
Abstract
After ischemic stroke, cell damage propagates from infarct core to surrounding tissues (penumbra). To reveal proteins involved in neurodegeneration and neuroprotection in penumbra, we studied protein expression changes in 2-mm ring around the core of photothrombotic infarct induced in the rat brain cortex by local laser irradiation after administration of Bengal Rose. The ultrastructural study showed edema and degeneration of neurons, glia, and capillaries. Morphological changes gradually decreased across the penumbra. Using the antibody microarrays, we studied changes in expression of >200 neuronal proteins in penumbra 4 or 24 h after focal photothrombotic infarct. Diverse cellular subsystems were involved in the penumbra tissue response: signal transduction pathways such as protein kinase Bα/GSK-3, protein kinase C and its β1 and β2 isoforms, Wnt/β-catenin (axin1, GSK-3, FRAT1), Notch/NUMB, DYRK1A, TDP43; mitochondria quality control (Pink1, parkin, HtrA2); ubiquitin-mediated proteolysis (ubiquilin-1, UCHL1); axon outgrowth and guidance (NAV-3, CRMP2, PKCβ2); vesicular trafficking (syntaxin-8, TMP21, Munc-18-3, synip, ALS2, VILIP1, syntaxin, synaptophysin, synaptotagmin); biosynthesis of neuromediators (tryptophan hydroxylase, monoamine oxidase B, glutamate decarboxylase, tyrosine hydroxylase, DOPA decarboxylase, dopamine transporter); intercellular interactions (N-cadherin, PMP22); cytoskeleton (neurofilament 68, neurofilament-M, doublecortin); and other proteins (LRP1, prion protein, β-amyloid). These proteins are involved in neurodegeneration or neuroprotection. Such changes were most expressed 4 h after photothrombotic impact. Immunohistochemical and Western blot studies of expression of monoamine oxidase B, UCHL1, DYRK1A, and Munc-18-3 confirmed the proteomic data. These data provide the integral view on the penumbra response to photothrombotic infarct. Some of these proteins can be potential targets for ischemic stroke therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is the leading cause of disability and the third cause of mortality in the world. Acute ischemia caused by vascular occlusion induces cell death not only in the infarction core but also in the surrounding tissue (penumbra). Oxygen and glucose deficit within the ischemic locus very quickly, in a few minutes, induces ATP depletion, generation of reactive oxygen species in mitochondria, oxidative membrane injury, loss of ionic gradients, Ca2+ influx and K+ release, and tissue edema. Ca2+ activates hydrolytic enzymes and induces cell necrosis or apoptosis. K+-mediated depolarization induces opening of NMDA channels in neighboring neurons, Ca2+ influx, and release of glutamate and K+. Such self-developing excitotoxic process results in injury propagation. The tissue damage in penumbra develops slower, for several hours and days, and this “therapeutic window” provides time for neuroprotection therapy [1–4].
Among potential neuroprotectors, diverse blockers of calcium channels, glutamate antagonists, inhibitors of nitric oxide signaling, antioxidants, and radical scavengers have been studied. However, despite a significant research effort, the efficient neuroprotective drugs for stroke pharmacotherapy are still absent. Some drugs that demonstrated the neuroprotective activity in vitro and in animal experiments were inefficient in human treatment [5]. Therefore, novel approaches based on comprehensive studies of molecular mechanisms of neurodegeneration and neuroprotection are needed [6, 7]. The response to injury is determined by the proteins present in the cell. If these are insufficient, the expression of appropriate genes leads to synthesis of additional proteins. The proteomic techniques provide information on expression of hundreds proteins in biological samples [8, 9] including penumbra [10–13].
In the present work, we studied the morphological and biochemical consequences of the focal photothrombotic infarction (PTI) in the rat cerebral cortex induced by the local photodynamic treatment. Photodynamic effect is based on photoexcitation of the photosensitizing dye in stained cells, following energy transfer to oxygen, generation of singlet oxygen and other reactive oxygen species, oxidative stress, and, finally, cell death [14]. Cerebral PTI occurs after administration of Bengal Rose and following laser irradiation. This hydrophilic photosensitizer does not pass the blood-brain barrier and penetrate into neuronal cells. It accumulates in microvessels. The following local photoirradiation induces focal oxidative damage of the vascular endothelium, platelet aggregation, and occlusion of microvessels. This PTI model is slightly invasive. The injury location, size, and degree are well controlled and reproducible [15–18]. However, penumbra resulting from such impact is rather narrow. Using less intensive but prolonged irradiation, we got the wider penumbra, sufficient for biochemical analysis. Preliminary proteomic study using the antibody microarrays has revealed the early changes in the expression of various neuronal proteins in the penumbra around the PTI core in the rat cerebral cortex 1 h after photodynamic impact [19]. In the present work, we performed a complex histological, ultrastructural, and proteomic study of consequences of the local photothrombotic infarction in the rat cerebral cortex 4 h (“therapeutic window”) and 24 h after PTI (“late reperfusion period”).
Materials and Methods
Chemicals
Cy3™ or Cy5™ monofunctional reactive dyes were supplied by GE Healthcare. The Panorama Ab Microarray—Neurobiology Kits (NBAA5; Sigma-Aldrich Co) and other chemicals were obtained from Sigma-Aldrich-Rus (Moscow, Russia).
Animals
The experiments were performed on adult male Wistar rats (200–250 g). The animal holding room was maintained at a temperature of 22–25 °C, 12-h light/dark schedule, and an air exchange rate of 18 changes per hour. All experimental procedures were carried out in accordance with the European Union guidelines 86/609/EEC for the use of experimental animals and local legislation for ethics of experiments on animals. The animal protocols used in this work were evaluated and approved by the Animal Care and Use Committee of Southern Federal University (Approval No. 02/2014).
Focal Photothrombotic Ischemia in the Rat Cerebral Cortex
Unilateral focal photothrombotic infarction in the rat primary somatosensory cortex was induced according to [18, 19]. The rats were anesthetized with chloral hydrate (300 mg/kg, i.p.). After the longitudinal incision of the skull skin, the periosteum was removed. Bengal Rose (20 mg/kg) was injected in the v. subclavia. Then the rats were fixed in the stereotactic holder. The unilateral irradiation of the somatosensory cortex was performed through the cranial bone using a 532-nm diode laser (64 mW/cm2, Ø 3 mm, 30 min). The animals were euthanized with the chloral hydrate overdose (600 mg/kg, i.p.) and decapitated 4 or 24 h after PTI. The symmetric cortical region in the contralateral hemisphere served as a control.
Histology
For histological study, the rats were transcardially perfused with 10 % buffered formalin (pH 7.2) under chloral hydrate anesthesia 24 h after laser irradiation. The extracted brains were post-fixed with formalin. The paraffin sections were stained with hematoxylin and eosin and imaged on the Eclipse FN1 microscope (Nikon, Japan; objective lens Plan Fluor ELWD, ×40/0.60). The numbers of normal, hypochromic (lighter cytoplasm and increased nucleus), hyperchromic (darker cytoplasm, shrunk nuclei), and pyknotic (wrinkled cells with shrunk nuclei) cells were counted 24 h after the treatment in six randomized fields in each slide using an ocular grid [20]. Statistical analysis was performed with Student’s t test for three independent experiments (three rats). Quantitative data were presented as means ± SEM. The criterion of significance was set as p <0.05.
