
Abstract
Cdk5, a cyclin-dependent kinase family member, is a global orchestrator of neuronal cytoskeletal dynamics. During embryogenesis, Cdk5 is indispensable for brain development. In adults, it is essential for numerous neuronal processes, including higher cognitive functions such as learning and memory formation, drug addiction, pain signaling, and long-term behavior changes through long-term potentiation and long-term depression, all of which rely on rapid alterations in the cytoskeleton. Cdk5 activity becomes deregulated in various brain disorders, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, attention-deficit hyperactivity disorder, epilepsy, schizophrenia, and ischemic stroke; these all result in profound remodeling of the neuronal cytoskeleton. This Commentary specifically focuses on the pleiotropic contribution of Cdk5 in regulating neuronal microtubule remodeling. Because the vast majority of the physiological substrates of Cdk5 are associated with the neuronal cytoskeleton, our emphasis is on the Cdk5 substrates, such as CRMP2, stathmin, drebrin, dixdc1, axin, MAP2, MAP1B, doublecortin, kinesin-5, and tau, that have allowed to unravel the molecular mechanisms through which Cdk5 exerts its divergent roles in regulating neuronal microtubule dynamics, both in healthy and disease states.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cyclin-dependent kinases (Cdks) are proline-directed serine/threonine kinases that play critical roles in cell cycle progression. Cdks bind to cyclins, their specific protein partners, for activation. Their activity can be further regulated through phosphorylation by the kinases Cdk-activating kinase-1 (Cak1), membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase isoform 1 (Myt1) and Wee1. Phosphorylation of Cdks by Cak1 at T161 (Cdk1 numbering) results in activation, whereas phosphorylation by Myt1 and Wee1 at T14 and Y15 respectively inhibit their activity. The activity of Cdk is also inhibited by binding of the cell cycle proteins cyclin-dependent kinase inhibitor 1 (CDKN1A) and cyclin-dependent kinase inhibitor 1B (CDKN1B) (henceforth referred to as p21Cip1 and p27Kip1, respectively).
Cdk Family Members
The Cdk family member Cdk5 (Gene ID: 1020, mapped at chromosome 7q36) shares a high homology with other Cdks; however, it is distinguished from those by possessing unique activation pathways and distinct cellular functions [1, 2]. Unlike other Cdks, Cdk5 does not participate in cell cycle regulation in proliferating cells, although it can aberrantly activate different components of the cell cycle in post-mitotic neurons, resulting in death under pathological conditions [3]. Furthermore, Cdk5 is not activated by the canonical cyclins (cyclin A, cyclin D, and cyclin E), but binds to its own specific partners, CDK5R1 and CDK5R2 (henceforth referred to as p35 and p39, respectively) [4, 5]. Expression of p35 is nearly ubiquitous, whereas p39 is exclusively expressed in the central nervous system (CNS). Cdk5 is also activated by cyclin I (CCNI) in post-mitotic cells (e.g., neurons and podocytes) [6]. Cyclin I does not activate any other Cdks, suggesting it might be a Cdk5-specific activator. Thus, Cdk5 has vital role in CNS where it is the main Cdk expressed.
Cdk5 Regulators
Similar to its unique activation partners, Cdk5 also possesses its own set of negative regulators and is not inhibited by p21Cip1 and p27Kip1, which inhibit other family members. Instead, glutathione S-transferase P (GSTP1), cyclin D1 (CCND1), and cyclin E (CCNE) inhibit Cdk5 activity [7–9]. Likewise, phosphorylation of Cdk5 at Y15 by the kinases Abl, Ephrin receptor A (Eph A), or Fyn does not inhibit its activity as has been observed for other Cdks. Instead, phosphorylation of Cdk5 at Y15 has been shown to either increase or has no effect on its activity [10–12].
Role of Cdk5 in Neuronal Development and Learning and Memory Formation
Cdk5 also differs from other family members in that it has distinct functions in a variety of neuronal and non-neuronal tissues. Although most of the Cdk/cyclin complexes shuttle between the cytoplasm and the nucleus, the majority of active Cdk5-p35 complexes reside in close proximity to the membrane and cytoskeletal elements. Although Cdk5 is a cytosolic kinase, both p35 and p39 have membrane-targeting motifs, and this alters the subcellular localization of active Cdk5 complex [13]. The vast majority of the physiological Cdk5 substrates are associated with the neuronal cytoskeleton. This, in turn, allows Cdk5 to play a pivotal role in neuronal development by regulating neuronal migration, neurite outgrowth, axon guidance, and synapse formation [14]. Associated processes during development, such as the establishment of polarity, migration, neurite outgrowth, and synaptogenesis all depend on cytoskeletal dynamics. In adults, higher cognitive functions such as learning and memory formation also rely on rapid cytoskeletal alterations, making Cdk5 activity crucial for neurotransmission, synaptic plasticity and homeostasis, drug addiction, and long-term behavioral changes [15–19].
