
Abstract
Chronic stress is a risk factor for major depression. Social defeat stress is a well-validated murine model of depression. However, little is known about the gene activity dynamics during the development of a depression-like state. We analyzed the effects of social defeat stress of varying duration (10 and 30 days) on the behavioral patterns and prefrontal-cortex transcriptome of C57BL/6 mice. The 10-day exposure to social defeat stress resulted in a high level of social avoidance with no signs of depression-associated behavior. Most animals exposed to 30 days of social defeat stress demonstrated clear hallmarks of depression, including a higher level of social avoidance, increased immobility in the forced swimming test, and anhedonic behavior. The monitoring of transcriptome changes revealed widespread alterations in gene expression on the 10th day. Surprisingly, the expression of only a few genes were affected by the 30th day of stress, apparently due to a reversal of the majority of the early stress-induced changes to the original basal state. Moreover, we have found that glucocorticoid-sensitive genes are clearly stimulated targets on the 10th day of stress, but these genes stop responding to the elevated corticosterone level by the 30th day of stress. The majority of genes altered by the 30-day stress were downregulated, with the most relevant ones participating in chromatin modifications and neuroplasticity (e.g., guanine nucleotide exchange factors of the Rho-family of GTPases). Very different molecular responses occur during short-term and long-term social stress in mice. The early-stress response is associated with social avoidance and with upregulation and downregulation of many genes, including those related to signal transduction and cell adhesion pathways. Downregulation of a few genes, in particular, genes for histone-modifying methyltransferases, is a signature response to prolonged stress that induces symptoms of depression. Altogether, our data show that the development of depression under social stress conditions is correlated with suppression of the overactive molecular response to induced stress, involving gene regulatory resistance to glucocorticoid molecules, potentially via a chromatin remodeling mechanism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Depression is a major burden on society and currently affects an estimated 350 million people globally. Depressive disorders were recently found to be the third leading financial burden and the leading cause of disability [1]. Depression can develop in the course of a severe illness or can be a consequence of posttraumatic stress disorder or chronic social stress. The latter cause is gaining significance under modern living conditions. The prefrontal cortex (PFC) is responsible for complex cognitive functions, regulation of emotion, and adaptation to stress. In this brain region, the integration of input signals takes place, with the subsequent regulation of the activity of the brain areas underneath it [2]. PFC activity is reduced in depressive-like states in rodents, and activation of PFC has an antidepressant effect [3,4,5]. Imaging and human postmortem studies indicate an altered activity in discrete regions within the prefrontal cortex, and their atrophy is associated with depression [6, 7]. Deep brain stimulation of discrete sites within the prefrontal cortex elicits a remarkable improvement in depressive symptoms in some patients, supporting the role of hypoactivity in various regions of the prefrontal cortex in depression [8,9,10].
Chronic defeat stress induces a depression-like state in mice, similar to depressive states in humans, with similarities in symptoms, etiology, and sensitivity to antidepressants, as well as brain neurochemical changes [11,12,13,14,15]. To elucidate the mechanism of the depressed state, transcriptome studies of certain brain regions of animals during social stress have been conducted [11, 16,17,18,19,20,21]. However, only a few studies dealt with the murine prefrontal cortex, and the reported transcriptomic patterns were not consistent across these studies [19,20,21]. Furthermore, there has not been a transcriptomic investigation of the development of depressive disease over time.
The aim of our work was to analyze the changes in gene expression profiles under social defeat stress of varying duration: 10 and 30 days. Studies on social defeat stress are usually limited to 10–14 days of stress, and only some sensitive animals exhibit the symptoms of depressive behavior [11, 16, 17, 22]. Longer stress periods, during which almost all studied animals develop depressive-like state, have been evaluated in only a few studies [12, 23,24,25,26]. Thus, genome-wide profiling at various stages during the development of the depression-like state should allow for the identification of the key genes involved in these pathological changes.
It is well known that the hypothalamus–pituitary–adrenal axis is the major system involved in stress response, and the altered glucocorticoid signaling is one of the major contributors for stress-induced psychopathologies [27,28,29]. Notwithstanding this, there are not so many investigations [30,31,32] concerning the role of glucocorticoids in chronic social defeat stress in mice. In this regard, in our analysis we included the search for potential targets of glucocorticoid receptor (GR).
Methods
Animals
Adult male mice of the C57BL/6 and ICR strains were provided by the Center for Genetic Resources of Laboratory Animals at the Institute of Cytology and Genetics, SB RAS, Novosibirsk, Russia (RFMEFI61914X0005 and RFMEFI62114Х0010). The animals were housed under standard conditions (12:12 h light/dark cycle, lights on at 8.00 a.m.; feed—pellets—and water were available ad libitum). The mice were weaned at 1 month of age and housed in groups of 8–10 in plastic cages (36 × 23 × 12 cm). Experiments were performed on mice 10–12 weeks of age. All procedures were approved by the Ethics Committee of the Institute of Cytology and Genetics, SB RAS (protocol 20.1, March 11, 2014).
Chronic Social Defeat Stress
Prolonged experience of social defeat in male mice was induced using the sensory contact model [14] with some modifications. Mice were housed in a steel cage (14 × 28 × 10 cm) bisected by a perforated transparent partition allowing the animals to see, hear, and smell each other but preventing physical contact. A C57BL/6 mouse was placed into the empty compartment adjacent to an aggressive ICR mouse. The animals were left undisturbed for 2 days to adapt to new housing conditions and sensory contact before they were subjected to encounters. Every afternoon (14:00–17:00 p.m. local time), the partition was removed for 10 min to encourage aggressive interactions. As a rule, aggressive confrontations between males are discontinued by lowering the partition if the strong aggression has lasted more than 3 min. During each defeat session, the C57BL/6 mouse was attacked by the aggressive ICR mouse and showed defensive behavior (sideways postures, upright postures, withdrawal, or freezing). Once a day, after the defeat session, each C57BL/6 mouse was placed in an unfamiliar cage with a new aggressive ICR mouse behind the partition. Each ICR mouse remained in its original cage. This procedure was performed once a day for 10 days (S10 group) or 30 days (S30 group).
Behavior of the Defeated Mice During Social Defeat
Behavior of the defeated mice was videotaped for 10 min during two consecutive encounters with different aggressors (days 7 and 8 for the S10 group, days 27 and 28 for the S30 group) (Fig. 1). For the analysis, we used the average parameters from the two sessions to minimize the influence of individual differences in the aggressors’ behavior. During a 10-min test, the following behavioral domains were analyzed in the defeated mice [14, 33]: (1) active defense—sideways and upright defensive postures during the aggressor’s attack and repulsion with one or two paws; (2) avoidance/flight—remaining at a prudential distance from the opponent, running away when opponent approaches, movement of the anterior part of the body and head away from the partner; (3) passive defense—freezing, remaining immobile when the aggressor was near the mouse or attacked it, position “on the back” after persecution by the aggressor; (4) freezing—complete lack of movement of the defeated mouse, without any attention to the aggressor; (5) waiting—the defeated mouse sits at the corner or at the cage’s wall and watches the aggressors movements; and (6) individual behavior—self-grooming, digging up and scattering the sawdust.