Immunohistochemistry
For immunohistochemical study, 5-μm sections of the paraffin-embedded experimental and control cortex tissues were used. The sections were demasked in Tris-EDTA buffer with 0.05 % Tween-20 (pH 9.0) in the programmed barocamera Pascal (Dako). Reactions were visualized using the REVEAL Polyvalent HRP-DAB Detection System (Spring Bioscience, SPD-060, USA) according to the manufacturer’s instructions. The primary antibodies against dual-specificity tyrosine-phosphorylated regulated kinase 1A (DYRK1A, D1694, Sigma-Aldrich, 1:6000), Munc-18-3 (M7695, Sigma-Aldrich, 1:6000), monoamine oxidase B (MAO-B, M1946, Sigma-Aldrich, 1:6000), and ubiquitin C-terminal hydrolase L1 (UCHL1, U5258, Sigma-Aldrich, 1:6000) were used. The sections were counterstained with Mayer’s hematoxylin and imaged on the Eclipse FN1 microscope (Nikon, Japan; objective lens Plan Fluor ELWD, ×40/0.60). Omission of primary antibody was routinely used to certify the absence of nonspecific labeling. All observations were performed in lamina granularis externa, lamina pyramidalis, and lamina granularis interna. Twelve images were acquired per section and studied using ImageJ software (NIH, USA) like in [19]. For quantification, the immunoreactivity coefficients: I = N ip/N t × 100 %, where N ip and N t are the numbers of immunopositive pixels and total number of pixels, respectively, were determined in each section in control (n = 3) and experimental (n = 3) samples. Statistical analysis was performed with Student’s t test for independent experiments. Quantitative data were presented as means ± SEM. The criterion of significance was set as p <0.05.
Electron Microscopy
At 4 h after PTI, the cerebral cortex slices about 1 mm thick were cut out as a triangular sector with a vertex at the center of the irradiated area so that it comprised a part of penumbra and undamaged area. These tissue pieces were fixed for 2 h with 2.5 % glutaraldehyde/0.1 M PBS at room temperature. Then they were washed with PBS, incubated for 1 h in 1 % OsO4/0.2 M PBS solution, dehydrated by ethanol, contrasted by 1 % uranyl acetate, and embedded in EPON resin. Ultrathin sections were prepared on the ultramicrotome Leica EM UC6 (Leica, Germany) and examined on the transmission electron microscope Tecnai G2 Spirit Bio TWIN (FEI Co., Netherlands).
Proteomic Study
The Panorama Antibody Array—Neurobiology kit (NBAA5; Sigma-Aldrich Co) contains two identical microarrays, which are the nitrocellulose-coated glass slides containing 448 microdroplets with immobilized antibodies against 144 neuronal proteins and different isoforms and epitopes of some of them (in total 224 variants; Supplement 1). Each of the 32 microdroplet sub-arrays had duplicate spots of 7 antibodies plus a single spot with non-labeled bovine serum albumin (BSA) as a negative control and a single spot with Cy3 and Cy5-conjugated BSA as a positive control.
At 4 or 24 h after photodynamic treatment, the rat cortex was extracted, and the red round region of photothrombotic infarct was excised using the Ø3-mm circular knife. Its diameter and the diameter of penumbra had been determined preliminarily in the histological and ultrastructural experiments. Then the ring-shape cortex area (2 mm width) around the PTI core (penumbra) was cut out by another Ø7-mm circular knife (the experimental sample). The similar piece from the non-irradiated contralateral somatosensory cortex was used as the control sample. Each sample contained the cortical tissue from two similarly treated rats in order to get enough material for analysis. These pieces were weighed, homogenized, and lysed in the Extraction/Labeling Buffer supplemented with protease and phosphatase inhibitor cocktails and nuclease bensonase (components of NBAA5). Then the control and experimental lysates were centrifuged for 4 min at 10,000 rpm in the cooled centrifuge Eppendorf 5417 C/R (Hamburg, Germany). The supernatants were frozen in liquid nitrogen and stored at −80 °C for further analysis. After thawing, the protein contents in both experimental and control samples were determined using Bradford reagent. Then samples were diluted to 1 mg/ml protein content and incubated for 30 min in darkness at room temperature with Cy3 or Cy5, respectively. The unbound dye was removed by centrifugation (4000 rpm; 4 min) of the SigmaSpin Columns (GRAA2 components) filled with 200 μl of the labeled protein samples. The eluates were collected, and protein concentration was detected again. The samples with dye-to-protein ratio >2 were used. In another set, these samples were stained oppositely, by Cy5 and Cy3, respectively.
The mixture of the control and experimental samples (10 μg/ml each) labeled with Cy3 and Cy5, respectively, was added into a tube with 5 ml of the array incubation buffer (NBAA5 component) and incubated for 40 min at room temperature on a rocking shaker. The same procedure was performed with the oppositely labeled samples: Cy5 and Cy3, respectively. Such swapped staining provides verification of results and compensation of a potential bias in binding of Cy3 or Cy5 dyes to protein samples. This provides the double test and full control of the experiment. After following triple washing of the microarrays in the washing buffer (NBAA5 component) and triple washing in the pure water, the microarray slides were air dried overnight in darkness.
The microarrays were scanned using the GenePix 4100A Microarray Scanner (Molecular Devices, USA) at 532 and 635 nm (fluorescence maximums of Cy3 and Cy5, respectively). The integrated fluorescence intensity in each antibody spot was proportional to the quantity of the bound protein. The fluorescence images of the antibody microarrays were analyzed and normalized (ratio-based normalization) using the software GenePix Pro 6.0. Local fluorescence values in the rings around each spot were used as a background. The median fluorescence value determined over all spot pixels was used for estimation of the protein content in each spot, and the ratios of the experimental to control values characterized the difference in the protein level between photothrombotic and normal cortical tissue in the contralateral cortex. Two samples labeled independently and reversely in duplicate provided four experimental values for each protein. Each experiment was repeated four times (eight rats) for 4 h reperfusion and two times (four rats) for 24 h reperfusion intervals, and the ratios were averaged. The standard statistical treatment based on Student’s t test and 95 % significance level was used. Quantitative data in Table 2 were presented as means ± SD. Although the mean experiment/control ratios significantly differed from 1 (p < 0.05), only >30 % differences (the cut-off level) in the protein levels between irradiated and control animals (i.e., experimental/control ratios > 1.3) are displayed in Table 2 and discussed. The reliability of the proteomic microarray data was provided by the self-control experiment using the swapped staining in the second microarray [19, 21].
Results
Morphological Changes in the Rat Cerebral Cortex after Photothrombotic Infarction
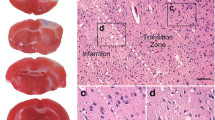
The 3-mm photothrombotic infarction core was surrounded by the 1.5–2-mm width penumbra at 24 h after PTI (Fig. 1). The number of cells with altered morphology in the control sensomotory cortex of the contralateral brain hemisphere was about 9 % (Table 1). Morphological changes observed 24 h after light exposure within PTI core were characteristic for necrosis. They included local neuropil vacuolization, swelling of neuronal cells, and edema (Fig. 2c). Significant fraction of neuronal cells were hyperchromic (41 %) or pyknotic (9 %); 12 % of cells were hypochromic (Fig. 2c, Table 1). Tissue alterations in penumbra were weaker (Fig. 2b, Table 1). Eighty percent of the neuronal cells looked undamaged; 11 % were hyperchromic, whereas the percentage of hypochromic and pyknotic nuclei did not differ from control. Somewhere, perineuronal edema and swelling of neuronal cells occurred.

Local photothrombotic infarction in the rat cerebral cortex occurred 4 h after photodynamic treatment. a Infarction core in the sensomotory cortex. Scale bar: 3 mm. b The frontal section through the infarction core and penumbra.The Infarct core is shown by black arrow; the ring-shape penumbra is shown by grey arrow.

Morphological changes in the rat cerebral cortex 24 h after PTI. Hematoxylin-eosin. a Control cortical tissue in the non-treated hemisphere. b Penumbra, 24 h after light exposure. Black arrows—pericellular edema; white arrows—hyperchromic neurons. c Infarction core, 24 h after light exposure. Asterisks—vacuolated neuropil; black arrows—nucleus deformation; white arrows—pyknotic cells. Scale bar: 50 μm
Ultrastructural Changes in the Rat Cerebral Cortex after Photothrombotic Infarction
The ultrastructural alterations in the rat cerebral cortex 4 h after PTI corresponded to that observed in histological experiments.