Role of Cdk5 in Neurodevelopmental and Neurodegenerative Diseases
In several brain disorders, such as mental retardation, attention-deficit/hyperactivity disorder (ADHD) [20], epilepsy [21], and schizophrenia [22], Cdk5 activity is significantly reduced and is believed to play a causal role in their development. By contrast, Cdk5 is also hyperactivated in many neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and ischemic stroke, and it is destructive and neurotoxic ([23–26]. A variety of neurotoxic insults such as exposure to β-amyloid (Aβ), excitotoxicity, ischemia, and oxidative stress disrupt the intracellular calcium homeostasis in neurons, thereby leading to the activation of calpain, which cleaves p35 into p25 and p10. Cdk5-p25 complex exhibits higher kinase activity in vitro than Cdk5-p35 complex. Furthermore, p25 has a six-fold longer half-life compared to p35 and lacks the membrane anchoring signal, which results in its constitutive activation and, most importantly, mislocalization of the Cdk5-p25 complex to the cytoplasm and the nucleus. There, Cdk5-p25 is able to access and phosphorylate a variety of atypical targets, such as tau, GM130, peroxiredoxins, lamin A, and lamin B, triggering a cascade of neurotoxic pathways that culminate in neuronal death [27–31]. Interestingly, Maccioni et al. revealed that Cdk5 can also be deregulated via increased stability of Cdk5-p35 complex when exposed to fibrillary β-amyloid toxicity [32].
Despite the adverse effects when it is misregulated, Cdk5 is a vital component of ongoing healthy neuronal function. Here, we highlight the functions of Cdk5 when it is “good” and when it is “bad” in neuronal signaling, with a specific emphasis on its direct targets and the molecular mechanisms by which it regulates microtubule (MT) cytoskeleton in the brain.
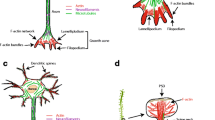
Similar to in any eukaryotic cell, the neuronal cytoskeletal network includes actin filaments and tubulin microtubules (MTs) but also contain neurofilaments (NFs), a class of intermediate filaments that are unique to neuronal cells. Actin, tubulin, and neurofilaments differ physiochemically in protein constituents, subcellular localization, diameter, mechanical stiffness, polarity, assembly dynamics, and the kind of molecular motors they associate with, which translate to their unique architecture and functions in the neurons [33]. Actin filaments form sheet-like structures and are highly enriched at the leading edge of dendrites and axons, including the lamellipodia and filopodia (Fig. 1). MTs are dynamic tubulin polymers, which switch stochastically between shrinking and growing phases. MTs form track-like structures in the axon and transport materials from the cell body to the axon terminals at the synapse [34] (Fig. 1). Neurofilaments form a structural matrix in the axon that nestles MTs and resists mechanical stresses [35]. This review focuses on Cdk5-mediated regulation of microtubules and we will also briefly discuss how Cdk5 deregulation causes cytoskeletal abnormalities in various neurological disorders.

Components of neuronal cytoskeleton. The neuronal cytoskeleton is composed of actin filaments (red), microtubules (MTs; green), and neurofilaments (NFs; purple). The axonal growth cone is comprised of lamellipodia and filopodia, and is highly enriched in actin filaments. Lamellipodia consist of a dense F-actin network, and filopodia contain bundled F-actin, whereas MTs emanate from axons. Axonal microtubules have an unidirectional arrangement with their plus ends facing the axon tip, thus facilitating directional axonal growth, whereas dendritic microtubules have a mixed orientation. NFs are highly enriched in the axons and maintain axonal integrity, caliber, and conduction velocity. MTs are approximately three times thicker in diameter compared to NFs, which are three times thicker than actin
Role of Cdk5 in MT Dynamics
Microtubules consist of 10–15 (usually 13) protofilaments of α/β-tubulin heterodimers that associate laterally to form a polarized hollow tube approximately 24 nm in diameter. The “plus” end of MTs favor polymerization and their “minus” end disassembly, resulting in MTs usually growing from their plus ends. Their polarity enables MTs to enter various subcellular locations and form productive interactions with various signaling molecules. MTs can rapidly switch from growth mode to shrinkage and vice versa. Thus, MTs are constantly assembling and disassembling, which allows them to play a major role in intracellular transport, organelle distribution, mitosis, and growth and maintenance of axons and dendrites (Fig. 1) [34].
MTs are present in both axons and dendrites, where they, however, differ in two major aspects. First, axonal MTs have a unidirectional arrangement, with their plus ends always facing the growth cone, thereby facilitating directional axonal growth, whereas mammalian dendritic MTs have a mixed orientation (Fig. 1). Second, despite a variety of microtubule-associated proteins (MAPs) present in both axons and dendrites, dendritic MTs are mostly associated with MAP2 and axonal MTs with tau. MAPs not only regulate MT dynamics but also aid in MT-mediated long-range transport of cellular cargos. Cdk5 has been shown to be particularly enriched in MT fractions and employs pleiotropic mechanisms to promote axonal generation and axonal transport [36, 37].