Experimental design. Detailed explanation in text
The Sucrose Preference Test
This test was used to assess stress-induced anhedonia. The test consists of a two-bottle choice where mice are given the choice between consuming water and a 1% solution of sucrose. The sucrose solution and water were provided on the 8th day of defeat stress for S10 group and on the 28th day of defeat stress for S30 group (30 min after the videotaping behaviors of mice during defeat session). On the next day after the partition test and daily defeat stress procedure, the sucrose solution and water were again provided to defeated mice. The mice were not food deprived and were exposed to the sucrose solution and water from 6:00 p.m. until 1:00 p.m. the next day. On the first day of provision of the sucrose solution, the mice were allowed to adapt to the testing conditions, and parameters of consumption were recorded on the second day of sucrose provision. The animals and the bottles were weighed to estimate consumption of the liquids. Sucrose solution preference (percentage of the consumed sucrose solution relative to the total amount of liquid intake) and sucrose solution intake/body weight (g/g) were calculated.
The Partition Test
This test [34] was employed for estimation of behavioral responses of mice to a conspecific individual behind the transparent perforated partition dividing the experimental cage. The number of approaches to the partition and the total time spent near it were scored during 5 min as indices of reacting to the partner. The first trial was carried out with the familiar aggressive ICR mouse, then the familiar partner was replaced by an unfamiliar one (a nonaggressive ICR mouse) and second trial were recorded for 5 min. The test was conducted on the 9th day of defeat stress for group S10 and on the 29th day of defeat stress for group S30.
The Forced Swimming Test
For quantification of depression-like behavior, we used the forced swimming test [35, 36]. Mice were individually placed into the Porsolt Forced Swim apparatus (Med Associates, Inc., USA): glass cylinders (height 45 cm, diameter 10 cm) containing water (27 cm deep, 24–25 °C). Immobility, which is indicative of helpless behavior, was recorded for 5 min. The test was conducted on the 10th day of defeat stress for the S10 group and on the 30th day of defeat stress for the S30 group.
Experimental Design
All of the mice were randomly subdivided into three groups: control, stress for 10 days (S10 group), and stress for 30 days (S30 group). A prolonged experience of social defeat in male mice was induced as described above. As a control for behavioral testing, we used male mice after 5 days of individual housing without aggressive interactions. The rationale for this choice is that it gives the best trade-off between group housing and social isolation: five days is sufficient for group housing to no longer be a factor and insufficient for social isolation to become a factor. Special investigations confirmed strong rationality of this control in the sensory contact model [37, 38]. Behavioral testing was performed consecutively: social defeat sessions, the partition test, the sucrose preference test, and the forced swimming test (Fig. 1). Mice were still undergoing the social stress protocol when these tests were carried out. The partition test and the forced swimming test were conducted the day after the last social defeat and shortly before the next one. The sucrose solution and water were provided immediately after the last social defeat until the start of the behavior test next day (consumption during 19 h). The experimental groups contained 16–24 animals. The animals were euthanized 48 h after the last behavioral test (forced swimming test) and 24 h after the last social defeat. All animals were sacrificed by decapitation, and blood samples and prefrontal cortices were collected for analysis. The prefrontal cortex was rapidly dissected and placed in RNALater solution. The samples were stored at −80 °C until RNA isolation.
Corticosterone Immunoassays
Quantitative determination of serum steroid levels was performed by means of commercially available assays for corticosterone (IDS, UK). Total trunk blood was collected and centrifuged at 3000 rpm for 10 min, and then the supernatant (serum) was stored at −70 °C until analysis. Appropriate dilutions of serum samples were prepared, and grossly hemolytic serum samples were excluded. The immunoassays were performed with strict adherence to manufacturer’s guidelines.
Analysis of the Behavioral Data
The test sessions were videotaped and scored by a trained observer using free open-source software BORIS (Behavioral Observation Research Interactive Software; http://www.boris.unito.it) [39]. The forced swimming test was scored by means of the Ethovision 10.0 software (Noldus Information Technology, the Netherlands).
The behavioral data were tested by either one-way or repeated-measures analysis of variance (ANOVA; with the post hoc Bonferroni test). The statistical-significance threshold was set to p <0.05. The behavior of defeated mice during social defeat was analyzed by the Mann–Whitney U test.
Total RNA Extraction
Total RNA was extracted from each prefrontal cortex. The tissue specimens were homogenized in TRI-Reagent (Sigma, USA), and RNA was isolated following the manufacturer’s protocol. The quantity and quality of total RNA were assessed by means of an Agilent 2100 Bioanalyzer and the Total RNA Nano kit (Agilent Technologies, USA). Only samples with an RNA integrity number greater than 8.0 were used for gene expression analysis.
Library Preparation
Four mice from each group (control, S10, and S30) were chosen randomly for high-throughput mRNA sequencing (RNA-seq) analysis. RNA-seq libraries of the murine prefrontal cortex were prepared in accordance with standard New England Biolab protocols (NEBNext mRNA Library Prep Master Mix Set for Illumina, NEB, USA). Briefly, polyA-tailed mRNA was purified from 1 μg of total RNA using the NEBNext® Poly(A) mRNA Magnetic Isolation Module (NEB, USA) and then broken into small fragments using divalent cations and heating. Using a reverse transcriptase and random primers, we synthesized first- and second-strand cDNAs. The cDNA was subjected to an end repair reaction with T4 DNA polymerase and Klenow DNA polymerase in order to blunt the termini. After that, an A base was added to the 3′ end of the blunt phosphorylated DNA fragments, and an Illumina adaptor with a single T overhang at its 3′ end was then ligated to the end of the DNA fragment. Size selection of DNA fragments was performed by means of Agencourt AMPure XP Beads (Beckman Coulter) and then PCR enrichment of the adapter-ligated library was conducted (six cycles of PCR). The size and quantity of the library were verified on the Agilent Bioanalyzer, and these fragments were subjected to nonstranded paired-end (2 × 100) sequencing on the Illumina HiSeq 2000 platform. Raw data were deposited in NCBI Sequence Read Archive (SRA—SRP075894).