Control cortex tissue
In the untreated contralateral hemisphere, cortical neurons contained numerous ribosomes, rough endoplasmic reticulum (ER), dictyosomes, mitochondria with well-developed cristae, and nuclei with decondensed heterochromatin (Fig. 3a). This indicates the high level of biosynthetic and bioenergetic processes. The neuropil contained dendrites, myelinated and non-myelinated axons, synapses (Fig. 3b), and blood vessels with well-developed endothelium and basal lamina (Fig. 3c).

The ultrastructure of the rat somatosensory cortex in the untreated hemisphere (control). a Two neurons (N), oligodendrocyte (OD), and surrounding neuropil. The neuronal nuclei contain the decondensed heterochromatin that indicates high transcriptional activity. The presence of numerous ribosomes, rough endoplasmic reticulum, dictyosomes, and mitochondria with moderately dense matrix and well-developed cristae indicates the high level of biosynthetic and bioenergetic processes. b The neuropil region containing numerous neurites, myelinated axons (MA), and synapses (S). c The blood capillary (Cap) contains the thick basal membrane (*) and endothelium, rich in organelles: G Golgi apparatus; M mitochondria. Scale bars: 2.5 μm in (a), 0.5 μm in (b) and (c)
PTI core
Massive vacuolization of the rat cortex tissue, edema, and swelling of neurites occurred within the infarction core (Fig. 4a–c). The cytoplasm of shrunk neurons was vacuolated due to swelling of endoplasmic reticulum. The nuclear chromatin was condensed (Fig. 4b). Synapses were destructed and had only the electron-dense contact structures but not vesicles (Fig.4a, inset). Myelin was locally disorganized (Fig. 4a, arrowheads). Bodies of glial cells were almost not observed. All capillary components were impaired (Fig. 4c): the basal membranes became thin, the endothelium—vacuolated; its mitochondria swelled and lost cristae. Thus, the edema and necrotic alterations of the neurons, and strong destruction of glial cells and blood vessels were characteristic for PTI core.

The ultrastructure of the photothrombotic infarction core in the rat somatosensory cortex 4 h after light exposure. a Massive tissue vacuolization. Big vacuoles (V) are the results of edema and swelling of neuronal and glial processes. Black arrowheads indicate myelin destruction regions. The destructed synapse (S) with disorganized vesicles (open arrowheads) but saved a synaptic contact is shown in the inset. b One of the saved but shrunk neurons is surrounded by big vacuoles. Neuronal cytoplasm contains numerous vacuolated ER cisterns and mitochondria. c Impairment of the blood capillary (Cap): thinning the basal membrane and endothelium. Swollen endothelial mitochondria lost cristae. ER endoplasmic reticulum, M mitochondria, NN neuron nucleus. Scale bars: 2.5 μm in (a), 1 μm in (b) and inset, 2 μm in (c)
Penumbra near PTI core
The ultrastructure of the penumbra region adjacent to the PTI core (about 2 mm from the core center) was similar to that in PTI but more salvaged (Fig. 5). Big empty vacuoles surrounded neurons and capillaries (Fig. 5a). Some neurons were shrunk, dark, and vacuolated (Fig. 5a) as in the infarction zone, but some of them looked much lighter (Fig. 5b). Neuronal mitochondria saved the inner structure with cristae and dense (Fig. 5a, inset) or light matrix (Fig. 5b). Their cytoplasm was only slightly vacuolated, but chromatin was more condensed as compared with control neurons. The cytoplasm of many axons and dendrites contained ribosomes, mitochondria, and ER elements (Fig. 5b, c). Synapses contained pre- and postsynaptic densities and synaptic cleft. Synaptic vesicles were present in some but not all synapses (Fig. 5c). Myelin was locally disorganized in almost all myelinated axons. Glial cells were observed rarely, and their cytoplasm was destructed. The structure of capillaries was not so altered as in the PTI core. The endothelium cytoplasm contained relatively saved organelles (Fig. 5a).

The ultrastructure of the penumbra region near PTI core in the rat somatosensory cortex (about 2 mm from the PTI core center) 4 h after light exposure. a The shrunk dark neuron (N) similar to that in the infarction core and the capillary (Cap) are surrounded by big empty vacuoles. b The light neuron (N) similar to that in the control area. The cytoplasm of these neurons is vacuolated but many mitochondria (M) saved almost normal structure inner structure with cristae and dense (a, inset, b). c The neuropil region. The axo-dendritic synapse (S) in the center with two contact zones. The myelin envelope of some axons is locally destructed. A axon; M mitochondria; thin arrows—synaptic clefts. Scale bars: 2 μm in (a), 1 μm in (b), 0.5 μm in inset and in (c)
Peripheral penumbra regions
At the penumbra periphery (about 3.5 mm from the PTI core center), the tissue was much less vacuolated. The clusters of normal neurons were observed within the vacuolated zone. Neuronal bodies contained both swollen and normal mitochondria. Some neurons contained rough ER (Fig. 6a) that indicated biosynthetic processes. The dendrites were filled with numerous microtubules (Fig.6b, inset). Oligodendrocytes also looked undamaged. Their cytoplasm contained numerous organelles and ribosomes. The chromatin was moderately condensed (Fig. 6a). Numerous synapses had the normal structure (Fig. 6b). However, some neurons and capillaries were surrounded by extended edema areas (Fig. 6a, c). The capillary ultrastructure was similar to that in control regions, but the endothelial mitochondria were a bit swollen, and some of them lost cristae and matrix. Present glycogen granules are a significant energy store (Fig. 6c).

The ultrastructure of the peripheral penumbra region (about 3.5 mm from the PTI core center) 4 h after light exposure. a The nucleus (NN) of the large neuron (N) contains decondensed heterochromatin. The neuronal cytoplasm is abundant with normal and swollen mitochondria and developed ER. This neuron and adjacent oligodendrocyte (OD) that has a normal ultrastructure are surrounded by the extended edema. b The neuropil region. The ultrastructure of neuropil components look normal except the big vacuole in the center and locally destructed myelin envelope (arrowhead). Numerous synapses (S) are present in this region. Inset: the large axo-dendritic synapse (S). Thin arrow points to dendritic microtubules. c The ultrastructure of the capillary in the penumbra periphery. Inset: the general view of this region. Arrowheads—glycogen granules, asterisks—basal membrane, D dendrite, M mitochondria, MA myelinated axon, Sp dendritic spine. Scale bars: 2 μm in (a) and in insets, 0.5 μm in (b) and (c)
Thus, the ultrastructure of neuronal bodies, neurites, glial cells, and capillaries gradually changed across the penumbra from almost so strongly damaged as in the PTI core to almost normal at the edge of penumbra.
PTI-Induced Changes in Expression of Neuronal Proteins in Penumbra
The proteomic study of expression of 144 neuronal and signaling proteins and their different isoforms and epitopes (in total 224 antibodies) showed >30 % upregulation of 22 proteins in the penumbra 4 h after PTI (Table 2). Their list included:
-
Signal transduction proteins: protein kinases Bα and Cβ2 (+38 and 47 %, respectively), and the total level of phosphorylated threonines (+43 %);
-
Proteins regulating axon outgrowth and guidance: NAV3 (neuron navigator 3) and its N-terminal and C-terminal regions (+53, +91, and 40 %, respectively) and CRMP2 (collapsin response mediator protein 2) (+59 %);
-
Proteins involved in the intracellular vesicular transport, docking, and fusion with a membrane: TMP21 (transmembrane protein 21) (+2.1 times), syntaxin-8 (+71 %), and Munc-18-3 (+31 %);
-
Enzymes involved in biosynthesis of neuromediators serotonin, dopamine, and GABA: tryptophan hydroxylase (+2.14-fold), monoamine oxidase B (+2.75-fold), and glutamate decarboxylase (+40 %);
-
Proteins that control the quality of mitochondria: PINK1 (PTEN-induced mitochondrial protein kinase) and its N-terminal region (+42 and 51 %, respectively); parkin, a ubiquitin ligase (+34). Another protein involved in the mitochondria quality control HtrA2 and its C-terminal region were, however, downregulated (−69 and −33 %, respectively);
-
Proteins regulating ubiquitin-mediated proteolysis: UCHL1 (ubiquitin C-terminal hydrolase L1) (+47 %) and ubiquilin-1 (+39 %);
-
Proteins involved in cell-cell interactions: PMP22 (peripheral myelin protein 22) that mediates myelin formation (+2.43 times) and N-cadherin participating in cell-cell adhesion (+50 %);
-
Miscellaneous proteins: tau, which regulates their polymerization of microtubules (+36 %); prion protein (+60 %); and LRP1 (low-density lipoprotein receptor-related protein-1), which regulates lipoprotein metabolism (+2.8-fold).
