
Abstract
Among five members of the K+-dependent Na+/Ca2+ exchanger (NCKX) family (NCKX1–5), only NCKX2 is highly expressed in mouse brain. NCKX2 in plasma membranes mediates cytosolic calcium excretion through electrogenic exchange of 4 Na+ for 1 Ca2+ and 1 K+. Here, we observed significantly decreased levels of NCKX2 protein and mRNA in the CA1 region of APP23 mice, a model of Alzheimer’s disease. We also found that, like APP23 mice, heterozygous NCKX2-mutant mice exhibit mildly impaired hippocampal LTP and memory acquisition, the latter based on novel object recognition and passive avoidance tasks. When we addressed underlying mechanisms, we found that both CaMKII autophosphorylation and CaMKIV phosphorylation significantly decreased in CA1 regions of NCKX2+/− relative to control mice. Likewise, phosphorylation of GluA1 (Ser-831) and CREB (Ser-133), respective downstream targets of CaMKII and CaMKIV, also significantly decreased in the CA1 region. BDNF protein and mRNA levels significantly decreased in CA1 of NCKX2+/− relative to control mice. Finally, CaN activity increased in CA1 of NCKX2+/− mice. Our findings suggest that like APP23 mice, NCKX2+/− mice may exhibit impaired learning and hippocampal LTP due to decreased CaM kinase II and CaM kinase IV activities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maintenance of calcium homeostasis is critical for activity-dependent neuronal plasticity and synaptic transmission in the brain [1,2,3,4]. Moreover, machineries enabling neuronal Ca2+ efflux, such as Na+/Ca2+ exchangers, mediate calcium homeostasis underlying synaptic transmission [5,6,7]. In addition, K+-dependent Na+/Ca2+ exchangers (NCKXs) mediate calcium excretion from the cytosol of neurons through electrogenic exchange of 4 Na+ for 1 Ca2+ and 1 K+, depending on the electrochemical gradient across the plasma membrane [8]. In mammals, NCKXs are found in five different isoforms (NCKX1–5) encoded by distinct genes [9, 10]. NCKX1 is expressed only in the retina, NCKX2 is highly expressed in the brain [11], and NCKX3 and NCKX4 are widely expressed in the brain, aorta, lung, and intestine. NCKX5 is mainly expressed in the skin and retina [12].
NCKX2 is predominantly expressed in neurons [8, 11], especially in the cerebral cortex, striatum, and hippocampus [12]. Lee et al. reported immunohistochemical studies conducted in cultured hippocampal neurons showing that NCKX2 is expressed in somatodendritic regions but not axon terminals [13]. Li et al. reported that NCKX2 is highly expressed in hippocampus and locates from distal dendrites to cell bodies of pyramidal neurons [6]. Moreover, others have reported that NCKX2 homozygous mice survive and exhibit mildly impaired spatial memory-related behaviors and hippocampal long-term potentiation (LTP) [6], although molecular mechanisms underlying these deficits remain unclear.
Hippocampal LTP is considered as a model of the molecular basis of synaptic plasticity and hence of learning and memory [14, 15]. We have reported that calcium/calmodulin-dependent protein kinase II (CaMKII) activity is essential for LTP induction in the hippocampal CA1 region [16, 17]. Heterozygous CaMKIIα-mutant mice show impaired spatial learning and memory [18]. CaMKII is highly localized in post-synaptic densities of excitatory synapses and becomes constitutively active through autophosphorylation following hippocampal LTP [16, 19,20,21,22]. CaMKII activity facilitates synaptic efficacy by direct phosphorylation of the post-synaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor (AMPAR) subunit GluA1 (Ser-831) [21, 23].
By contrast, calcium/calmodulin-dependent protein kinase IV (CaMKIV) is found primarily in neuronal nuclei [24, 25] and functions in maintenance of hippocampal LTP through phosphorylation of the cyclic AMP-responsible element-binding protein (CREB) (Ser-133) [26]. Therefore, CaMKIV null mice exhibit impaired hippocampal LTP and CREB (Ser-133) phosphorylation in CA1 [27], and some have reported behavioral deficits including impaired eye-blink memory and fear memory [28, 29]. However, others, such as Takao et al. [30], report that CaMKIV null mice exhibit normal memory-related behaviors.
The APP23 mouse, a transgenic model of Alzheimer’s disease (AD), expresses mutant human-type APP (the Swedish double mutation) under control of the murine brain and neuron-specific Thy-1 promoter [31]. APP23 mice exhibit abnormal amyloid-β aggregation and tau protein hyperphosphorylation in the brain [32, 33]. We also previously reported that APP23 mice exhibit cognitive deficits and decreased hippocampal LTP in CA1 [34]. Since NCKX2 protein levels are significantly reduced in the APP23 mouse brain, we asked whether reduced NCKX2 levels seen in NCKX2+/− mice were associated with cognitive deficits or impaired LTP. Here, we demonstrate decreased activities of both CaMKII and CaMKIV concomitant with increased calcineurin (CaN) activity in CA1 of NCKX2+/− mice. We conclude that unbalanced activities of Ca2+-dependent kinases and phosphatases underlie impaired hippocampal LTP and learning behaviors in these mice.