Gene Expression Analysis
On average, ~34 million paired-end reads (25–67 million) were obtained for each sample of prefrontal cortex RNA by means of Illumina nonstranded sequencing. The sequencing data were preprocessed with the Trimmomatic 0.32 tool [40] to remove adapters and low-quality sequences. After barcode trimming, we assessed the quality of the sequencing data in the FastQC software package and mapped them onto the Mus musculus reference genome assembly mm9 (Ensemble release 67) using TopHat version 2.0.4 [41]. The free programming language, R, was used for the final data processing and visualization. The data were then converted into gene count tables by means of ENSEMBL gene annotation data. The resulting tables were subjected to an analysis of differential gene expression in the DESeq2 software package [42]. The Benjamini-Hochberg correction for multiple testing was applied to the resulting p values, and the genes with an adjusted p value <0.05 were designated as differentially expressed (DE) for further analysis. Genes that failed to converge to the Generalized Linear Model in the differential expression analysis were excluded. RNA-seq quality metrics showed that within each library, more than 93% of the reads were uniquely mapped to the genome, with less than 1% being rRNA (Fig. S1).
Pathway Analysis
Gene Ontology (GO) enrichment analysis was conducted using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) [43, 44]. The set of genes from our RNA-seq study with counts ≥10 served as a reference set. Enrichment of GOs was assessed using a hypergeometric test corrected for multiple testing (Benjamini-Hochberg, p adj < 0.05). Only statistically significant enriched terms were reported and interpreted.
Real-Time Quantitative PCR (RT-qPCR)
RT-qPCR was performed to confirm the differential expression of genes identified by the RNA-seq process. The six selected genes were analyzed by qPCR, covering multiple fold changes and p values: three of them were downregulated only in the S30 group (Grin2c, Mcf2l, Trank1), one of them was downregulated in the S10 group (Nptx2), and two genes were changed both in the S10 and S30 groups (Arhgef7, Sparc). A primer set for each gene was designed using Primer-BLAST. Forward and reverse primer sequences are listed in Table S1. In order to verify the RNA-seq results, RT-qPCR was done on the same libraries, extended by additional biological replicates up to a total of five to seven samples per group. The PCR cycling conditions were 95 °C for 5 min followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Data were collected at 60 °C. A melt curve analysis was performed at the end of each qPCR experiment. All real-time qPCRs were performed on the Bio-Rad CFX platform (Bio-Rad, Hercules, CA) using qPCR set with Eva Green I (Syntol, Russia). The qPCR data were analyzed using the ΔΔCt method and normalized to the housekeeping genes Hprt1 and 16S. All qPCR reactions were performed in duplicates.
Analysis of Glucocorticoid-Sensitive Genes and ChIP-seq Datasets for GR Binding Sites
The dataset of dexamethasone-affected genes in mouse primary cortical astrocyte cultures was downloaded from a publicly available depository [45]. Genes that were differentially expressed at 2, 4, 6, 12, 18, or 24 h after dexamethasone treatment were used for Pearson correlation analysis with the whole set of genes from our RNA-seq study.
In the analysis of mouse ChIP-seq data for glucocorticoid receptor (GR) binding sites [46,47,48,49,50], we first assessed the genomic loci for the promoters (the 2-kb region upstream of the transcription start site, TSS) of 22,698 annotated protein-coding genes (Ensembl v67). For each dataset, we quantified the number of annotated genes that have at least one promoter overlapping with GR binding sites and how many of these genes are present in the DEG_S10 or DEG_S30 gene sets. Finally, we estimated the significance of the overlap with a hypergeometric test. For the analysis of rat PC12 cells [51] and rat hippocampus [52] datasets, we utilized two alternative approaches. First, we identified regions in the mouse reference genome that were homologous to reported ChIP-seq peaks in the rat reference genome and then analyzed them similarly to the mouse ChIP-seq dataset. Second, we identified the mouse genes that were orthologs to the rat genes with a GR binding site in the promoter. In all the analyses of orthologous genes, we used only the 20,956 protein-coding mouse genes which have orthologs in the rat, as reported in Ensembl v67.
Results
Serum Corticosterone Levels and the Behavioral Response to Chronic Social Defeat Stress
The behavioral response was analyzed in a battery of tests, which included the partition test, the sucrose preference test, the forced swimming test, and analysis of behavior in a social defeat session.
The partition test evaluates sociability of mice in a home environment. Similar to the previous studies, the partition test revealed a reliable segregation between the control and depressive (S30 group) animals and an intermediate diffuse distribution of the S10 group (Fig. 2e). One-way ANOVA revealed a significant effect of the groups on the total time spent near the partition in reaction to aggressive partner [F(2,58) = 17.8, p < 0.001] and unfamiliar nonaggressive partner [F(2,58) = 17.9, p < 0.001]. Response to aggressive partner was lower in the S10 (p < 0.01) and S30 (p < 0.001) groups, but response to unfamiliar partner was lower only in the S30 group (p < 0.001) (Fig. 2a, b). This result indicated that mice from the S30 group actively avoid a partner and demonstrated a social avoidance to both aggressive and nonaggressive unfamiliar mice.

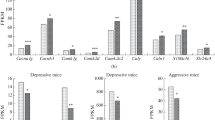
The behavioral response to chronic social stress and serum corticosterone levels. a, b Total time spent near the partition in response to an aggressive (a) and unfamiliar nonaggressive (b) partner. c, d Immobility time and latency of immobility in the forced swimming test. e Frequency distribution histogram for the absolute time spent near partition in reaction to the aggressive partner. f The sucrose solution preference test. g Serum levels of corticosterone. *p < 0.05, **p < 0.01, ***p < 0.001 as compared with control, ## p < 0.01 as compared with group S10 (assessed by one-way ANOVA, post hoc Bonferroni test)
A sucrose solution is a natural reward, and a decrease in sucrose consumption reflects of anhedonia: one of the core symptoms of major depressive disorder. One-way ANOVA revealed that chronic stress exposure influenced the preference for sucrose solution [Fig. 2f; F(2,42) = 8.08, p < 0.001]: the S30 group showed a lower sucrose solution preference in comparison with the controls and S10 group (p < 0.05). We did not observe changes in total liquid intake; therefore, the intake of decreased sucrose solution reflects a change in the desire to consume a sweet drink rather than a change in liquid intake under stress (Fig. S2).
To assess the influence of stress on depressive behavior, the mice were analyzed by the forced swimming test. Figure 2c and d shows that the total time spent immobile [F(2,58) = 5.66; p < 0.01] and latency of immobility [F(2,58) = 5.54; p = 0.01] varied as a function of stress duration. Exposure to 30-day but not 10-day stress decreased the latency of immobility (p < 0.01) and increased the total duration of immobility (p < 0.01) as compared to the control mice, thus reflecting aggravation of depressive symptoms under the influence of chronic social defeat stress.
Behavioral analysis during a social defeat session revealed a significant reduction in the manifestation of individual behavior (p < 0.001) in the S30 group as compared to the S10 group (Table S2). During the 10-min interaction with an aggressive partner, the mice in the S30 group spent more time frozen in the corner in the absence of an aggressor (p < 0.05) or watching the aggressor (p < 0.05), in comparison with the S10 group.