Overexpression of these proteins could be directed to cell survival and tissue recovery rather than to destructive processes. On the other hand, overexpression of DYRK1A (dual-specificity tyrosine-phosphorylated regulated kinase 1A) (+84 and +53 % for its N- and C-terminal regions, respectively) or β-amyloid peptide (fragment 13–28) (+47 %) could be neurotoxic.
At the same time, 21 proteins were downregulated (Table 2). Tissue damage could be associated with downregulation of:
-
Cell signaling proteins such as protein kinase C (−4.7 times) and protein kinase Cβ1 (−81 %); TDP-43 (transactivation response DNA-binding protein) and its N-terminal region (−2.33 and −3.28, respectively); GSK-3 (glycogen synthase kinase-3) (−33 %); FRAT1 (frequently rearranged in advanced T-cell lymphomas-1) and its C-terminal region (−64 and −94 %, respectively); and axin1, components of the Wnt signaling pathway (C-terminal region, −35 %).
-
Cytoskeleton elements: microtubule-associated protein doublecortin and its N-terminal region (−31 and −99 %, respectively), neurofilament 68 (−65 %), and o-glycosylated neurofilament-M (−41 %).
-
Regulators of vesicular transport: VILIP-1 (visinin-like protein-1), C-terminal (−71 %), ALS2 (amyotrophic lateral sclerosis 2) and its homolog ALS2CL (ALS2 C-terminal like) (−55 and −54 %, respectively), and synip (N-terminal region) (−58 %).
-
Synaptic vesicle proteins: syntaxin (−84 %), synaptophysin (−60 %), and synaptotagmin (−46 %) and proteins involved in the synthesis of neuromediators: tyrosine hydroxylase that mediates l-DOPA synthesis from l-tyrosine (−48 %), DOPA decarboxylase involved in dopamine synthesis (−39 %), and dopamine transporter responsible for dopamine reuptake from the synaptic cleft (−39 %).
On the other hand, downregulation of GSK-3 (glycogen synthase kinase-3) (−33 %) and NUMB, antagonist of the Notch signaling (−85 %), could lead to neuroprotection.
At 24 h after PTI, the changes in the protein profile in were considerably weaker (Table 2): only ten proteins were overexpressed comparing with the untreated contralateral cortex: syntaxin 8 (+2.2 times); monoamine oxidase B (+2.0 times); DYRK1A, N-terminal region (+49 %); tau (+44 %); β-amyloid (+39 %); PMP22 (+34 %); PINK1, N-terminal region (+34 %); tryptophan hydroxylase (+33 %); glutamate decarboxylase (+30 %); and UCHL1 (+30 %).
At the same time, nine proteins were downregulated: protein kinases C (−3.1 times) and Cβ1 (−2 times); FRAT1, C-terminal region (−2.18 times), syntaxin (−46 %); synip, N-terminal region (−42 %), neurofilament 68 (−40 %); synaptophysin (−39 %); tyrosine hydroxylase (−38 %); TDP-43 and its C-terminal region (−34 and −37 %, respectively).
Immunohistochemistry
In the untreated rat cortex, UCHL1 localizes mainly in the neuronal perikaryon, and less staining was observed in dendrites (Fig. 7a, c). In penumbra, it was concentrated mainly in the neuronal nuclei at 4 h after PTI (Fig. 7b), whereas at 24 h it was redistributed from the nuclei to the cytoplasm (Fig. 7d). Its mean immunoreactivity increased by 70 % (p < 0.05) 4 h after PTI (Fig. 7e). The similar tendency was observed 24 h after PTI (+40 %, p > 0.05).

Expression of UCHL1 in the untreated contralateral (CL) rat brain cortex (a and c) and in penumbra 4 or 24 h after PTI (b and d, respectively). e Mean index of UCHL1 immunoreactivity in control (CL) cortex areas and in penumbra 4 and 24 h after PTI ± standard error (Student’s t test for independent groups; *p < 0.05; n = 3). Scale bar: 50 μm
Both in penumbra and in control cortex, DYRK1A was localized mainly in the cell nuclei and less in the cytoplasm (Fig. 8a–d). Its mean level in penumbra increased more than twice for 4 h after PTI (Fig. 8b, e, p < 0.05). After 24 h, the increase in the DYRK1A immunoreactivity was not significant (+36 %, p > 0.05; Fig. 8d, e).

Expression of DYRK1A in the untreated contralateral (CL) rat brain cortex (a and c) and in penumbra 4 or 24 h after PTI (b and d, respectively). e Mean index of DYRK1A immunoreactivity in control (CL) cortex areas and in penumbra 4 and 24 h after PTI ± standard error. Student’s t test for independent groups; *p < 0.05; n = 3). Scale bar: 50 μm
The mean level of Munc-18-3 in penumbra (Fig. 9b, d), at first glance, was higher than in the control cortex 4 and 24 h after PTI (Fig. 9a, c). However, this overexpression was not significant (+40 and +30 %, p > 0.05; Fig. 9e).

Expression of Munc18-1-3 in the untreated contralateral (CL) rat brain cortex (a and c) and in penumbra 4 or 24 h after PTI (b and d, respectively). e Mean index of Munc18-1-3 immunoreactivity in control (CL) cortex areas and in penumbra 4 and 24 h after PTI ± standard error (Student’s t test for independent groups; n = 3). Scale bar: 50 μm
In the control preparations, monoaminoxidase B was localized mainly in dendrites (Fig. 10a, c). After PTI, it was also observed in the cytoplasm of the neuronal perikaryon. Its mean level in penumbra increased by about 90 and 80 %, respectively, 4 and 24 h after PTI (p < 0.05; Fig. 10b, d, and e).

Expression of MAO-B in the untreated contralateral (CL) rat brain cortex (a and c) and in penumbra 4 or 24 h after PTI (b and d, respectively). e Mean index of MAO-B immunoreactivity in control (CL) cortex areas and in penumbra 4 and 24 h after PTI ± standard error (Student’s t test for independent groups; *p < 0.05; n = 3). Scale bar: 50 μm
Discussion
The photothrombotic infarct reproduces morphological alterations in the cerebral cortex after occlusion of small blood vessels [1–4]. The used impact regime provided a 1.5–2-mm penumbra around the PTI core. This was confirmed by the histological and electron microscopic data. Morphological alterations in the penumbra were similar to that in the PTI core but were lesser and decreased gradually across the penumbra. At 4 h after light exposure, local photodynamic impact induced typical ischemic alterations in the PTI core: massive tissue vacuolization, edema, and degenerative changes in neurons, glia, and blood vessels. Swelling and destruction of mitochondria, ER and dictyosomes, degradation of synapses, disorganization of myelin, swelling of neuronal and glial processes, edema, and destruction of capillary components were observed at the ultrastructural level. These changes could be the consequences of impairment of energy metabolism and protein biosynthesis. Similar alterations have been observed in the photothrombotic “ring” stroke model 4 h after spontaneous reperfusion [22]. At the border between the PTI core and penumbra, we observed almost the same morphological changes. However, at a distance of 1.5–2 mm from the PTI core, the cell morphology was almost normal except edema around some neuronal cells and vessels. On the basis of these data, we defined the approximate width of penumbra.