Materials and Methods
Animals
NCKX2+/− Mice
NCKX2 heterozygotes were generated using the Sleeping Beauty (SB) transposon system, as described [35]. Briefly, we generated a doubly transgenic mouse containing the SB transposon vector and the SB transposase expression vector. The former was mobilized by the latter in the germline and transposed into various genomic loci. Mating of doubly transgenic with wild-type (WT) mice generated multiple types of progeny. The fact that the SB transposon vector contained a green fluorescent protein (GFP) expression cassette flanked by a constitutively active promoter and splice donor enabled selection gene insertions based on fluorescence of newborn progeny [35]. We determined transposon insertion sites by splinkerette-PCR using genomic tail DNA from GFP-positive mice [35]. An insertion into NCKX2 was identified in one GFP-positive mouse, based on mapping of a sequence flanking the insertion site (TATAAGAATGAAAACTCCAAAAATCCTCCTGA) to NCKX2 intron 8 (NCBI accession number NM_172426, registered as Slc24a2, transcript variant 1). Since the SB transposon vector contained a LacZ cassette flanked by a splice acceptor and a polyadenylation signal, NCKX2 transcription was disrupted by splicing of exon 8 and the SB transposon vector splice acceptor. Genotyping of mutant mice was performed by PCR using the following primers: 5′-ATGGAGTCAAATTCCAAACTAGATCTCAGG-3′ for the WT allele, 5′-ATACATGACCCGTTAAGATGTTTCCTGGTC-3′ for the WT and mutant alleles, and 5′-CTTGTGTCATGCACAAAGTAGATGTCC-3′for the mutant allele. Bands of 193 and 363 bp were predicted from mutant and WT alleles, respectively. NCKX2+/− mice were backcrossed to C57BL/6J mice for more than nine generations. For some experiments, NCKX2+/− and WT mice were used at 8–10 weeks of age. Mice were housed in cages with free access to food and water at a constant temperature (23 ± 1 °C) and humidity (55 ± 5%) with a 12-h light/dark cycle (09:00–21:00). All animal protocols were approved by the Committee on Animal Experiments at Tohoku University.
APP23 Transgenic Mice
APP23 transgenic mice (male), which express mutant human-type ΑPP with the Swedish mutation under control of the murine brain and neuron-specific Thy-1 promoter, were provided by Novartis Pharma Inc. (Nervous System Research, Basel, Switzerland). For details relevant to construction of these mice, see [31]. Mice were housed in cages with free access to food and water at a constant temperature (23 ± 1 °C) and humidity (55 ± 5%) with a 12-h light/dark cycle (09:00–21:00). For experiments assessing NCKX2 levels, 14-month-old APP23 and WT mice were used.
Behavioral Analyses
In the following behavioral analyses, evaluators were blinded to treatment conditions.
-
1.
Y-maze task: A detailed protocol of this task has been reported [36]. In brief, spontaneous alternation behavior in a Y-maze serves as an indicator of spatial reference memory. Testing is conducted in an apparatus consisting of three identical arms (50 × 16 × 32 cm) of black plexiglas. A mouse is placed at the end of one arm and allowed to move freely through the maze during an 8-min session, and alternation behaviors are scored.
-
2.
Novel object recognition task: A detailed protocol of this task has been reported [36]. The task is based on the tendency of normal rodents to discriminate a novel from a familiar object and is a test of memory. Mice are individually habituated to an open-field box (35 × 25 × 35 cm) for two consecutive days. The experimenter scoring behavior is blinded to the treatment. During acquisition phases, two objects of the same material are placed in a symmetric position in the center of the chamber for 5 min. One hour later, one object is replaced by a novel object, and exploratory behavior is again analyzed for 5 min. The number of approaches to the two objects is scored.
-
3.
Step-through passive avoidance task: A detailed protocol of this task has been reported [36]. The test is based on rodents’ inherent preference for a dark compartment and is a test of memory based on fear conditioning. Briefly, training and retention trials are conducted in a box consisting of dark (25 × 25 × 25 cm) and light (14 × 10 × 25 cm) compartments. The floor is constructed of stainless steel rods, and those in the dark compartment are connected to an electronic stimulator (Nihon Kohden, Tokyo, Japan). Mice are habituated to the apparatus the day before passive avoidance acquisition. On training trials, a mouse is placed in the light compartment, and when it enters the dark compartment, the door is closed to prevent escape and the animal receives an electric shock (1 mA for 500 ms) from the floor for 30 s. The mouse is then removed from the apparatus after a 30 s period without shock. The identical procedure without footshock is repeated 24 h later, and the time (latency) is seconds required for the mouse to enter the dark compartment is measured as an indicator of retention (memory).
-
4.
Elevated-plus maze task: The elevated plus maze task measures anxiety behavior in mice and employs an apparatus consisting of a central 6 × 6 cm platform and four arms radiating like a plus sign from the platform: two open and two enclosed arms, all 30 cm long, 6 cm wide, and 15 cm high with nontransparent side and end walls. The maze is elevated 70 cm above floor level. At the beginning of a 5-min test session, mice are placed in the center facing an enclosed arm, and the following parameters are scored: (1) the number of total, open, and closed arm entries, and (2) time spent in different parts of the maze (open and closed arms, and central platform). For the test, time spent open and closed arm is recorded, with longer periods in closed arm associated with increased anxiety behavior.
-
5.
Marble-burying task: This task is based on the tendency of mice to exhibit increased digging behavior in high anxiety states. A cage is filled approximately 5–10 cm deep with wood chips lightly tamped down to make a flat surface. Glass marbles, evenly spaced about 4 cm apart, are placed on the surface. Animals are then placed in the cage for 30 min, and at the end of the test, the number of marbles covered by bedding is measured.
-
6.
Light-dark task: This task is based on the tendency mice to prefer a dark room in states of high anxiety. The light/dark box apparatus consists of a wooden box (48 × 24× 27 cm) divided into two equal-size compartments by a barrier containing a doorway (10 cm height × 10 cm width). One compartment is painted black and covered with a lid, and the other is painted white and illuminated with a 60-W light bulb positioned 40 cm above the upper edge of the box. Mice are placed in the dark compartment initially, and the time to enter the light compartment is recorded.
-
7.
Open-field task: This task is based on the tendency mice to stay close to a wall in states of high anxiety. Mice are placed in a Plexiglas box (60 × 60 × 60 cm) for 10 min, and their movements are recorded with an overhead video camera. A circle with a 15 cm radius marks the center of the test floor, and time spent in that zone is recorded.
-
8.
Tail-suspension task: A detailed protocol of this task, which evaluates depressive behaviors, is described [37]. The test is based on the fact that rodents become immobile when subjected to short-term, inescapable stress such as suspension by the tail. For the test, mice are suspended by the tail and time spent immobile is recorded, with longer periods associated with increased depressive behavior.
-
9.
Forced-swim task: The forced-swim task, described elsewhere [37], is also a test of depressive behavior. Mice are placed individually in glass cylinders (height, 20 cm; diameter, 15 cm) filled with 25 °C water and, like the tail-suspension test, the duration of immobile periods within a test period (5 min) is scored. Longer immobile periods correlate with increased depressive behaviors.