We assessed the influence of chronic exposure to stress on the basal level of serum corticosterone. Serum corticosterone concentrations increased as a function of the duration of stress [F(2,33) = 6.41, p < 0.01] among the three groups. Both S30 group and S10 group showed corticosterone levels higher than those in the control group of mice (p < 0.05 both; Fig. 2g).
Thus, the results show that 30 days of social defeat stress causes a pronounced depression-like state, whereas 10 days of stress is insufficient for the development of this pathology.
Opposing Effects of Chronic Social Defeat Stress on the Prefrontal Cortex Transcriptome
In accordance with the results of the behavioral tests, the analysis of RNA-seq data revealed differences in the transcriptomic profiles between the control and stressed mice, both after 10 and 30 days of stress (Fig. 3). The most prominent changes were observed in the S10 group, with 360 genes being upregulated and 113 genes downregulated, as compared to the controls (Table S3, subset “DEG_S10”). In the S30 group versus the controls, only 6 upregulated and 29 downregulated genes were differentially expressed (Table 1 and Table S4, subset “DEG_S30”). The expression of nine genes was altered in both stressed groups with comparable fold changes (Table 1). Meanwhile, a total of 177 genes were differentially expressed between S10 and S30 groups (Table S5, subset “DEG_S30vsS10”), with about half of them (90 genes) also being altered in the S10 group versus the controls. Figure 4 shows that the majority of the genes altered by 10-day stress tended to return toward their control levels of expression by the 30th day of stress, with some of them even reverting beyond the control values (panel c). This latter cluster of genes (87 genes) showed differential expression only between the two stressed groups and was characterized by a profound reversal in their expression level trend and modulating back toward the control levels. Thus, the main trend in the mRNA expression dynamics observed from the transcriptome data is an induction of stress-responsive genes followed by a partial recovery of their expression with an increased duration of social defeat stress.

A Venn diagram of the differentially expressed genes (adjusted p < 0.05)

Relative per-library expression of genes differentially expressed in the S10 group versus the control and S30 groups. Left panels: heatmaps showing the scaled rlog-transformed gene counts across 12 analyzed libraries. Right panels: the results of principal component analysis applied to the respective subsets of genes in order to assess expression similarities among the samples; x- and y-axes correspond to the first and second principal components. The genes are split into subsets (a–d) according to their status of differential expression in three possible comparisons between the S10, S30, and control groups (accounting for intersections): (a) only in S10 versus control, 383 genes; (b) in S10 versus both S30 and control, 90 genes; (c) only in S30 versus S10, 87 genes; (d) only in S30 versus control, 25 genes
Pathway Analysis Links Stress to Neuronal Plasticity
To decipher the functional processes affected by the stress conditions under study, we used several widely used programs for functional enrichment analysis and gene network construction. The broadest and most tightly connected functional group of genes responding to 10-day stress were related to the structural maintenance and regulation of the extracellular matrix, intercellular contacts, and cell adhesion (Table S6). A schematic representation of STRING Protein-Protein Interaction Network of DEG_S10 genes is shown in Fig. S3 which demonstrates the tightly functional connections between the genes in the discussed GO categories. This group is coupled with the alteration of genes from the PI3K-Akt signaling pathway (Ptk2, Sgk1), as well as several key genes related to Wnt/Bmp signaling (Bmp4, Bmp5, Bmp6, Bmp7, Wnt4, Wnt6, etc.), and is involved in the development and maintenance of brain structures. Thus, the observed changes on the 10th day of stress point to the modulation of cortical cell plasticity.
In contrast, the group of genes affected by 30-day stress did not produce any significantly enriched functional relationships when compared to the controls, in part due to its small size. However, the differentially expressed genes in the S30 group related to cell communication, in particular, for the regulation of small GTPase-mediated signal transduction (Arhgef7, Mcf2l, Myo9b) and calcium ion binding (Sparc, Plch2, Otof) (Table S7). We also analyzed the subset of 87 genes showing differential expression between the two stressed groups alone (Fig. 4, bottom panel). Intriguingly, this subset was significantly enriched with chromatin remodeling proteins (Table S8), including the three enzymes of histone H3 methylation, Kmt2a, Kmt2d, and Setd1b, making it tempting to speculate that the observed systemic reversal of stress-induced changes may involve epigenetic mechanisms.
RT-qPCR Validation of RNA-seq Results
Comparison of fold change in differential expression values determined by RNA-seq and qPCR shows similar level and direction of changes for most genes (Fig. 5). Downregulation of three of five genes from DEG_S30 and one of three genes from DEG_S10 was confirmed by RT-qPCR: Arhgef7, Grin2c, Mcf2l, Nptx2. In addition, downregulation of Grin2c in the S10 group was also confirmed (nominal p = 0.059 in the RNA-seq data). Each of the confirmed fold changes was greater than that indicated by the RNA-seq. The other gene expression changes were not significant.

RT-qPCR validation of RNA-seq results. a mRNA level of Arhgef7, Grin2c, Mcf2l, Nptx2, Sparc, Trank1 normalized to Hprt1 and 16S (one-way ANOVA, LCD post hoc test, values represent mean ± SEM, n = 5–6 animals/group, *p < 0.05). b Fold changes of the genes revealed by RNA-seq
Glucocorticoid Regulation of the Differentially Expressed Genes
Given the well-known influence of glucocorticoids on the manifestation of the stress response and the elevated levels of corticosterone observed in both S10 and S30 groups, it is compelling to investigate whether glucocorticoid signaling is responsible for the gene expression changes seen in these groups. Because there were no available data on the expression changes in the murine prefrontal cortex following glucocorticoid treatment, we used microarray data derived from murine cortical astrocytes [45] to perform the correlation analysis with our RNA-seq data. As shown in Fig. 6, significant correlations were found between the log fold changes (LFCs) obtained for the S10 group and the LFCs of dexamethasone-affected genes, with the strongest correlation at 12 and 24 h after treatment. Contrariwise, the S30 group seemed to lack a similar relationship. Significant negative correlations were also revealed for LFCs obtained for S10vsS30 comparison. Similarly, in terms of the enrichment analysis, dexamethasone-responsive genes were significantly enriched in the DE genes of S10 but not S30 group (Table S9). Although the observed correlation coefficients were not very pronounced, this could be partially attributed to the limitations of comparing RNA-seq data to a more error-prone microarray dataset. In this respect, increasing the p-value cutoff for microarray data resulted in a stronger correlation between the stress-induced and dexamethasone-induced expression changes (Fig. 6b). The results suggest that, in spite of the elevated levels of corticosterone in both S10 and S30 groups, the glucocorticoid-responsive genes follow the same overall trend of gene expression changes, namely an alteration after 10 days of stress and recovery to control levels at 30th day of stress.