In order to resist neurodegeneration, diverse neuroprotective and recovery processes are simultaneously initiated in the penumbra. A number of signaling pathways have been already tested in order to find the potential molecular targets for treatment of cerebral ischemia. But none of the tested modulators of signaling proteins or ion channels have demonstrated the neuroprotective activity without deleterious side effects [1–6]. We think that the study of a broader range of proteins involved in various cellular subsystems allows to find more effective neuroprotectors. The proteomic approach provides information on possible involvement of hundreds of proteins in such complex multifactor process as stroke. The previous proteomic studies of the stroke mechanisms already revealed changes in the expression of diverse bioenergetic [13]; antiapoptotic, inflammatory, antioxidant, and mitochondrial heat shock proteins [11]; protein kinase CβII and CRMP2 [10]; and some extracellular proteins [12] in the penumbra. Using the proteomic antibody microarrays, we have recently shown the early changes (1 h after PTI) in expression of 24 neuronal proteins in the penumbra in the rat cerebral cortex: upregulation of 18 and downregulation of 6 proteins [19].
The present work has expanded the reperfusion time interval for 4 and 24 h after PTI. The highest changes in the neuronal protein profile were observed 4 h after PTI: 22 proteins were upregulated and 21 proteins downregulated. At 24 h after PTI, the alterations of the protein profile were lesser: ten proteins were upregulated and nine proteins downregulated (Table 2). Some proteins were overexpressed only 1 h after PTI and the levels of others changed only 4 h after PTI, whereas the expression of several proteins changed during all intervals from 1 to 24 h (Table 2 and [19]).
The changes in the expression of some proteins could lead to neuroprotection and recovery. The increased level of phosphothreonines indicated the intensification of protein kinase activity in signal transduction processes. Phosphothreonines are rare in the normal tissue, but their level increased significantly after brain injury. The observed upregulation of the antiapoptotic protein kinase Bα (Akt-α) was apparently aimed to cell protection against stroke-induced apoptosis. These data correlate with the increased phosphorylation of Akt and rise in the Akt mRNA level in the ischemic brain of rats [23, 24]. Downregulation of proapoptotic kinase GSK-3, which is negatively controlled by protein kinase B/Akt, could be also neuroprotective.
Brain ischemia leads to deterioration of the ubiquitin-proteasome system (UPS) that cannot cope with overproduction of misfolded proteins and metabolism deregulation [25]. We observed the upregulation of ubiquilin-1 in penumbra 1 h [19] and 4 h after PTI. Ubiquilin-1 is a ubiquitin-like protein also known as UBQLN1. It mediates degradation of misfolded proteins and contributes to brain protection from oxidative stress and ischemic stroke [26]. Surprisingly, ubiquitin C-terminal hydrolase L1, which removes ubiquitin and prevents protein degradation, was also overexpressed. Simultaneous overexpression of UCHL1, ubiquitin, and parkin in the rabbit spinal cord 3 and 6 but not 24 h after 15-min transient ischemia [27] corresponded to our data. UCHL1 is a neuron-specific and abundant protein in the brain. It maintains recycling of ubiquitin monomers and thereby sustains protein degradation [27, 28]. Its overexpression is related to ischemic tolerance, whereas its mutations or downregulation occur in neurodegenerative diseases [28]. Thus, the observed upregulation of UPS components could be associated with neuroprotection. Appearance of UCHL1 in the blood serum and cerebral spinal fluid is a biomarker of ischemic brain injury [29].
The PINK1/HtrA2/parkin complex controls the mitochondria quality and protects neuronal cells from stress-induced mitochondrial dysfunction. Upon mitochondria depolarization, ubiquitin kinase PINK1 either phosphorylates serine protease HtrA2, which destroys misfolded proteins (molecular quality control), or recruits cytoplasmic ubiquitin ligase parkin, which mediates ubiquitination and proteasomal degradation of defective mitochondrial proteins and stimulates mitophagy (organelle quality control) [30, 31]. Overexpression of PINK1 contributes to neuroprotection under cerebral ischemia [32]. So, the observed upregulation of PINK1 and parkin (Table 2) also mediated the protective processes in penumbra. However, the simultaneous downregulation of HtrA2 showed failure of the organelle quality control pathway. The reasons of this inconsistency remain to be studied.
The levels of neuron navigator protein NAV-3, CRMP2, a component of the collapsin/semaphorin signaling pathway, and protein kinase Cβ2 increased in penumbra 4 h after PTI. NAV-3 and CRMP2 mediate axon outgrowth and guidance under ischemic and glutamate neurotoxicity [33, 34]. CRMP2 was shown to be upregulated in the ischemic mouse cerebral cortex [35]. Protein kinase Cβ2 controls these processes. The simultaneous upregulation of CRMP2, PKCβ2, and UCHL1 occurred also in the brain of mice subjected to hypoxic preconditioning (HPC). The authors suggested the central role of PKCβ2/CRMP2 interaction in HPC-induced neuroprotection [10].
N-cadherin, which forms the intercellular adhesive contacts [36], and peripheral myelin protein PMP22, which mediates formation of myelin sheaths and neuroglial interactions [37], were upregulated in penumbra 4 h after PTI. The increased expression of N-cadherin on cerebral microvessels contributes to formation of new vessels, increase of blood flow, and survival of the cerebral tissue in penumbra [36]. PMP22 overexpression could be associated with recovery of myelin envelopes. Upregulation of these proteins in penumbra 1 h after PTI was observed previously [19]. This suggests the involvement of these proteins in recovery of the cortical tissue in the first 4 h of reperfusion.
Prion protein (PrP) is highly expressed in the brain: in neurons, glial and microvascular endothelial cells. It is involved in cell trafficking, adhesion, protection against oxidative stress, and survival [38–40]. In our experiments, PrP was upregulated in penumbra 4 but not 24 h after PTI. Overexpression of PrP was also observed in mice 4 and 8 h but not 24 h after focal ischemia [40] that correlated with our data. Upregulation of PrP and PrP mRNA after cerebral ischemia leads to three protective effects: neuroprotection, neurogenesis, and angiogenesis [38–40].
The changes in expression of proteins that participate in the intracellular vesicular transport and synaptic transmission in penumbra could be associated with recovery processes. The observed upregulation of syntaxin-8, TMP21, and Munc-18-3 could activate vesicular transport and reparation of injured membranes. Syntaxin-8 (STX8) mediates vesicle docking and fusion with ER, Golgi, and surface membranes [41]. A transmembrane protein TMP21 participates in vesicle trafficking between endoplasmic reticulum and Golgi [42]. The syntaxin-binding protein Munc-18-3 is involved in the vesicle-recognition complex SNARE [43]. Synip, oppositely, prevents the assembly of the SNARE complex and inhibits docking of vesicles and mixing of lipids and vesicle content in the SNARE-dependent vesicle fusion [44]. Its downregulation in penumbra 4 and 24 h after PTI could play a protective role.
Proteins ALS2 and its homolog ALS2CL were downregulated in penumbra 4 h after PTI. ALS2 is a guanine nucleotide exchange factor for the small GTPase Rab5. It mediates endolysosomal trafficking via regulation of fusion between endosomes and autophagosomes. These processes are necessary for neurite outgrowth and neuroprotection. Loss of these functions in neuronal cells leads to accumulation of insoluble misfolded proteins, multivesicular bodies and autophagosomes, preclusion of the long-range trafficking of axonal vesicles, swelling, and degeneration of neurites, susceptibility to oxidative stress, NMDA-mediated excitotoxicity, and motor dysfunction [45].
The level of VILIP1 decreased in penumbra 4 h after PTI. This Ca2+-binding protein serves as a Ca2+ sensor. It is abundant in brain. It influences different signaling cascades including cyclic nucleotide and MAPK signaling through a Ca2+/myristoyl switch mechanism. After [Ca2+]i increase, VILIP1 moves to the cellular membranes and regulates vesicular trafficking and recycling of receptors and ion channels [46]. Upon stroke, it is released from damaged neuronal cells into the cerebrospinal fluid and blood serum being a stroke marker [47].