-
10.
Rotarod task: The rotarod task measures motor coordination [38]. The apparatus consists of a base platform and an iron rod of 3 cm diameter and 30 cm long with a non-slippery surface. This rod is divided into three equal sections by two disks, enabling three mice to walk simultaneously on the rod rotating at 4 turns/min. Animals are placed on the rotating drum up to 5 min, and intervals between mounting and falling from the rod are recorded as performance time.
-
11.
Beam-walking task: The beam-walking task, which also assesses motor coordination, is described elsewhere [38]. The apparatus consists of a rectangular base (870 mm × 200 mm × 17 mm) of medium density fiberboard with a Formica cover. A vertical stand (310 mm × 160 mm × 5 mm; Perspex) is fixed to the left side of the base supporting a black “goal box” (155 mm × 160 mm × 5 mm; Perspex) with a matte surface on all inside faces. A horizontal rod (500 mm × 5 mm diameter; dowelled) is fixed between the base of the “goal box” and a vertical stainless steel pole (315 mm × 5 mm) placed at a 90° angle. Further support is provided the steel pole by a Perspex rod, preventing sideways movement of the beam. The dowelled rod is marked in graduated cm from 0 to 50 cm. For the test, number of missteps is counted, with increased missteps associated with motor coordination deficit.
Electrophysiology
Hippocampal slices were prepared as described [36]. Transverse slices (400 μm thick) cut with a vibratome (Microslicer DTK-1000) were incubated for 2 h in continuously oxygenized (95% O2, 5% CO2) artificial cerebrospinal fluid at room temperature. Slices were transferred to an interface recording chamber and perfused at a flow rate of 2 ml/min with artificial cerebrospinal fluid warmed to 34 °C. Field excitatory post-synaptic potentials (fEPSPs) were evoked by a test stimulus (0.05 Hz) through a bipolar stimulating electrode placed on the Schaffer collateral/ commissural pathway and recorded from the stratum radiatum of CA1using a glass electrode filled with 3 M NaCl. High-frequency stimulation (HFS) of 100 Hz with a 1-s duration was applied twice with a 10-s interval, and test stimulation was continued for indicated periods.
Biochemical Analysis
Immunoblotting and immunohistochemistry were performed as described [36], using the following antibodies: anti-phospho CaMKII (1:5000 [39]), anti-CaMKII (1:5000 [17]), anti-phospho-CaMKIV (Thr-196) (1:2000, Abcam, Cambridge, MA, USA), anti-phospho-synapsin I (Ser-603) (1:2000, Millipore, Billerica, MA, USA), anti-phospho-GluA1 (Ser-831) (1:1000, Millipore), anti-phospho-GluA1 (Ser-845) (1:1000, Millipore), anti-phospho-MAP kinase (di-phosphorylated ERK 1/2) (1:2000, Sigma-Aldrich, St. Louis, MO, USA), anti-phospho-CREB (Ser-133) (1:1000, Millipore), anti-NCKX2 (1:1000), anti-BDNF (1:1000, Santa Cruz Biotechnology Inc., Dallas, TX, USA), and anti-β-tubulin (1:5000, Sigma-Aldrich). Bound antibodies were visualized using the enhanced chemiluminescence detection system (Amersham Life Science, Buckinghamshire, UK) and analyzed semiquantitatively using the National Institutes of Health Image program.
Real-Time PCR
Quantification of BDNF or NCKX2 mRNAs was undertaken in 48-well PCR plates (Mini Opticon Real Time PCR System, Bio-Rad) using iQ SYBR Green Supermix 2× (Bio-Rad). Primer sequences used were as follows: BDNF-IF (CCTGCATCTGTTGGGGAGAC), BDNF-IR (GCCTTGTCCGTGGACGTTTA), BDNF-IVF (CAGAGCAGCTGCCTTGATGTT), BDNF-IVR (GCCTTGTCCGTGGACGTTTA), NCKX2-forward (CTGGAGGAGCGAAGGAAAGG), NCKX2-reverse (TGTGAAAGTTCTGGGGCTGAC), GAPDH-F (TGTGTCCGTCGTGGATCTGA), and GADPH-R (CACCACCTTCTTGATGTCATCATAC). Relative quantities of target transcripts were determined by the comparative threshold cycle (ΔCT) method and normalized to GAPDH. Product purity and specificity were confirmed by omitting the template and performing a standard melting curve analysis.
Calcineurin Assay
CaN activity was measured as described [40]. Hippocampal tissues (100–200 mg) were homogenized in 1.5 ml of trypsin-EDTA (GIBCO/BR L), pelleted, and lysed in 100 μl CaN assay buffer (Quantizyme Assay System AK-804, BioMol, Plymouth Meeting, PA). CaN activity was assessed using 3 μg of protein from extracts according to the manufacturer’s procedure (BioMol). Phosphatase activity was monitored spectrophotometrically by detecting phosphate released from a CaN-specific RII phosphopeptide.
Immunohistochemistry
Immunohistochemistry was performed as described [37]. WT mice at 10 weeks of age or APP23 mice and WT mice at 14 months of age were anesthetized with sevoflurane and perfused via the ascending aorta with phosphate-buffered saline (PBS; pH 7.4) until the outflow became clear. The perfusate was then switched to phosphate buffer (pH 7.4) containing 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 15 min. The brain was removed, post-fixed in the same solution for 2-h at 4 °C, embedded in 2% agarose, and sliced at 50 μm using a vibratome (Dosaka EM Co. Ltd., Kyoto, Japan, or Leica VT1000S, Nussloch, Germany). Coronal brain sections were permeabilized with 0.3% Triton X-100 in PBS, blocked with 5% normal goat or donkey serum for 3 h, and incubated overnight with mouse anti-MAP2 monoclonal antibody (1:500) (Millipore), anti-NCKX2 polyclonal antibody (1:500), anti-PSD95 monoclonal antibody (1:500) (Abcam), anti-synaptophysin monoclonal antibody (1:500) (Sigma-Aldrich), or anti-GFAP monoclonal antibody (1:500) (Sigma-Aldrich) in blocking solution at 4 °C. After thorough washing in PBS, sections were incubated 3 h in Alexa 488-labeled anti-rabbit IgG or Alexa 594-labeled anti-mouse IgG. After several PBS washes, sections were mounted on slides with Vectashield (Vector Laboratories, Burlingame, CA, USA). Immunofluorescent images were analyzed using a confocal laser-scanning microscope (Nikon EZ-C1, Nikon, Tokyo, Japan or LSM 710, Zeiss, Oberkochen, Germany).