Correlation between the changes of gene expression in response to social defeat stress (RNA-seq, prefrontal cortex) and in response to dexamethasone treatment (microarray, primary cortical astrocytes). a Scatterplots of log2-fold-changes of gene expression, observed between three experimental groups (no p value cutoff) versus six time points after dexamethasone treatment (p value < 0.01). Straight lines show linear fits of the data. Each plot is accompanied by Pearson correlation coefficient and p value. b Dependency between the level of correlation coefficients and a p-value cutoff used for choosing differentially expressed genes in each time point of microarray study (shown by grayscale gradient). Correlation coefficients with significance p ≥ 0.01 are marked by filled circles
To further investigate whether the differentially expressed genes may be directly modulated by the glucocorticoid receptor, we performed an enrichment analysis of GR binding sites (GRBS) within the promoter regions of DE genes using several published ChIP-seq datasets for the GR across different mouse cell types [46,47,48,49,50]. These data, however, lacked brain or neuron-related cell types; therefore, we additionally analyzed ChIP-seq data in rat neuronal PC12 cells [51] and rat hippocampus [52]. For the S10 group, we found a significant overrepresentation of GR binding sites (as compared to all annotated genes) in DE gene promoters for all mouse ChIP-seq datasets (nominal p value <0.05, hypergeometric test; Table 2), except for erythroid progenitor cells. This trend was also apparent when we combined the five experimental datasets. In the rat datasets, hippocampal GR binding sites are overrepresented in group S10 genes. In contrast to group S10, the promoters of DE genes in the S30 group demonstrated almost no overlap with GR binding sites.
Discussion
Social defeat stress is the most relevant mouse model of depression and exhibits features of construct, face, and predictive validity, although the intensity of the stress used is more severe than that seen in most humans [53]. Using this model with some modifications gives stable results from different groups of researchers. However, these studies are usually limited to 10–14 days of stress and only 50–60% of the mice exhibited depressive symptoms [11, 16, 17, 22]. In our work, we additionally used a more prolonged stress period—30 days, when almost all animals studied develop a severe depressive state.
In this study on male mice, we demonstrated that after 30 days of social defeat stress (S30 group), the mice developed a pronounced depression-like state, whereas the mice in the S10 group showed only some signs of behavioral aberrations. In the depressive animals, we observed a stable social avoidance regardless of the type of partner. Also, these animals showed an increased immobility in the forced swimming test and a decreased preference for sucrose solution, which are indicative of helpless behavior and stress-induced anhedonia—one of the core symptoms of depression. During a 10-min interaction with an aggressive partner, the mice in the S30 group preferred to spend time in the corner of their compartment, in complete immobility, or watching the aggressor from afar, and did not represent individual behaviors (rears, self-grooming). Overall, it appears that under the influence of chronic social defeat stress, the animals develop behavioral deficits and show a weaker reaction toward any external stimuli. In the majority of situations, mice in the S30 group preferred to remain sedentary, expressing no interest in their surroundings. These observed effects are in agreement with other studies involving social stress of similar duration (20–30 days), which showed the development of a pronounced behavioral deficit in stressed animals [12, 24, 54, 55].
Mice in the S10 group showed only social avoidance to the aggressive partner behind the partition and no significant differences in the other tests. The level of response to the aggressive partner in the S10 group (Fig. 3a, e) indicated that the disturbances in the behavior under the influence of the stress had already begun but not yet reached the level of depressive animals. Other studies of 10-day social defeat stress showed that behavioral disturbances are not observed in the whole group. So, investigators have to divide the animals into a stress-susceptible group and a stress-resistant group. The basis of this separation may be the defeat-induced social avoidance [11, 16], development of anhedonic behavior [56], or differences in the forced swimming test [57]. In the present study, using the most variable parameter between groups, the level of response to the aggressive partner, we divided the S10 group into two subgroups: similar to the response of the control animals or similar to the response of the S30 group. However, these subgroups did not show any differences in other tests (Fig. S4). This finding suggests that despite the weakened reaction to the partner, all other signs of the depression-like state did not appear in the S10 group.
Our study seems to be the first report of effect of long (30 days) social defeat stress on murine prefrontal cortex transcriptome. The analysis revealed an interesting relation between the stress duration and the number of genes affected by social defeat stress: 10 days of stress changed the expression of 473 genes, whereas 30 days of stress changed the expression of only 35 genes. The overlap of these gene sets is small (only nine genes), highlighting a qualitative difference between the depression-like state and the stress response. Comparison of the gene expression changes among the groups of mice showed that the expression of the majority of genes that were affected by 10 days of stress returned almost to the baseline level after continued stress (30 days). Thus, we see a seemingly paradoxical response: an increase in stress duration reverses the changes in transcriptome, against the background of the pronounced depression-like state in the mice. However, one can assume that transcriptomic changes that occurred at day 10 may lead indirectly to the behavioral changes observed at day 30, possibly through some mechanisms associated with neuroplasticity. It is worth mentioning also that studies of 10-day social defeat stress received less transcriptomic changes in stress-susceptible mice with symptoms of depressive-like state than in stress-resilient mice [16, 20]. So, it seems that the depressive animals have a reduced reaction to stress and other external unpleasant stimuli in terms of stress-related gene expression.
In addition, in terms of gene expression, our work confirmed the findings that the development of a depressive state is accompanied by a decrease in responsiveness to glucocorticoids (glucocorticoid resistance) [58, 59]. So, in patients with depression despite the often high glucocorticoid concentrations were found a reduction in expression of GR-sensitive genes and an inability of GR to suppress inflammation [60], confirming defective signaling through glucocorticoid receptors. In our model, we have found that the targeted genes stop responding to the elevated corticosterone levels. Using data on the dexamethasone-induced gene expression, we found significant correlations between expression of DE genes in the S10 group and dexamethasone-affected genes, while S30 group seemed to lack a similar relationship. Also in group S10, most of the DE genes can be targets of glucocorticoids, as demonstrated by the significant enrichment GR binding sites, with no similar enrichment in the S30 group. All of this suggests that the differential expression in S30 group does not depend on glucocorticoids, despite the elevated level of corticosterone. It is known that chronic stress leads to a disturbance of the negative feedback system and dysregulation of the normal hypothalamic–pituitary–adrenocortical axis functionality [61]. In our study, the level of GR mRNA remained unchanged in the S10 and S30 groups; however, in the S10 group, we found the increased level of Fkpb5 mRNA (Fig. S4), indicating activation of GR-signaling pathway in the S10 group. The glucocorticoid resistance may be caused by changes in the functional activity of the GR, including post-transcriptional receptor modifications or different GR isoforms [27, 62, 63]. Another possible mechanism of glucocorticoid resistance under chronic stress is epigenetic modification of the regulatory regions of the target genes [27]. We found that depressive mice (S30 group) had significantly reduced expression of Dot1l, a histone H3K79 methyltransferase, indicating active gene transcription. The analysis of DE genes between the two stressed groups revealed the downregulation of lysine N-methyltransferases (Kmt2a, Kmt2d, Setd1b) (Fig. S5), which are essential for synaptic plasticity and might be involved in cortical dysfunction [64]. It is well known that disruption of methyltransferase expression occurs in various neurodevelopmental disorders and syndromes [65]. The data showing expression changes in genes relating to epigenetic modifications suggest that an epigenetic mechanism for the stress-related glucocorticoid resistance is most likely.