At the same time, syntaxin, synaptophysin, and synaptotagmin involved in exocytosis of synaptic vesicles and neurotransmitter release were downregulated. This corresponded to the ultrastructural data on destruction and disorganization of synaptic vesicles. Likewise, the ischemia-induced downregulation of synaptophysin and SNAP-25 observed in hippocampal neurons of gerbils could be due to presynaptic degeneration that preceded the delayed neuronal death [48]. In rats, synaptotagmin mRNA was downregulated during early recirculation after focal cerebral ischemia [49].
Synaptic enzymes involved in the metabolism of neuromediators such as tryptophan hydroxylase that mediates biosynthesis of serotonin, monoamine oxidase B that performs oxidative deamination of serotonin and dopamine in neurons and astrocytes, and glutamate decarboxylase that converts l-glutamate into GABA were upregulated in the penumbra 4 h after PTI. Overexpression of the gene encoding glutamate decarboxylase was observed after MCAO in the human and rat brain [50]. The levels of monoamine oxidase B and syntaxin 8 remained increased at 24 h after PTI. These processes could be aimed to restore lost synaptic functions. However, tyrosine hydroxylase that synthesized DOPA from tyrosine, DOPA decarboxylase, and dopamine transporter that mediate dopamine synthesis and reuptake from the synaptic cleft were downregulated 4 h after PTI. This indicated the impairment of the dopamine metabolism.
Protein kinase DYRK1A phosphorylates diverse transcription factors and signaling proteins. However, its overexpression could deregulate multiple signaling pathways and induce neurodegeneration, neuronal loss, and dementia in Down syndrome, aging, or Alzheimer type pathology [51, 52]. DYRK1A stimulates the pro-apoptotic ASK1-JNK signaling pathway [53]. The observed upregulation of DYRK1A in penumbra 4 and 24 h after PTI could be neurotoxic. β-Amyloid is produced from amyloid precursor protein (APP) by TMP21, a member of the presenilin complex [42]. The observed accumulation of β-amyloid peptide (13–28) in penumbra could be also neurotoxic.
Protein kinase C and its β1 isoform were significantly downregulated in penumbra in the post-ischemic period (1, 4, and 24 h), whereas PKC β2 was upregulated (Table 2, [19]). The levels of other PKC isoforms: α, γ, δ, ε, and η did not differ more than 20 % from that in the untreated contralateral cortex. These results are in agreement with rapid loss of total PKC level and activity after ischemic brain injury, suggesting degradation of PKC [54]. PKC inhibitors protected neurons from post-ischemic [55] and excitotoxic injury [56] that indicated involvement of PKC in neurodegeneration. The observed decrease in the PKC level could reduce the damage of neuronal cells in penumbra.
TDP-43 normally regulates transcription, splicing, transport, and stability of mRNA. In the acute ischemic stroke caused in a rat by occlusion of the middle cerebral artery, the level of TDP-43 progressively decreased in the ischemic core but not in penumbra from 10 min to 24 h post-treatment [57]. We observed the significant fall in the TDP-43 level in penumbra 1, 4, and 24 h after focal photothrombotic infarct ([19]; Table 2). Such decrease in TDP-43 level could suppress the protein synthesis and become neurotoxic.
The cytoskeleton elements neurofilament 68 and o-glycosylated neurofilament-M, which are transported along the axon towards synaptic terminals, and microtubule-associated protein doublecortin were downregulated 4 but not 24 h after PTI. Doublecortin is known to be involved in vesicle trafficking, growth of neuronal processes, and migration of cortical neurons during neurogenesis [58]. Ablation of doublecortin-immunopositive neurons in newborn mice inhibited brain recovery after experimental stroke [59]. Downregulation of doublecortin also reduced the migration of neural progenitor cells into the injured regions [60]. Therefore, its downregulation could reduce the recovery potential in penumbra.
Cell survival and apoptosis are indirectly controlled by the Wnt/β-catenin and Notch signaling pathways [61, 62]. β-Catenin can act as a transcription factor responsible for expression of various prosurvival genes. In the cytoplasm, it is a part of the axin/APC/CSK-3β/β-catenin complex, in which axin1 facilitates GSK-3-mediated phosphorylation and UPS-mediated degradation of β-catenin. Wnt stimulates the disassembly of this complex and liberation of β-catenin, which moves into the nucleus and induces expression of some prosurvival genes. Inhibition of the Wnt pathway under focal ischemia exacerbated neuronal death [63]. The ubiquitous protein FRAT1 mediates dissociation of GSK-3 from axin and prevents phosphorylation and degradation of β-catenin [64]. Wnt signaling and FRAT1 play a neuroprotective role in the brain. We observed downregulation of FRAT1, GSK-3, and axin1 in the penumbra after PTI. The downregulation of GSK-3 and axin1 in penumbra could be neuroprotective. However, the downregulation of FRAT1 could reduce its neuroprotective potential.
The Notch signaling pathway controls the differentiation, maturation, and survival of neuronal and glial cells. The protein NUMB is an intrinsic antagonist of Notch-mediated signal transduction. Upregulation of NUMB in penumbra after cerebral ischemia and following caspase-3-mediated apoptosis have been reported [65]. In our experiments, however, the level of NUMB in penumbra decreased 1–4 h after PTI, indicating the prosurvival character of this process. Notch signaling also regulates angiogenesis [66]. The downregulation of Notch antagonist NUMB could facilitate the reparative neovascularization in penumbra.
The observed overexpression of LRP1 in penumbra 4 h after PTI corresponded to its upregulation in the ischemic tissue after middle cerebral artery occlusion, where it contributed to development of edema [62]. LRP1 is found in astrocytes, microglia, and neurons [67]. Importantly, it regulates the expression of thrombolytic tissue plasminogen activator (tPA) [68], which is the only approved medication for treatment of patients with acute ischemic stroke. The LRP1/t-PA interaction leads to permeability of the blood-brain barrier and activation of microglia in the ischemic brain [68, 69]. However, the data of different authors on the role of microglial activation during cerebral ischemia are controversial: it may be either deleterious or neuroprotective [69].
The same cellular subsystems were involved in the penumbra tissue response 24 h after PTI. However, the changes in expression of neuronal proteins were lesser and included fewer proteins than after 4 h (Table 2).
Thus, the following proteins and cellular subsystems were involved in penumbra response to PTI: signal transduction pathways (PKBα/GSK-3, PKC and its isoforms, Wnt/β-catenin (axin1, GSK-3, FRAT1), Notch/NUMB, DYRK1A, and TDP43); mitochondria quality control (Pink1, parkin, HtrA2); ubiquitin-proteasomal proteolysis (ubiquilin-1, UCHL1); axon outgrowth and guidance (NAV-3, CRMP2, PKCβ2); vesicular trafficking including docking and fusion of synaptic vesicles (syntaxin-8, TMP21, Munc-18-3, synip, ALS2, VILIP1, syntaxin, synaptophysin, and synaptotagmin); biosynthesis of neuromediators (tryptophan hydroxylase, monoamine oxidase B, glutamate decarboxylase, tyrosine hydroxylase, DOPA decarboxylase, and dopamine transporter); intercellular interactions and tissue integrity (N-cadherin, PMP22); cytoskeleton elements (neurofilament 68, o-glycosylated neurofilament-M, doublecortin); and miscellaneous proteins (LRP1, prion protein, β-amyloid). The changes in their expression in penumbra could be directed either to neurodegeneration or to neuroprotection (Fig. 11). At 1 h after PTI, these changes were directed mainly to cell survival and tissue recovery [19]. Later, 4 h after PTI, both injurious and protective processes were observed.

The summary scheme of changes in the expression of neuronal and signaling proteins in penumbra 4 h after focal photothrombotic infarction in the rat cerebral cortex
These changes were averaged across the whole penumbra samples. But penumbra is inhomogeneous. The damaging processes dominate near the PTI core, whereas protective processes prevail at the penumbra periphery. The balance border between the predominance of either neurodegeneration or neuroprotection depends on the distance from the infarct core. The movement of this border within penumbra determines the fate of neurons and glial cells. The aim of the neuroprotective anti-stroke therapy is to limit the propagation of this border.