After incubation with anti-NCKX2 polyclonal antibody (1:500), sections were immersed in 3% H2O2 in PBS for 30 min at room temperature and processed using the Vectastain ABC kit (Vector Laboratories Inc., Burlingame, CA, USA) with secondary antibody (biotinylated anti-rabbit IgG, 1:200; Vector Laboratories Inc.). The peroxidase reaction product was detected using 3,3′-diaminobenzidine-tetrahydrochloride (DAB; Sigma-Aldrich). After washing in PBS, sections were dehydrated in a graded series of ethanols, immersed in xylene, mounted in Entellan (MERCK, Dannstadt, Germany) and cover-slipped.
Data Analysis
Data are expressed as means ± SEM. Statistical analysis was performed using Prism 6 software (GraphPad Software, San Diego, CA, USA). Comparisons between two experimental groups were made using the unpaired Student’s t-test. Statistical significance for differences among groups was tested by one-way or two-way analysis of variance, followed by a post hoc Bonferroni’s multiple comparison test between control and other groups. Asterisks in graphs denote statistical significance (*P < 0.05, **P < 0.01).
Results
APP23 Mice Exhibit Decreased Levels of NCKX2 mRNA and Protein in the CA1 Region
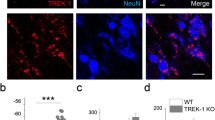
We first examined potential changes in NCKX2 levels in brains of APP23 mice, which express amyloid precursor protein (APP) with human “Swedish” KM670/671NL mutations [31]. NCKX2 mRNA and protein levels significantly decreased in homogenates of the hippocampal CA1 region of APP23 relative to WT mice but were comparable to WT in other brain regions tested (protein levels 64.3 ± 6.4%, n = 4; mRNA levels 46.5 ± 5.2%, n = 6) (Fig. 1a–c). Immunohistochemical analyses confirmed reduced NCKX2 protein expression in CA1 pyramidal neurons of APP23 relative to WT mice (Fig. 1d).

NCKX2 expression decreases in the CA1 of APP23 mice. a Representative images of immunoblots of extracts from CA1, dentate gyrus (DG), prefrontal cortex (PFC), cingulate cortex (CC), and striatum (STR) probed with antibodies to NCKX2 and β-tubulin in 14-month-old APP23 (A) and WT (W) mice. b Quantitation of NCKX2 protein levels shown in a. c Quantitative analyses of NCKX2 mRNA in regions shown in a. d Immunohistochemical analyses showing NCKX2 expression in hippocampus of 14-month-old APP23 and WT mice. Scale bars, 500 μm (low magnification, left) and 50 μm (high magnification, right). Error bars in b and c indicate SEM. **p < 0.01 versus WT mice
We next used immunofluorescence to ask whether NCKX2 in WT mice is expressed in neurons or astrocytes of the hippocampal CA1 region. We found that NCKX2 is predominantly expressed in cell bodies of hippocampal pyramidal neurons based on double-staining with antibodies to NCKX2 and anti-PSD95 (a marker of post-synaptic density), anti-synaptophysin (a marker of pre-synaptic terminals), anti-MAP2 (a marker of neuronal cell bodies and dendrites), or anti-GFAP (an astrocyte marker) (Fig. 2). Although NCKX2 protein was also faintly expressed in astrocytes, it was localized primarily to both post- and the pre-synaptic regions of pyramidal neurons.

NCKX2 is expressed at pre- and post-synaptic sites of neurons and in astrocytes. Confocal microscopy images showing double immunofluorescence staining of CA1 for NCKX2 (in green) and PSD95, a marker of post-synaptic densities, synaptophysin, a marker of pre-synaptic terminals, MAP2, a marker of neuronal cell bodies and dendrites, or GFAP, an astrocyte marker (all in red). Scale bars, 20 μm (low magnification) and 5 μm (high magnification) images. High-magnification images of boxes in column 3 are shown in column 4. Neural nuclei were stained by DAPI (in blue) (color figure online)
NCKX2+/− Mice Show Impaired Hippocampal LTP and Reduced NCKX Levels
To address the relevance of reduced hippocampal NCKX2 levels to neural plasticity, we examined hippocampal LTP induced by high-frequency stimulation (HFS; 100 Hz, two trains) of collateral/commissural pathways, using hippocampal slice preparations from NCKX2+/− and WT mice. In WT mice, HFS caused a stable and long-lasting increase in fEPSPs (60 min: 174.2 ± 4.5%, n = 5), as anticipated, whereas hippocampal LTP in NCKX2+/− mice was significantly reduced relative to WT mice (60 min: 143.2 ± 7.0%, n = 5) (Fig. 3a–c). Furthermore, reduced NCKX2 protein and mRNA levels in CA1 of NCKX2+/− mice resembled decreases seen in the APP23 mouse hippocampus (protein levels 52.0 ± 4.7%, n = 5; mRNA levels 20.7 ± 0.79%, n = 3) (Fig. 3d–f).