Gene ontology analyses revealed that the most significantly enriched gene categories in group S10 are related to the extracellular matrix and cell adhesion. The related genes are mostly upregulated. The extracellular matrix is produced intracellularly and secreted to form a dense network of proteins and glycans, occupying the parenchyma of virtually all tissues. The extracellular matrix supports cell adhesion, transmits signals to cell surface adhesion receptors, modulates cell functionality and plasticity, and participates in the regulation of brain architecture and synaptic plasticity [66, 67]. We uncovered a significant enhancement of expression of the genes linked to the formation of the basement membrane (Col8a1, Lama1, Lama2, Lama4, Lamc3, Mmrn2, Nid1, Sparc). Increased synthesis of collagens and laminins leads to increased density (compaction) of the extracellular matrix. Similarly enhanced expression of extracellular matrix genes was found after chronic stress in the rat [68] and mouse [18, 20]. Thus, enhanced expression of the extracellular matrix-related and cell adhesion genes in the prefrontal cortex may affect cortical neural plasticity, including morphological neuronal changes and the afferent and/or efferent neural pathways participating in stress-related emotional behavioral patterns.
Among the DE genes in the depressive animals (S30 group), the strongest downregulation was observed for the Robo3 gene. Furthermore, in the S10 group, the expression of Robo3 was not different from that in the control group. The Robo3 gene encodes a transmembrane receptor and plays a distinct role in axonal guidance [69]. In the adult brain, SLIT-ROBO signaling promotes cell adhesion by stimulating the interaction between E-cadherin and β-catenin at the plasma membrane [70]. In addition to the downregulation of Robo3 in the depressive mice, we also uncovered decreased expression of one type of ephrin receptor (Epha10) and one subunit of the NMDA receptor (Grin2c), both of which are involved with cell-to-cell communication and neuronal synaptic plasticity [71, 72]. We detected the downregulation of two guanine nucleotide exchange factors (GEFs) of the Rho-family of GTPases in our set of DE genes in depressive mice: Arhgef7 and Mcf2l. Decreased expression of these GEFs reduces the activity of their downstream effector, Rac1, and its regulatory processes. Rac1 is involved in the regulation of actin cytoskeletal spine dynamics, stress fiber formation, and actin polymerization and stabilization, as implied by the observed transcriptional downregulation of some relevant genes (Myo9b, Otof, Fyco1, and Daam2). A decrease in cytoskeletal spine density and aberrations in the cytoskeleton and cell adhesion are characteristic of depressive states [71, 73, 74]. Our results seem to support the idea that major depressive disorder is associated with defective cell adhesion and impaired neuronal plasticity.
Altogether, our data show that the development of depression under social defeat stress conditions is accompanied by suppression of the overactive molecular response to induced stress, involving gene regulatory resistance to glucocorticoids, potentially via a chromatin remodeling mechanism. These processes may lead to an observed impairment (at the transcriptional level) of cell adhesion and neuronal plasticity.
References
Ferrari AJ, Charlson FJ, Norman RE, Patten SB, Freedman G, Murray CJ, Vos T, Whiteford HA (2013) Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med 10(11):e1001547. doi:10.1371/journal.pmed.1001547
Challis C, Berton O (2015) Top-down control of serotonin systems by the prefrontal cortex: a path towards restored socioemotional function in depression. ACS Chem Neurosci 6(7):1040–1054. doi:10.1021/acschemneuro.5b00007
Covington HE 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, LaPlant Q, Mouzon E et al (2010) Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci 30(48):16082–16090. doi:10.1523/JNEUROSCI.1731-10.2010
Vialou V, Bagot RC, Cahill ME, Ferguson D, Robison AJ, Dietz DM, Fallon B, Mazei-Robison M et al (2014) Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: role of DeltaFosB. J Neurosci 34(11):3878–3887. doi:10.1523/JNEUROSCI.1787-13.2014
Lehmann ML, Herkenham M (2011) Environmental enrichment confers stress resiliency to social defeat through an infralimbic cortex-dependent neuroanatomical pathway. J Neurosci 31(16):6159–6173. doi:10.1523/JNEUROSCI.0577-11.2011
Albert PR, Vahid-Ansari F, Luckhart C (2014) Serotonin-prefrontal cortical circuitry in anxiety and depression phenotypes: pivotal role of pre- and post-synaptic 5-HT1A receptor expression. Front Behav Neurosci 8:199. doi:10.3389/fnbeh.2014.00199
Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ (2007) GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology 32(2):471–482. doi:10.1038/sj.npp.1301234
Hamani C, Mayberg H, Snyder B, Giacobbe P, Kennedy S, Lozano AM (2009) Deep brain stimulation of the subcallosal cingulate gyrus for depression: anatomical location of active contacts in clinical responders and a suggested guideline for targeting. J Neurosurg 111(6):1209–1215. doi:10.3171/2008.10.JNS08763
Berlim MT, McGirr A, Van den Eynde F, Fleck MP, Giacobbe P (2014) Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord 159:31–38. doi:10.1016/j.jad.2014.02.016
Holtzheimer PE, Mayberg HS (2011) Deep brain stimulation for psychiatric disorders. Annu Rev Neurosci 34:289–307. doi:10.1146/annurev-neuro-061010-113638
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM et al (2006) Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311(5762):864–868. doi:10.1126/science.1120972
Kudryavtseva NN, Avgustinovich DF (1998) Behavioral and physiological markers of experimental depression induced by social conflicts (DISC). Aggress Behav 24:271–286. doi:10.1002/(SICI)1098-2337(1998)24:4<271::AID-AB3>3.0.CO;2-M
Venzala E, Garcia-Garcia AL, Elizalde N, Delagrange P, Tordera RM (2012) Chronic social defeat stress model: behavioral features, antidepressant action, and interaction with biological risk factors. Psychopharmacology 224(2):313–325. doi:10.1007/s00213-012-2754-5
Kudryavtseva NN, Bakshtanovskaya IV, Koryakina LA (1991) Social model of depression in mice of C57BL/6J strain. Pharmacol Biochem Behav 38(2):315–320