The present proteomic data provided a list of proteins with the changed expression in penumbra. These were potentially involved either in infarct development or in tissue resistance and recovery. Protein downregulation occurs mainly due to proteolysis, whereas increase in their levels requires gene expression. The proteomic data, however, do not disclose the reasons of these changes because signaling pathways and transcription factors, which control expression of these proteins, remain unknown. Further mechanistic studies should answer these questions. Some of these proteins are novel in the context of the stroke mechanism research, and detailed investigation of the reasons and consequences of changes in their levels can shed light on the novel mechanism of the penumbra response to ischemia. Anyway, the obtained results may indicate the potential targets for diagnosis and treatment of the cortical infarction in the penumbra.
References
Chavez JC, Hurko O, Barone FC, Feuerstein GZ (2009) Pharmacologic interventions for stroke looking beyond the thrombolysis time window into the penumbra with biomarkers, not a stopwatch. Stroke 40:e558–e563
Iadecola C, Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14:1363–1368
Meisel A, Prass K, Wolf T, Dirnagl U (2004) Stroke. In: Bähr M (ed) Neuroprotection: models, mechanisms and therapies. Wiley-Blackwell, New York, pp 9–43
Moskowitz MA (2010) Brain protection: maybe yes, maybe no. Stroke 41:S85–S86
Ginsberg MD (2008) Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology 55:363–389. doi:10.1016/j.neuropharm.2007.12.007
Mehta SL, Manhas N, Raghubir R (2007) Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 54:34–66
Puyal J, Ginet V, Clarke PG (2013) Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol 105:24–48
Spisak S, Tulassay Z, Molnar B, Guttman A (2007) Protein microchips in biomedicine and biomarker discovery. Electrophoresis 28:4261–4273
Wingren C, Borrebaeck CA (2009) Antibody-based microarrays. Methods Mol Biol 509:57–84
Bu X, Zhang N, Yang X, Liu Y, Du J, Liang J, Xu Q, Li J (2011) Proteomic analysis of PKCβII-interacting proteins involved in HPC-induced neuroprotection against cerebral ischemia of mice. J Neurochem 117:346–356
Datta A, Park JE, Li X, Zhang H, Ho ZS, Heese K, Lim SK, Tam JP et al (2010) Phenotyping of an in vitro model of ischemic penumbra by iTRAQ-based shotgun quantitative proteomics. J Proteome Res 9:472–484
Dayon L, Turck N, Garcí-Berrocoso T, Walter N, Burkhard PR, Vilalta A, Sahuquillo J, Montaner J et al (2011) Brain extracellular fluid protein changes in acute stroke patients. J Proteome Res 10:1043–1051
Villa RF, Gorini A, Ferrari F, Hoyer S (2013) Energy metabolism of cerebral mitochondria during aging, ischemia and post-ischemic recovery assessed by functional proteomics of enzymes. Neurochem Int 63:765–781
Uzdensky AB (2010) Cellular and molecular mechanisms of photodynamic therapy. Nauka, Saint Petersburg (in Russian)
Dietrich WD, Watson BD, Busto R, Ginsberg MD, Bethea JR (1987) Photochemically induced cerebral infarction. I. Early microvascular alterations. Acta Neuropathol 72:315–325
Schmidt A, Hoppen M, Strecker JK, Diederich K, Schäbitz WR, Schilling M, Minnerup J (2012) Photochemically induced ischemic stroke in rats. Exp Transl Stroke Med 4:13
Shanina EV, Redecker C, Reinecke S, Schallert T, Witte OW (2005) Long-term effects of sequential cortical infarcts on scar size, brain volume and cognitive function. Behav Brain Res 158:69–77
Pevsner PH, Eichenbaum JW, Miller DC, Pivawer G, Eichenbaum KD, Stern A, Zakian KL, Koutcher JA (2001) A photothrombotic model of small early ischemic infarcts in the rat brain with histologic and MRI correlation. J Pharmacol Toxicol Methods 45:227–233
Demyanenko SV, Panchenko SN, Uzdensky AB (2015) Expression of neuronal and signaling proteins in penumbra around a photothrombotic infarction core in rat cerebral cortex. Biochemistry (Mosc) 80:790–799. doi:10.1134/S0006297915060152
Brown AW, Brieley JB (1973) The earliest alterations in rat neurones and astrocytes after anoxia-ischemia. Acta Neuropathol 23:9–22
Demyanenko SV, Uzdensky AB, Sharifulina SA, Lapteva TO, Polyakova LP (2014) PDT-induced epigenetic changes in the mouse cerebral cortex: a protein microarray study. Biochim Biophys Acta 1840:262–270
Jiang W, Gu W, Hossmann KA, Mies G, Wester P (2006) Establishing a photothrombotic ‘ring’ stroke model in adult mice with late spontaneous reperfusion: quantitative measurements of cerebral blood flow and cerebral protein synthesis. J Cereb Blood Flow Metab 26:927–936
Zhao H, Sapolsky RM, Steinberg GK (2006) Phosphoinositide-3-kinase/Akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol 34:249–270
Liu BN, Han BX, Liu F (2014) Neuroprotective effect of pAkt and HIF-1α on ischemia rats. Asian Pac J Trop Med 7:221–225. doi:10.1016/S1995-7645(14)60025-0
Caldeira MV, Salazar IL, Curcio M, Canzoniero LM, Duarte CB (2014) Role of the ubiquitin-proteasome system in brain ischemia: friend or foe? Prog Neurobiol 112:50–69
Liu Y, Lü L, Hettinger CL, Dong G, Zhang D, Rezvani K, Wang X, Wang H (2014) Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J Neurosci 34:2813–282. doi:10.1523/JNEUROSCI.3541-13.2014
Yamauchi T, Sakurai M, Abe K, Matsumiya G, Sawa Y (2008) Ubiquitin-mediated stress response in the spinal cord after transient ischemia. Stroke 39:1883–1889
Gong B, Leznik E (2007) The role of ubiquitin C-terminal hydrolase L1 in neurodegenerative disorders. Drug News Perspect 20:365–370
Siman R, Roberts VL, McNeil E, Dang A, Bavaria JE, Ramchandren S, McGarvey M (2008) Biomarker evidence for mild central nervous system injury after surgically-induced circulation arrest. Brain Res 1213:1–11
de Castro IP, Martins LM, Loh SH (2011) Mitochondrial quality control and Parkinson’s disease: a pathway unfolds. Mol Neurobiol 43:80–86. doi:10.1007/s12035-010-8150-4
McKeon JE, Sha D, Li L, Chin LS (2015) Parkin-mediated K63-polyubiquitination targets ubiquitin C-terminal hydrolase L1 for degradation by the autophagy-lysosome system. Cell Mol Life Sci 72:1811–1824. doi:10.1007/s00018-014-1781-2
Zhao Y, Chen F, Chen S, Liu X, Cui M, Dong Q (2013) The Parkinson’s disease-associated gene PINK1 protects neurons from ischemic damage by decreasing mitochondrial translocation of the fission promoter Drp1. J Neurochem 127:711–722. doi:10.1111/jnc.12340
Hou ST, Jiang SX, Aylsworth A, Ferguson G, Slinn J, Hu H, Leung T, Kappler J et al (2009) CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. J Neurochem 111:870–881
Maes T, Barceló A, Buesa C (2002) Neuron navigator: a human gene family with homology to unc-53, a cell guidance gene from Caenorhabditis elegans. Genomics 80:21–30
Chen A, Liao WP, Lu Q, Wong WS, Wong PT (2007) Upregulation of dihydropyrimidinase-related protein 2, spectrin alpha II chain, heat shock cognate protein 70 pseudogene 1 and tropomodulin 2 after focal cerebral ischemia in rats—a proteomics approach. Neurochem Int 50:1078–1086
Zechariah A, El Ali A, Doeppner TR, Jin F, Hasan MR, Helfrich I, Mies G, Hermann DM (2013) Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke 44:1690–1697. doi:10.1161/STROKEAHA.111.000240