LTP is impaired in the hippocampal CA1 region of NCKX2 +/− mice. a Representative fEPSPs were recorded from CA1 in NCKX2+/− and WT mice. b–c Changes in slope of fEPSPs following HFS recorded in CA1 were attenuated in NCKX2+/− relative to WT mice at 1 min (post-tetanic potential (PTP)) and 60 min (LTP). d Representative images of immunoblots of hippocampal CA1 probed with NCKX2 and β-tubulin antibodies. e Quantitative analyses of NCKX2 protein levels. f Quantitative analyses of NCKX2 mRNA levels. Error bars in c and e indicate SEM. **p < 0.01 versus WT mice
NCKX2+/− Mice Exhibit Deficits in Memory-Related Behaviors
To confirm relevance of impaired hippocampal LTP seen in NCKX2+/− mice, we examined memory-related behaviors in these and WT mice. Although we observed comparable performance in a Y-maze task (Fig. 4a, b), in the novel object recognition task, NCKX2+/− mice failed to discriminate a novel object (51.0 ± 3.0%, n = 6 compared to 61.3 ± 2.1% of WT mice, n = 6) (Fig. 4c). In the step-through passive avoidance task, which assesses contextual memory, the latency time to enter the dark compartment in NCKX2+/− mice was significantly decreased relative to WT mice starting at 2 days after stimulation (2 days: 185.1 ± 53.2%, n = 6; 3 days: 126.0 ± 56.7%, n = 6; 4 days: 112.8 ± 46.6%, n = 6; 5 days: 106.9 ± 43.6%, n = 6) (Fig. 4d). These findings suggest that some memory-related behaviors are partially impaired in NCKX2+/− mice.

a–d NCKX2+/− mice exhibit abnormal memory-related behaviors. Total arm entries (a) or alternations (b) in a Y-maze task were measured in NCKX2+/− and WT mice. c The number of times a mouse recognized a novel object in a novel object recognition (NOR) task was assessed in NCKX2+/− and WT mice. In test sessions, that number significantly decreased in NCKX2+/− relative to WT mice. d Latency time in first and retention trials after 1–5 days of a passive-avoidance task. Latency time on retention trials significantly decreased in NCKX2+/− relative to WT mice. e–i NCKX2+/− mice exhibit normal anxiety-like behaviors. Number (e) or duration in terms of time (f) of arm entries in an elevated-plus maze task. g Number of marbles buried in a marble-burying task. h Time spent in an open compartment in a light-dark task. i Time spent in center circle in an open-field task. j–k NCKX2+/− and WT exhibit comparable depressive-like behaviors. Immobility time as measured by tail-suspension (j) or forced-swim (k) tasks. l–m NCKX2+/− mice show normal locomotor activity. Latency time in a rotarod task (l) and the number of missteps as evaluated by a beam-walking task (m). Error bars indicate SEM. **p < 0.01 versus familiar object, +p < 0.05 versus WT mice
To rule out anxiety as a confounding influence, we conducted four behavioral tests of anxiety. Both NCKX2+/− and WT mice (n = 6 each) showed comparable performance in the elevated-plus maze task (Fig. 4e, f), exhibited similar tendencies to bury marbles in a marble burying task (n = 6 each) (Fig. 4g), and spent similar amounts of time in an open compartment in a light-dark task (n = 6 each) (Fig. 4h). NCKX2+/− and WT mice also spent comparable time in the center circle in the open-field task (n = 6 each) (Fig. 4i). Overall, we conclude that NCKX2+/− mice did not exhibit anxiety behaviors to any greater level than did WT mice.
Likewise, NCKX2+/− mice did not exhibit depressive-like behaviors, as assessed by tail-suspension (n = 6) (Fig. 4j) and forced-swim (n = 6) (Fig. 4k) tasks. Finally, NCKX2+/− mice showed no motor deficits, as assessed by rotarod (n = 6) (Fig. 4l) and beam-walking (n = 6) (Fig. 4m) tasks.
Taken together, among behavioral analyses evaluated, only memory-related behaviors, as measured in the novel object recognition task and step-through passive avoidance test, were impaired in NCKX2+/− mice.
CaMKII and CaMKIV Activities are Downregulated in CA1 of NCKX2+/− Mice
To address mechanisms underlying LTP impairment and abnormal memory-related behaviors, we examined CaMKIIα (Thr-286) autophosphorylation and phosphorylation of CaMKV (Thr-196) and ERK (Thr-202/Tyr-204) in CA1 extracts from NCKX2+/− and WT mice. CaMKIIα (Thr-286) autophosphorylation and CaMKIV (Thr-196) phosphorylation were significantly decreased in NCKX2+/− compared to WT mice (pCaMKII (Thr-286) 73.8 ± 8.0%, n = 5; pCaMKIV 68.6 ± 7.6%, n = 5) (Fig. 5a, b). By contrast, ERK (Thr-202/Tyr-204) phosphorylation was unchanged in NCKX2+/− mice (Fig. 5a, b). Both reduced CaMKII and CaMKIV activities correlated with perturbed phosphorylation of their endogenous substrates: phosphorylation of GluA1 Ser-831 (76.1 ± 6.2%, n = 5) (Fig. 5c, d) and CREB Ser-133 (64.5 ± 6.7%, n = 5) (Fig. 5c, d) significantly decreased in CA1 of NCKX2+/− mice. Phosphorylation of synapsin I (Ser-603), a pre-synaptic CaMKII substrate, was unchanged in NCKX2+/− mice (n = 5) (Fig. 5c, d), as was GluA1 (Ser-845) phosphorylation by PKA (n = 5) (Fig. 5c, d).

NCKX2+/− mice show decreased CaMKII and CaMKIV activities in hippocampal CA1. a Representative images of immunoblots probed with antibodies against autophosphorylated CaMKII, CaMKII, phosphorylated CaMKIV (Thr-196), phosphorylated ERK (Thr-202/Tyr-204), and β-tubulin. b Quantitative analyses of data shown in a. c Representative images of immunoblots probed with antibodies against phosphorylated GluA1 (Ser-831), phosphorylated GluA1 (Ser-845), phosphorylated Synapsin I (Ser-603), phosphorylated CREB (Ser-133), and β-tubulin. d Quantitative analyses of data shown in c. Quantitative analyses of BDNF mRNAs containing exons I (e) or IV (f). g, h Representative images of immunoblots probed with NCKX2 and β-tubulin antibodies (g). Quantitative analyses of NCKX2 and WT protein levels (h). i Analysis of active CaN levels in hippocampal slices of WT and NCKX2+/− mice. Error bars indicate SEM. **p < 0.01 versus WT mice
BDNF mRNA and Protein Levels Decrease in CA1 of NCKX2+/− Mice
To address the relevance of reduced CaMKII and CaMKIV phosphorylation, we examined brain-derived neurotrophic factor (BDNF) mRNA and protein levels, as CaMKII and CaMKIV activities are required to induce expression of BDNF mRNA containing exon I or exon IV [41, 42]. Levels of BDNF transcripts containing exons I or IV significantly decreased in CA1 of NCKX2+/− compared with WT mice (exon I 56.8 ± 6.7%, n = 7; exon IV 59.5 ± 6.8%, n = 7) (Fig. 5e, f). Similarly, BDNF protein levels significantly decreased in CA1 of NCKX2+/− compared with WT mice (60.9 ± 5.7%, n = 4) (Fig. 5g, h).