Bartolomucci A, Leopardi R (2009) Stress and depression: preclinical research and clinical implications. PLoS One 4(1):e4265. doi:10.1371/journal.pone.0004265
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A et al (2007) Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131(2):391–404. doi:10.1016/j.cell.2007.09.018
Warren BL, Vialou VF, Iniguez SD, Alcantara LF, Wright KN, Feng J, Kennedy PJ, Laplant Q et al (2013) Neurobiological sequelae of witnessing stressful events in adult mice. Biol Psychiatry 73(1):7–14. doi:10.1016/j.biopsych.2012.06.006
Stankiewicz AM, Goscik J, Majewska A, Swiergiel AH, Juszczak GR (2015) The effect of acute and chronic social stress on the hippocampal transcriptome in mice. PLoS One 10(11):e0142195. doi:10.1371/journal.pone.0142195
Stankiewicz AM, Goscik J, Swiergiel AH, Majewska A, Wieczorek M, Juszczak GR, Lisowski P (2014) Social stress increases expression of hemoglobin genes in mouse prefrontal cortex. BMC Neurosci 15(1):130. doi:10.1186/s12868-014-0130-6
Bagot RC, Cates HM, Purushothaman I, Lorsch ZS, Walker DM, Wang J, Huang X, Schluter OM et al (2016) Circuit-wide transcriptional profiling reveals brain region-specific gene networks regulating depression susceptibility. Neuron 90(5):969–983. doi:10.1016/j.neuron.2016.04.015
Tordera RM, Garcia-Garcia AL, Elizalde N, Segura V, Aso E, Venzala E, Ramirez MJ, Del Rio J (2011) Chronic stress and impaired glutamate function elicit a depressive-like phenotype and common changes in gene expression in the mouse frontal cortex. Eur Neuropsychopharmacol 21(1):23–32. doi:10.1016/j.euroneuro.2010.06.016
Dias C, Feng J, Sun H, Shao NY, Mazei-Robison MS, Damez-Werno D, Scobie K, Bagot R et al (2014) Beta-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature 516(7529):51–55. doi:10.1038/nature13976
Bondar NP, Kovalenko IL, Avgustinovich DF, Smagin DA, Kudryavtseva NN (2009) Anhedonia in the shadow of chronic social defeat stress, or when the experimental context matters. The Open Behavioral Science Journal 3:17–27. doi:10.2174/1874230000903010017
Avgustinovich DF, Kovalenko IL, Kudryavtseva NN (2005) A model of anxious depression: Persistence of behavioral pathology. Neurosci Behav Physiol 35(9):917–924. doi:10.1007/s11055-005-0146-6
Kudryavtseva NN, Bondar NP, Boyarskikh UA, Filipenko ML (2010) Snca and Bdnf gene expression in the VTA and raphe nuclei of midbrain in chronically victorious and defeated male mice. PLoS One 5(11):e14089. doi:10.1371/journal.pone.0014089
Boyarskikh UA, Bondar NP, Filipenko ML, Kudryavtseva NN (2013) Downregulation of serotonergic gene expression in the raphe nuclei of the midbrain under chronic social defeat stress in male mice. Mol Neurobiol 48(1):13–21. doi:10.1007/s12035-013-8413-y
Merkulov VM, Merkulova TI, Bondar NP (2017) Mechanisms of brain glucocorticoid resistance in stress-induced psychopathologies. Biochemistry (Mosc) 82(3):351–365. doi:10.1134/S0006297917030142
Pariante CM, Lightman SL (2008) The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31(9):464–468. doi:10.1016/j.tins.2008.06.006
Cattaneo A, Riva MA (2015) Stress-induced mechanisms in mental illness: a role for glucocorticoid signalling. J Steroid Biochem Mol Biol. doi:10.1016/j.jsbmb.2015.07.021
Wu X, Wu J, Xia S, Li B, Dong J (2013) Icaritin opposes the development of social aversion after defeat stress via increases of GR mRNA and BDNF mRNA in mice. Behav Brain Res 256:602–608. doi:10.1016/j.bbr.2013.09.034
Lehmann ML, Brachman RA, Martinowich K, Schloesser RJ, Herkenham M (2013) Glucocorticoids orchestrate divergent effects on mood through adult neurogenesis. J Neurosci 33(7):2961–2972. doi:10.1523/JNEUROSCI.3878-12.2013
Koch CE, Bartlang MS, Kiehn JT, Lucke L, Naujokat N, Helfrich-Forster C, Reber SO, Oster H (2016) Time-of-day-dependent adaptation of the HPA axis to predictable social defeat stress. J Endocrinol 231(3):209–221. doi:10.1530/JOE-16-0163
Brain PF, McAllister KH, Walmsley SV (1989) Drug effects on social behaviour: methods in ethopharmacology. In: Boulton AA, Baker GB, Greenshaw AJ (eds) Neuromethods. psychopharmacology. The Humana Press Inc, Clifton, NJ, pp. 687–739
Kudryavtseva NN (2003) Use of the “partition” test in behavioral and pharmacological experiments. Neurosci Behav Physiol 33(5):461–471. doi:10.1023/A:1023411217051
Castagne V, Porsolt RD, Moser P (2009) Use of latency to immobility improves detection of antidepressant-like activity in the behavioral despair test in the mouse. Eur J Pharmacol 616(1–3):128–133. doi:10.1016/j.ejphar.2009.06.018
Porsolt RD, Bertin A, Jalfre M (1977) Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther 229(2):327–336
Avgustinovich DF, Kovalenko IL, Bondar NP (2005) Choice of “control” in experimental researches of animal social interactions in mice. Ross Fiziol Zh Im I M Sechenova 91(12):1454–1468
Kudryavtseva NN (2011) Sensory contact model: protocol, control, applications. Nova Science Publishers, Inc, New York
Friard O, Gamba M (2016) BORIS: a free, versatile open-source event-logging software for video/audio coding and live observations. Methods Ecol Evol. doi:10.1111/2041-210X.12584
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. doi:10.1093/bioinformatics/btu170
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14(4):R36. doi:10.1186/gb-2013-14-4-r36
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550. doi:10.1186/s13059-014-0550-8
Wang J, Duncan D, Shi Z, Zhang B (2013) WEB-based GEne SeT AnaLysis toolkit (WebGestalt): Update 2013. Nucleic Acids Res 41 (web server issue):W77–83. doi:10.1093/nar/gkt439
Zhang B, Kirov S, Snoddy J (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33 (web server issue):W741–748. doi:10.1093/nar/gki475
Carter BS, Meng F, Thompson RC (2012) Glucocorticoid treatment of astrocytes results in temporally dynamic transcriptome regulation and astrocyte-enriched mRNA changes in vitro. Physiol Genomics 44(24):1188–1200. doi:10.1152/physiolgenomics.00097.2012
John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA (2011) Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43(3):264–268. doi:10.1038/ng.759
Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC (2012) Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci U S A 109(28):11160–11165. doi:10.1073/pnas.1111334109
