2015.06.001
Eyre HA, Air T, Pradhan A, Johnston J, Lavretsky H, Stuart MJ, Baune BT (2016) A meta-analysis of chemokines in major depression. Prog Neuro-Psychopharmacol Biol Psychiatry 68:1–8. doi:10.1016/j.pnpbp.2016.02.006
Krishnan V, Nestler EJ (2010) Linking molecules to mood: new insight into the biology of depression. Am J Psychiatry 167(11):1305–1320. doi:10.1176/appi.ajp.2009.10030434
Riazi K, Galic MA, Kentner AC (2015) Microglia-dependent alteration of glutamatergic synaptic transmission and plasticity in the hippocampus during peripheral inflammation. 35(12):4942–4952. doi:10.1523/jneurosci.4485-14.2015
Eyre H, Baune BT (2012) Neuroplastic changes in depression: a role for the immune system. Psychoneuroendocrinology 37(9):1397–1416. doi:10.1016/j.psyneuen.2012.03.019
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, Haroon E, Miller AH (2013) A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA psychiatry 70(1):31–41. doi:10.1001/2013.jamapsychiatry.4
Miller AH, Haroon E, Felger JC (2016) Therapeutic implications of brain-immune interactions: treatment in translation. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. doi:10.1038/npp.2016.167
Wohleb ES, Delpech JC (2016) Dynamic cross-talk between microglia and peripheral monocytes underlies stress-induced neuroinflammation and behavioral consequences. Prog Neuro-Psychopharmacol Biol Psychiatry. doi:10.1016/j.pnpbp.2016.04.013
Mills CD (2015) Anatomy of a discovery: m1 and m2 macrophages. Front Immunol 6:212. doi:10.3389/fimmu.2015.00212
Durairaj H, Steury MD, Parameswaran N (2015) Paroxetine differentially modulates LPS-induced TNFalpha and IL-6 production in mouse macrophages. Int Immunopharmacol 25(2):485–492. doi:10.1016/j.intimp.2015.02.029
Ge S, Song L, Serwanski DR, Kuziel WA, Pachter JS (2008) Transcellular transport of CCL2 across brain microvascular endothelial cells. J Neurochem 104(5):1219–1232. doi:10.1111/j.1471-4159.2007.05056.x
Wohleb ES, Franklin T, Iwata M, Duman RS (2016) Integrating neuroimmune systems in the neurobiology of depression. Nat Rev Neurosci 17(8):497–511. doi:10.1038/nrn.2016.69
Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010) FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10(7):490–500. doi:10.1038/nri2785
Maes M, Berk M, Goehler L, Song C, Anderson G, Galecki P, Leonard B (2012) Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med 10:66. doi:10.1186/1741-7015-10-66
Uher R, Mors O, Hauser J, Rietschel M, Maier W, Kozel D, Henigsberg N, Souery D et al (2009) Body weight as a predictor of antidepressant efficacy in the GENDEP project. J Affect Disord 118(1–3):147–154. doi:10.1016/j.jad.2009.02.013
Orenes-Pinero E, Pineda J, Roldan V, Hernandez-Romero D, Marco P, Tello-Montoliu A, Sogorb F, Valdes M et al (2015) Effects of body mass index on the lipid profile and biomarkers of inflammation and a fibrinolytic and prothrombotic state. J Atheroscler Thromb 22(6):610–617. doi:10.5551/jat.26161
Slyepchenko A, Brunoni AR, McIntyre RS, Quevedo J, Carvalho AF (2016) The adverse effects of smoking on health outcomes in bipolar disorder: a review and synthesis of biological mechanisms. Curr Mol Med 16(2):187–205
Liu CS, Carvalho AF, McIntyre RS (2014) Towards a “metabolic” subtype of major depressive disorder: shared pathophysiological mechanisms may contribute to cognitive dysfunction. CNS & neurological disorders drug targets 13(10):1693–1707
Slyepchenko A, Maes M, Machado-Vieira R, Anderson G, Solmi M, Sanz Y, Berk M, Kohler CA et al (2016) Intestinal dysbiosis, gut hyperpermeability and bacterial translocation: missing links between depression, obesity and type 2 diabetes. Curr Pharm Des 22(40):6087–6106
Slyepchenko A, Maes M, Jacka FN, Kohler CA, Barichello T, McIntyre RS, Berk M, Grande I et al (2017) Gut microbiota, bacterial translocation, and interactions with diet: pathophysiological links between major depressive disorder and non-communicable medical comorbidities. Psychother Psychosom 86(1):31–46. doi:10.1159/000448957
Gold PW (2015) The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry 20(1):32–47. doi:10.1038/mp.2014.163
Eller T, Vasar V, Shlik J, Maron E (2009) The role of IL-2 and soluble IL-2R in depression and antidepressant response. Current opinion in investigational drugs (London, England : 2000) 10(7):638–643
Jarventausta K, Sorri A, Kampman O, Bjorkqvist M, Tuohimaa K, Hamalainen M, Moilanen E, Leinonen E et al (2017) Changes in interleukin-6 levels during electroconvulsive therapy may reflect the therapeutic response in major depression. Acta Psychiatr Scand 135(1):87–92. doi:10.1111/acps.12665
Guloksuz S, Rutten BP, Arts B, van Os J, Kenis G (2014) The immune system and electroconvulsive therapy for depression. The journal of ECT 30(2):132–137. doi:10.1097/yct.0000000000000127
Fernandes BS, Williams LM, Steiner J, Leboyer M, Carvalho AF, Berk M (2017) The new field of ‘precision psychiatry’. BMC Med 15(1):80. doi:10.1186/s12916-017-0849-x
Acknowledgements
CAK is supported by a postdoctoral fellowship award from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil). MM is supported by a visiting research fellowship from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; Brazil). AFC is supported by a research fellowship award from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; Brazil).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
In the past 12 months, CLR has served on the scientific advisory board for Usona Institute. All other authors report no conflicts of interest.
Electronic supplementary material
ESM 1
(DOCX 688 kb)
Rights and permissions
About this article

Cite this article
Köhler, C.A., Freitas, T.H., Stubbs, B. et al. Peripheral Alterations in Cytokine and Chemokine Levels After Antidepressant Drug Treatment for Major Depressive Disorder: Systematic Review and Meta-Analysis. Mol Neurobiol 55, 4195–4206 (2018). https://doi.org/10.1007/s12035-017-0632-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0632-1