CaN Activity Increases in CA1 of NCKX2+/− Mice
To identify mechanisms underlying reduced CaMKII and CaMKIV activities, we assessed Ca2+-dependent protein phosphatase CaN activity [43, 44]. As expected, CaN activity significantly increased in NCKX2+/− mouse hippocampus compared to hippocampus of WT mice (131.1 ± 3.0%, n = 6) (Fig. 5i).
CaMKII and CaMKIV Activities Decrease in CA1 of APP23 Mice
Finally, we asked whether CaMKII and CaMKIV phosphorylation decreased in the CA1 region of APP23 mice. As expected, CaMKII (Thr-296) autophosphorylation and CaMKIV (Thr-196) phosphorylation significantly decreased in CA1 of APP23 mice without changes in total protein levels (Fig. 6).

Reduced CaMKII and CaMKIV activities in CA1 of APP23 mice. a Representative images of immunoblots probed with antibodies against autophosphorylated CaMKII, CaMKII, phosphorylated CaMKIV (Thr-196), CaMKIV, and β-tubulin. b Quantitative analyses of data shown in a. Error bars indicate SEM. **p < 0.01 versus WT mice
Discussion
In excitable cells such as neurons, Ca2+ influx and efflux through ion channels and transporters, respectively, are critical to maintain calcium homeostasis [1, 3]. Cation/Ca2+ exchanger proteins primarily contribute to Ca2+ extrusion from the cytosol. These types of proteins have been classified into Na+/Ca2+ exchangers (NCXs; NCX1–3) and K+-dependent Na+/Ca2+ exchangers (NCKXs: NCKX1–5) [9, 45]. Notably, we found that among NCKXs, NCKX2 levels are partially reduced in CA1 of APP23 mice. Therefore we used NCKX2 heterozygotes to evaluate effects of partial loss of NCKX2 function in the brain. Although others have reported impaired hippocampal LTP induction in NCKX2 homozygous null mice [6], those mice did not survive until adulthood in our studies, and hence, we used heterozygotes for analysis. Here, we show that both CaMKII and CaMKIV activities are reduced in the CA1 of NCKX2+/− mice. Interestingly, CaN activity increased ~130% in CA1 of NCKX2+/− mice, suggesting mildly elevated Ca2+ in hippocampal neurons in those mice. Thus, an imbalance in activity of protein kinases and phosphatases likely underlies cognitive deficits seen in NCKX2+/− mice.
We also confirmed that signaling downstream of CaMKII and CaMKIV is dysregulated in NCKX2+/− mice. CaMKIV directly phosphorylates CREB at Ser-133 and is critical for consolidation of hippocampus-dependent memory in mice [46]. Furthermore, CaMKIV mutant mice show impaired CREB activity and LTP induction in hippocampus [26], and BDNF gene expression is mediated by CREB phosphorylation [47]. Thus, CREB mutant mice exhibit impaired memory-related behaviors [48], and BDNF mutant mice exhibit deficits in spatial learning and memory and hippocampal LTP [49, 50]. Likewise, CaMKII is essential for CREB activity through additional CREB phosphorylation at Ser-142 [51]. We recently confirmed that CaMKII is required for CREB phosphorylation at Ser-133 and BDNF expression in the hippocampus of CaMKIV null mice [37]. Thus, we propose that both CaMKIV and CaMKII activities synergistically activate CREB/BDNF pathways in rodent hippocampus. Reduced CaMKII activity seen in NCKX2+/− mice also accounts for impaired LTP induction due to decreased phosphorylation of GluA1 at Ser-831. However, ERK phosphorylation at Thr-202/Tyr-204 was unchanged in CA1 of NCKX2+/− mice, suggesting that ERK activity is not essential for CREB phosphorylation.
We speculated increased CaN activity by a mild elevation of synaptic Ca2+ concentration in CA1 of NCKX2+/− mice. CaN is activated by low Ca2+ concentrations (KD = ~0.1 to 1 nM) relative to those of CaMKII (KD = ~40 to 100 nM) [52, 53]. CaN increases PP1 activity through dephosphorylation of dopamine- and cAMP-regulated protein of 32 kDa (Thr-34), inactivating CaMKII activity at post-synaptic densities [54, 55]. CaN is also abundant in CA1 cell bodies, where it dephosphorylates CaMKIV [56, 57]. Thus, reduced CaMKII and CaMKIV activity could be due in part to elevated CaN activity in CA1 (Figs. 6 and 7).

Proposed model showing mechanisms underlying memory deficits in NCKX2+/− mice. CaMKII and CaMKIV activity is downregulated in NCKX2+/− mice, possibly due to increased CaN phosphatase activity. As a result, CREB (Ser-133) phosphorylation and BDNF expression decrease. In addition, CaMKII downregulation decreases GluA1 (Ser-831) phosphorylation. Downregulation of both CaMKII and CaMKIV activities promotes cognitive deficits seen in NCKX2+/− mice, possibly via enhancing CaN activity
In neurons, several proteins, including Ca2+-ATPase in plasma membranes, Ca2+-ATPase in the endoplasmic reticulum, NCXs, NCKXs, and mitochondrial Ca2+ uniporters mediate Ca2+ excretion from the cytosol in somata and at post-synaptic densities [58, 59]. Recently, Lee [14] reported that NCKX2 endocytosis in the somato-dendritic region but not at axon terminals is regulated by Src family kinase-dependent tyrosine phosphorylation of NCKX2 Tyr-365 in cultured mouse hippocampal neurons. Our data shows that NCKX2 is predominantly expressed in somata of hippocampal pyramidal neurons and only weakly expressed in pre- and post-synaptic regions. Thus, partial reduction in NCKX2 levels in both synaptic and somatic regions likely accounts for impaired hippocampal LTP and cognitive deficits seen in NCKX2+/− mice.