Quarles RH (2002) Myelin sheaths: glycoproteins involved in their formation, maintenance and degeneration. Cell Mol Life Sci 59:1851–1871
Doeppner TR, Kaltwasser B, Schlechter J, Jaschke J, Kilic E, Bähr M, Hermann DM, Weise J (2015) Cellular prion protein promotes post-ischemic neuronal survival, angioneurogenesis and enhances neural progenitor cell homing via proteasome inhibition. Cell Death Dis 6:e2024. doi:10.1038/cddis.2015.365
McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F et al (2004) Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 165:227–235
Weise J, Crome O, Sandau R, Schulz-Schaeffer W, Bähr M, Zerr I (2004) Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett 372:146–150
Chen B, Zhao L, Li X, Ji YS, Li N, Xu XF, Chen ZY (2014) Syntaxin 8 modulates the post-synthetic trafficking of the TrkA receptor and inflammatory pain transmission. J Biol Chem 289:19556–19569. doi:10.1074/jbc.M114.567925
Bromley-Brits K, Song W (2012) The role of TMP21 in trafficking and amyloid-β precursor protein (APP) processing in Alzheimer’s disease. Curr Alzheimer Res 9:411–424
Jahn R (2000) Sec1/Munc18 proteins: mediators of membrane fusion moving to center stage. Neuron 27:201–204
Yu H, Rathore SS, Shen J (2013) Synip arrests soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-dependent membrane fusion as a selective target membrane SNARE-binding inhibitor. J Biol Chem 288:18885–18893. doi:10.1074/jbc.M113.465450
Hadano S, Otomo A, Kunita R, Suzuki-Utsunomiya K, Akatsuka A, Koike M, Aoki M, Uchiyama Y et al (2010) Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS One 5:e9805. doi:10.1371/journal.pone.0009805
Braunewell KH, Klein-Szanto AJ (2009) Visinin-like proteins (VSNLs): interaction partners and emerging functions in signal transduction of a subfamily of neuronal Ca2+-sensor proteins. Cell Tissue Res 335:301–316. doi:10.1007/s00441-008-0716-3
Stejskal D, Sporova L, Svestak M, Karpisek M (2011) Determination of serum visinin like protein-1 and its potential for the diagnosis of brain injury due to the stroke: a pilot study. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 155:263–268. doi:10.5507/bp.2011.049
Ishimaru H, Casamenti F, Uéda K, Maruyama Y, Pepeu G (2001) Changes in presynaptic proteins, SNAP-25 and synaptophysin, in the hippocampal CA1 area in ischemic gerbils. Brain Res 903:94–101
Mitsios N, Saka M, Krupinski J, Pennucci R, Sanfeliu C, Wang Q, Rubio F, Gaffney J et al (2007) A microarray study of gene and protein regulation in human and rat brain following middle cerebral artery occlusion. BMC Neurosci 12(8):93
Schmidt-Kastner R, Zhang B, Belayev L, Khoutorova L, Amin R, Busto R, Ginsberg MD (2002) DNA microarray analysis of cortical gene expression during early recirculation after focal brain ischemia in rat. Brain Res Mol Brain Res 108:81–93
Abbassi R, Johns TG, Kassiou M, Munoz L (2015) DYRK1A in neurodegeneration and cancer: molecular basis and clinical implications. Pharmacol Ther 151:87–98. doi:10.1016/j.pharmthera.2015.03.004
Wegiel J, Gong CX, Hwang YW (2011) The role of DYRK1A in neurodegenerative diseases. FEBS J 278:236–245. doi:10.1111/j.1742-4658.2010.07955.x
Choi HK, Chung KC (2011) DYRK1A positively stimulates ASK1-JNK signaling pathway during apoptotic cell death. Exp Neurobiol 20:35–44
Bright R, Mochly-Rosen D (2005) The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke 36:2781–2790
Hara H, Onodera H, Yoshidomi M, Matsuda Y, Kogure K (1990) Staurosporine, a novel protein kinase C inhibitor, prevents postischemic neuronal damage in the gerbil and rat. J Cereb Blood Flow Metab 10:646–653
Felipo V, Miñana MD, Grisolía S (1993) Inhibitors of protein kinase C prevent the toxicity of glutamate in primary neuronal cultures. Brain Res 604:192–196
Kanazawa M, Kakita A, Igarashi H, Takahashi T, Kawamura K, Takahashi H, Nakada T, Nishizawa M et al (2011) Biochemical and histopathological alterations in TAR DNA-binding protein-43 after acute ischemic stroke in rats. J Neurochem 116:957–965
Friocourt G, Koulakoff A, Chafey P, Boucher D, Fauchereau F, Chelly J, Francis F (2003) Doublecortin functions at the extremities of growing neuronal processes. Cereb Cortex 13:620–626
Wang X, Mao X, Xie L, Sun F, Greenberg DA, Jin K (2012) Conditional depletion of neurogenesis inhibits long-term recovery after experimental stroke in mice. PLoS One 7:e38932. doi:10.1371/journal.pone.0038932
Osman AM, Porritt MJ, Nilsson M, Kuhn HG (2011) Long-term stimulation of neural progenitor cell migration after cortical ischemia in mice. Stroke 42:3559–3565. doi:10.1161/STROKEAHA.111.627802
Li F, Chong ZZ, Maiese K (2005) Vital elements of the Wnt-Frizzled signaling pathway in the nervous system. Curr Neurovasc Res 2:331–340
Mathieu P, Adami PV, Morelli L (2013) Notch signaling in the pathologic adult brain. Biomol Concepta 4:465–476. doi:10.1515/bmc-2013-0006
Mastroiacovo F, Busceti CL, Biagioni F, Moyanova SG, Meisler MH, Battaglia G, Caricasole A, Bruno V et al (2009) Induction of the Wnt antagonist, Dickkopf-1, contributes to the development of neuronal death in models of brain focal ischemia. J Cereb Blood Flow Metab 29:264–276. doi:10.1038/jcbfm.2008.111
Culbert AA, Brown MJ, Frame S, Hagen T, Cross DA, Bax B, Reith AD (2001) GSK-3 inhibition by adenoviral FRAT1 overexpression is neuroprotective and induces Tau dephosphorylation and beta-catenin stabilisation without elevation of glycogen synthase activity. FEBS Lett 507:288–294
Ma M, Wang X, Ding X, Teng J, Shao F, Zhang J (2013) Numb/Notch signaling plays an important role in cerebral ischemia-induced apoptosis. Neurochem Res 38:254–261. doi:10.1007/s11064-012-0914-y
Al Haj Zen A, Madeddu P (2009) Notch signalling in ischaemia-induced angiogenesis. Biochem Soc Trans 37:1221–1227. doi:10.1042/BST0371221
Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA (2003) Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest 112:1533–1540
Herz J (2001) The LDL receptor gene family: (un)expected signal transducers in the brain. Neuron 29:571–581
Zhang C, An J, Strickland DK, Yepes M (2009) The low-density lipoprotein receptor-related protein 1 mediates tissue-type plasminogen activator-induced microglial activation in the ischemic brain. Am J Pathol 174:586–594. doi:10.2353/ajpath.2009.080661
Acknowledgments
The work was supported by the Russian Science Foundation (grant 14-15-00068). A.B. Uzdensky work was also supported by the Ministry of Education and Science of Russian Federation (grant “Science organization” #790). The authors used the equipment of the Center for Collective Use of Southern Federal University “High technology” supported by the Ministry of Education and Science of Russian Federation (project RFMEFI59414X0002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Uzdensky, A., Demyanenko, S., Fedorenko, G. et al. Protein Profile and Morphological Alterations in Penumbra after Focal Photothrombotic Infarction in the Rat Cerebral Cortex. Mol Neurobiol 54, 4172–4188 (2017). https://doi.org/10.1007/s12035-016-9964-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9964-5