Yu CY, Mayba O, Lee JV, Tran J, Harris C, Speed TP, Wang JC (2010) Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS One 5(12):e15188. doi:10.1371/journal.pone.0015188
Lee HY, Gao X, Barrasa MI, Li H, Elmes RR, Peters LL, Lodish HF (2015) PPAR-alpha and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature 522(7557):474–477. doi:10.1038/nature14326
Grontved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, Aguilera G, Hager GL (2013) C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J 32(11):1568–1583. doi:10.1038/emboj.2013.106
Polman JA, Welten JE, Bosch DS, de Jonge RT, Balog J, van der Maarel SM, de Kloet ER, Datson NA (2012) A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci 13:118. doi:10.1186/1471-2202-13-118
Polman JA, de Kloet ER, Datson NA (2013) Two populations of glucocorticoid receptor-binding sites in the male rat hippocampal genome. Endocrinology 154(5):1832–1844. doi:10.1210/en.2012-2187
Nestler EJ, Hyman SE (2010) Animal models of neuropsychiatric disorders. Nat Neurosci 13(10):1161–1169. doi:10.1038/nn.2647
Bartolomucci A, Pederzani T, Sacerdote P, Panerai AE, Parmigiani S, Palanza P (2004) Behavioral and physiological characterization of male mice under chronic psychosocial stress. Psychoneuroendocrinology 29(7):899–910. doi:10.1016/j.psyneuen.2003.08.003
Duque A, Vinader-Caerols C, Monleon S (2017) Indomethacin counteracts the effects of chronic social defeat stress on emotional but not recognition memory in mice. PLoS One 12(3):e0173182. doi:10.1371/journal.pone.0173182
Veeraiah P, Noronha JM, Maitra S, Bagga P, Khandelwal N, Chakravarty S, Kumar A, Patel AB (2014) Dysfunctional glutamatergic and gamma-aminobutyric acidergic activities in prefrontal cortex of mice in social defeat model of depression. Biol Psychiatry 76(3):231–238. doi:10.1016/j.biopsych.2013.09.024
Kanarik M, Alttoa A, Matrov D, Koiv K, Sharp T, Panksepp J, Harro J (2011) Brain responses to chronic social defeat stress: effects on regional oxidative metabolism as a function of a hedonic trait, and gene expression in susceptible and resilient rats. Eur Neuropsychopharmacol 21(1):92–107. doi:10.1016/j.euroneuro.2010.06.015
Pariante CM, Miller AH (2001) Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry 49(5):391–404. doi:10.1016/S0006-3223(00)01088-X
Holsboer F (2000) The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23(5):477–501. doi:10.1016/S0893-133X(00)00159-7
Menke A, Arloth J, Putz B, Weber P, Klengel T, Mehta D, Gonik M, Rex-Haffner M et al (2012) Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology 37(6):1455–1464. doi:10.1038/npp.2011.331
Fuchsl AM, Reber SO (2016) Chronic psychosocial stress and negative feedback inhibition: enhanced hippocampal glucocorticoid signaling despite lower cytoplasmic GR expression. PLoS One 11(4):e0153164. doi:10.1371/journal.pone.0153164
Merkulov VM, Merkulova TI (2012) Glucocorticoid receptor isoforms generated by alternative splicing and alternative translation initiation. Russ J Genetics 2(3):205–213
Oakley RH, Cidlowski JA (2011) Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286(5):3177–3184. doi:10.1074/jbc.R110.179325
Akbarian S, Huang HS (2009) Epigenetic regulation in human brain-focus on histone lysine methylation. Biol Psychiatry 65(3):198–203. doi:10.1016/j.biopsych.2008.08.015
Shen E, Shulha H, Weng Z, Akbarian S (2014) Regulation of histone H3K4 methylation in brain development and disease. Philos Trans R Soc Lond B Biol Sci 369(1652). doi:10.1098/rstb.2013.0514
Bonneh-Barkay D, Wiley CA (2009) Brain extracellular matrix in neurodegeneration. Brain Pathol 19(4):573–585. doi:10.1111/j.1750-3639.2008.00195.x
Kerrisk ME, Cingolani LA, Koleske AJ (2014) ECM receptors in neuronal structure, synaptic plasticity, and behavior. Prog Brain Res 214:101–131. doi:10.1016/B978-0-444-63486-3.00005-0
Li XH, Chen JX, Yue GX, Liu YY, Zhao X, Guo XL, Liu Q, Jiang YM et al (2013) Gene expression profile of the hippocampus of rats subjected to chronic immobilization stress. PLoS One 8(3):e57621. doi:10.1371/journal.pone.0057621
Barber M, Di Meglio T, Andrews WD, Hernandez-Miranda LR, Murakami F, Chedotal A, Parnavelas JG (2009) The role of Robo3 in the development of cortical interneurons. Cereb Cortex 19(Suppl 1):i22–i31. doi:10.1093/cercor/bhp041
Dickinson RE, Duncan WC (2010) The SLIT-ROBO pathway: a regulator of cell function with implications for the reproductive system. Reproduction 139(4):697–704. doi:10.1530/REP-10-0017
Aasheim HC, Patzke S, Hjorthaug HS, Finne EF (2005) Characterization of a novel Eph receptor tyrosine kinase, EphA10, expressed in testis. Biochim Biophys Acta 1723(1–3):1–7. doi:10.1016/j.bbagen.2005.01.011
Bouvier G, Bidoret C, Casado M, Paoletti P (2015) Presynaptic NMDA receptors: roles and rules. Neuroscience 311:322–340. doi:10.1016/j.neuroscience.2015.10.033
Sandi C (2004) Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci 5(12):917–930. doi:10.1038/nrn1555
Dityatev A, Schachner M, Sonderegger P (2010) The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat Rev Neurosci 11(11):735–746. doi:10.1038/nrn2898
Acknowledgments
This work was supported by grants #14-04-01707 from the Russian Foundation for Basic Research (behavioral study), a grant from the Government of the Russian Federation #14.B25.31.0033 (HiSeq Illumina sequencing), a grant from the Russian Science Foundation #14-44-00077 (analysis of ChIP-seq datasets), and a grant from the Russian Science Foundation #16-15-10131 (analysis of RNA-seq data and dataset on dexamethasone-affected genes).
All behavioral experiments were conducted in the Laboratory of Experimental Models of Neurodegenerative Processes at the Institute of Physiology and Basic Medicine, Novosibirsk, Russia. We would like to thank Pavlov K.S. for technical assistance with the behavioral testing.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors report no biomedical financial interests or potential conflicts of interest.
Rights and permissions
About this article

Cite this article
Bondar, N., Bryzgalov, L., Ershov, N. et al. Molecular Adaptations to Social Defeat Stress and Induced Depression in Mice. Mol Neurobiol 55, 3394–3407 (2018). https://doi.org/10.1007/s12035-017-0586-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0586-3