NCKX2 protein and mRNA levels are significantly decreased in CA1 of APP23 mice. Our immunohistochemical analysis indicates that the expression levels of NCKX2 dramatically decrease in CA1 of APP23 relative to WT mice (Fig. 1d). NCKX2 expression also slightly decreased but not significant in the DG of APP23 mice (Fig. 1d). Since CaMKIIα-containing neurons are selectively lost in CA1 of AD patients [60], reductions in both CaMKII and NCKX2 may be associated with cognitive deficits seen in these individuals.
In conclusion, NCKX2 expression decreases in CA1 of APP23 mice, and NCKX2 decreases are associated with reduced CaMKII and CaMKIV activity in hippocampus and mild impairment of hippocampal LTP and learning behaviors. A question to be addressed in future studies is whether NCKX2 overexpression could rescue memory dysfunction and related behaviors in APP23 mice.
Abbreviations
- AD:
-
Alzheimer’s disease
- AMPAR:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor
- BDNF:
-
Brain-derived neurotrophic factor
- CaMKII:
-
Calcium/calmodulin-dependent protein kinase II
- CaMKIV:
-
Calcium/calmodulin-dependent protein kinase IV
- CaN:
-
Calcineurin
- CREB:
-
cAMP-responsive element binding protein
- DG:
-
Dentate gyrus
- ERK:
-
Extracellular signal-regulated kinase
- fEPSPs:
-
Field excitatory post-synaptic potentials
- GFP:
-
Green fluorescent protein
- HFS:
-
High-frequency stimulation
- LTP:
-
Long-term potentiation
- NCKXs:
-
K+-dependent Na+/Ca2+ exchangers
- NCXs:
-
Na+/Ca2+ exchangers
- PP1:
-
Protein phosphatase 1
- WT:
-
Wild-type
References
Berridge MJ (1998) Neuronal calcium signaling. Neuron 21:13–26
Berrie MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21
Augustine GJ, Santamaria F, Tanaka K (2003) Local calcium signaling in neurons. Neuron 40:331–346
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signaling: dynamics, homeostasis and remodeling. Nat Rev Mol Cell Biol 4:517–529
Jeon D, Yang YM, Jeong MJ et al (2003) Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron 38:965–976
Li XF, Kiedrowski L, Tremblay F et al (2006) Importance of K+-dependent Na+/Ca2+-exchanger 2, NCKX2, in motor learning and memory. J Biol Chem 281:6273–6282
Lee SH, Park KH, Ho WK et al (2007) Postnatal developmental changes in Ca2+ homeostasis in supraoptic magnocellular neurons. Cell Calcium 41:441–450
Dong H, Light PE, French RJ et al (2001) Electrophysiological characterization and ionic stoichiometry of the rat brain K+-dependent Na+/Ca2+ exchanger, NCKX2. J Biol Chem 276:25919–25928
Cai X, Lytton J (2004) Molecular cloning of a sixth member of the K+-dependent Na+/Ca2+ exchanger gene family, NCKX6. J Biol Chem 279:5867–5876
Lytton J (2007) Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J 406:365–382
Tsoi M, Rhee KH, Bungard D et al (1998) Molecular cloning of a novel potassium-dependent sodium-calcium exchanger from rat brain. J Biol Chem 273:4155–4162
Lytton J, Li XF, Dong H et al (2002) K+-dependent Na+/Ca2+ exchangers in the brain. Ann N Y Acad Sci 976:382–393
Lee KH, Ho WK, Lee SH (2013) Endocytosis of somatodendritic NCKX2 is regulated by Src family kinase-dependent tyrosine phosphorylation. Front Cell Neurosci 7:14
Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39
Malenka RC, Nicoll RA (1999) Long-term potentiation—a decade of progress? Science 285:1870–1874
Fukunaga K, Stoppini L, Miyamoto E et al (1993) Long-term potentiation is associated with a increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 268:7863–7867
Fukunaga K, Muller D, Miyamoto E (1995) Increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II and its endogenous substrates in the induction of long term potentiation. J Biol Chem 270:6119–6124
Silva AJ, Paylor R, Wehner JM et al (1992) Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257:206–211
Lledo PM, Hjelmstad GO, Mukherji S et al (1995) Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci U S A 92:11175–11179
Wang YJ, Chen GH, Hu XY et al (2005) The expression of calcium/calmodulin-dependent protein kinase II-alpha in the hippocampus of patients with Alzheimer’s disease and its links with AD-related pathology. Brain Res 1031:101–108
Barria A, Muller D, Derkach V et al (1997) Regulatory phosphorylation of AMPA-type glutamate receptor by CaM-KII during long-term potentiation. Science 276:2042–2045
Giese KP, Fedorov NB, Filipkowski RK et al (1998) Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279:870–873
McGlade-McCulloh E, Yamamoto H, Tan SE et al (1993) Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature 362:640–642
Jensen KF, Ohmstede CA, Fisher RS et al (1991) Nuclear and axonal localization of Ca2+/calmodulin-dependent protein kinase type Gr in rat cerebellar cortex. Proc Natl Acad Sci U S A 88:2850–2853
Miyano O, Kameshita I, Fujisawa H (1992) Purification and characterization of a brain-specific multifunctional calmodulin-dependent protein kinase from rat cerebellum. J Biol Chem 267:1198–1203
Kasahara J, Fukunaga K, Miyamoto E (2001) Activation of calcium/calmodulin-dependent protein kinase IV in long term potentiation in the rat hippocampal CA1 region. J Biol Chem 276:24044–24050
Ho N, Liauw JA, Blaeser F et al (2000) Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 20:6459–6472
Lee KH, Chatila TA, Ram RA et al (2009) Impaired memory of eyeblink conditioning in CaMKIV KO mice. Behav Neurosci 123:438–442
Wei F, Qiu CS, Liauw J et al (2002) Calcium calmodulin-dependent protein kinase IV is required for fear memory. Nat Neurosci 5:573–579
Takao K, Tanda K, Nakamura K et al (2010) Comprehensive behavioral analysis of calcium/calmodulin-dependent protein kinase IV knockout mice. PLoS One 5:e9460
Sturchler-Pierrat C, Abramowski D, Duke M et al (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer’s disease-like pathology. Proc Natl Acad Sci U S A 94:13287–13292
Sturchler-Pierrat C, Staufenbiel M (2000) Pathogenic mechanisms of Alzheimer’s disease analyzed in the APP23 transgenic mouse model. Ann N Y Acad Sci 920:134–139
Calhoun ME, Burgermeister P, Phinney AL et al (1999) Neuronal overexpression of mutant amyloid precursor proteins results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A 96:14088–14093
Moriguchi S, Ishizuka T, Yabuki Y et al (2016) Blockade of the KATP channel Kir6.2 by memantine represents a novel mechanism relevant to Alzheimer’s disease therapy. Mol Psychiatry. doi:10.1038/mp.2016.187
Keng VW, Yae K, Hayakawa T et al (2005) Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat Methods 2:763–769
Moriguchi S, Yamamoto Y, Ikuno T et al (2011) Sigma-1 receptor stimulation by dehydroepiandrosterone ameliorates cognitive impairment through activation of CaM kinase II, protein kinase C and extracellular signal-regulated kinase in olfactory bulbectomized mice. J Neurochem 117:879–891
Moriguchi S, Sakagami H, Yabuki Y et al (2015) Stimulation of sigma-1 receptor ameliorates depressive-like behaviors in CaMKIV null mice. Mol Neurobiol 52:1210–1222
Moriguchi S, Yabuki Y, Fukunaga K (2012) Reduced calcium/calmodulin-dependent protein kinase II activity in the hippocampus is associated with impaired cognitive function in MPTP-treated mice. J Neurochem 120:541–551
Fukunaga K, Horikawa K, Shibata S et al (2002) Ca2+/calmodulin-dependent protein kinase II-dependent long-term potentiation in the rat suprachiasmatic nucleus and its inhibition by melatonin. J Neurosci Res 70:799–807
Taigen T, De Windt LJ, Lim HW et al (2000) Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A 97:1196–1201
Zheng F, Zhou X, Luo Y et al (2011) Regulation of brain-derived neurotrophic factor exon IV transcription through calcium responsive elements in cortical neurons. PLoS One 6:e28441
Kidane AH, Heinrich G, Dirks RP et al (2009) Differential neuroendocrine expression of multiple brain-derived neurotrophic factor transcripts. Endocrinology 150:1361–1368
Soderling TR, Derkach VA (2000) Postsynaptic protein phosphorylation and LTP. Trend Neurosci 23:75–80
Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function synaptic and behavioral memory. Nat Rev Neurosci 3:175–190
Blaustein MP, Lederer WJ (1999) Sodium/calcium exchange: its physiological implications. Physiol Rev 79:763–854
Kang H, Sun LD, Atkins CM et al (2001) An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell 106:771–783
Tao X, Finkbeiner S, Arnold DB et al (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20:709–726
Gass P, Wolfer DP, Balschun D et al (1998) Deficits in memory tasks of mice with CREB mutations depend on gene dosage. Learn Mem 5:274–288
Montkowski A, Holsboer F (1997) Intact spatial learning and memory in transgenic mice with reduced BDNF. Neuroreport 8:779–782
Korte M, Kang H, Bonhoeffer T et al (1998) A role for BDNF in the late-phase of hippocampal long-term potentiation. Neuropharmacology 37:553–559
Sun P, Enslen H, Myung PS et al (1994) Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev 8:2527–2539
Schlman H, Lou LL (1989) Multifunctional Ca2+/calmodulin-dependent protein kinase: domain structure and regulation. Trends Biol Sci 14:62–66
Klee CB (1991) Concerted regulation of protein phosphorylation and dephosphorylation by calmodulin. Neurochem Res 16:1059–1065
Strack S, Barban MA, Wadzinski BE et al (1997) Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatase 1 and 2A. J Neurochem 68:2119–2128
Hemmings HC Jr, Greengard P, Tung HY et al (1984) DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature 310:503–505
Morioka M, Nagahiro S, Fukunaga K et al (1997) Calcineurin in the adult rat hippocampus: different distribution in CA1 and CA3 subfields. Neuroscience 78:673–684
Kasahara J, Fukunaga K, Miyamoto E (1999) Differential effects of a calcineurin inhibitor on glutamate-induced phosphorylation of Ca2+/calmodulin-dependent protein kinases in cultured rat hippocampal neurons. J Biol Chem 274:9061–9067
Thayer SA, Usachev YM, Pottorf WJ (2002) Modulating Ca2+ clearance from neurons. Front Biosci 7:D1255–D1279
Lee KH, Lee JS, Lee D et al (2012) KIF21A-mediated axonal transport and selective endocytosis underlie the polarized targeting of NCKX2. J Neurosci 32:4102–4117
Wang JH, Kelly PT (1995) Postsynaptic injection of Ca2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron 15:443–452
Acknowledgements
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology, and the Ministry of Health and Welfare of Japan (KAKENHI 22390109 to K.F.; 20790398 to S.M.; 23590319 to T.I.; 25460350 to S.K.), the Uehara Memorial Foundation (K.F.) and the Smoking Research Foundation (S.M.). We also thank Novartis Pharma for providing APP23 mice.
Author information
Authors and Affiliations
Contributions
S.M., Y.Y., R.I., H.I., Y.S., K.S., H.T., and T.I. performed experiments. S.K., K.H., J.T., and T.I. provided NCKX2 antibody and NCKX2 knockout mice, and critical advice. S.M. and K.F. wrote the manuscript and designed the study.
Corresponding authors
Ethics declarations
All animal protocols were approved by the Committee on Animal Experiments at Tohoku University.
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Moriguchi, S., Kita, S., Yabuki, Y. et al. Reduced CaM Kinase II and CaM Kinase IV Activities Underlie Cognitive Deficits in NCKX2 Heterozygous Mice. Mol Neurobiol 55, 3889–3900 (2018). https://doi.org/10.1007/s12035-017-0596-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0596-1