Cdk5 directly influences MT dynamics by regulating the acetylation levels of α-tubulin, the monomer of MTs. Mechanistically, α-tubulins are acetylated and deacetylated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively [38]. Acetylation of α-tubulin promotes MT stability and favors axonal growth, whereas deacetylation blocks growth cone dynamics. Cdk5 phosphorylates Sirtuin-2 (SIRT2), an HDAC, inhibiting its activity (Fig. 2) (Table 1). This event increases the pool of acetylated α-tubulin in neurons, leading to axonal growth and maintenance [39].

Cdk5-mediated regulation of MT dynamics and axonal growth. Cdk5 regulates MTs dynamics by phosphorylating several MT-associated proteins (MAPs), such as p35, CRMP2, axin, stathmin, doublecortin, TPPP, tau, and MAP1B. As shown on the left, Cdk5 phosphorylates p35 at T138 in fetal brains, which inhibits its MT polymerization activity and results in the dynamic reorganization of the MT architecture. Furthermore, Cdk5 regulates CRMP2 through several direct and indirect mechanisms (shown in the middle). Direct phosphorylation of CRMP2 by Cdk5 primes it to be phosphorylated by GSK3β, which inactivates CRMP2, resulting in growth cone collapse. Indirectly, Cdk5 activates CRMP2 by phosphorylating Axin. Phosphorylated axin binds to GSK3β, which inhibits its activity. Cdk5 also inhibits GSK3β activity by inhibiting PP1 through phosphorylation, which increases the pool of non-phosphorylated, active CRMP2, thus promoting axonal growth. Cdk5 increases the pool of acetylated α-tubulin by inhibiting SIRT2 resulting in MT stability and axonal growth. Moreover, Cdk5-mediated phosphorylation of stathmin prevents sequestration of free tubulin, facilitating axonal growth (shown on the right). Cdk5-mediated phosphorylation of MAP1B enhances its MT-binding affinity, whereas Cdk5-mediated phosphorylation of doublecortin, tau and TPPP lower their affinity toward MTs. Red and green arrows represent activating and inactivating pathways, respectively
Cdk5-Mediated Regulation of MT Dynamics Through MAPs
The dynamic polymerization and depolymerization of MTs is crucial for their biological functions as it allows them to rapidly reorganize and probe the surrounding space in response to guidance cues. Cdk5 has been shown to affect the association between MTs and MAPS by phosphorylating a number of MAPs, including collapsing response mediator protein 2 (CRMP2), axin, stathmin, doublecortin, TPPP, tau, and MAP1B (Fig. 2 and Table 1) as discussed below.
Cdk5 and CRMP2
CRMP2 is highly enriched in dendrites and axons and mediates signals from several environmental cues, such as ephrins, semaphorins, and neurotrophins. It facilitates MT assembly, cell migration [40], axonal growth and guidance [41], dendritic spine development [42], and synaptic plasticity [43].
Depending on the cellular context, Cdk5 can either activate or inhibit CRMP2, and so promote either axonal elongation or growth cone collapse, respectively (Fig. 2, Table 1). For instance, in dorsal root ganglion neurons, Cdk5 promotes Semaphorin 3A (Sema3A)-induced growth cone collapse through inhibiting CRMP2 and alpha2-chimaerin [44], a GTPase activating protein (GAP) for Rac1. Active alpha2-chimaerin acts as an adaptor protein and recruits CRMP2 through interactions with its SH2 domain and Cdk5-p35 through its GAP domain, which in turn allows Cdk5 to phosphorylate CRMP-2 at serine 522. CRMP2 phosphorylation at S522 primes it for glycogen synthase kinase-β (GSK3β) kinase-mediated phosphorylation at T514 and S518, resulting in inactivation and triggering growth cone collapse (Fig. 2). Importantly, Cdk5-mediated CRMP2 phosphorylation at S522 is essential for proper organization of the dendritic field and the precise bifurcation of apical dendrites of CA1 pyramidal neurons in vivo [42, 45].
In a reverse mechanism, in response to neurotrophins, such as brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT3), Cdk5 promotes axonal growth by inhibiting the inactivation of CRMP2 through directly phosphorylating Axin at T485 [46] (Fig. 2). Axin is a scaffolding protein; upon phosphorylation, it binds to and inhibits the activity of GSK3β, thereby increasing the pool of non-phosphorylated active CRMP2 in the growth cone and promoting axonal growth. Cdk5 also indirectly inhibits GSK3β activity via inactivating protein phosphatase 1 (PP1) [47]. PP1 activates GSK3β by dephosphorylating it at S9. Thus, Cdk5-mediated inhibition of GSK3β activity promotes axonal growth. GSK3β functions as a negative regulator of axon formation [48]. Intriguingly, a recent study showed that p25 (cleaved product of p35) preferentially binds GSK3β compared to Cdk5, which not only activates GSK3β but also alters its substrate specificity [49]. By contrast, GSK3β does not bind p35. These findings suggest that p25 upregulation may in part promote neurotoxicity via elevated GSK3β activity.
Effect of Cdk5 on Other MAPs
Cdk5-mediated phosphorylation of MAPs often affects their affinity of MT binding (Table 1). For instance, in cultured cerebellar macro neurons, Cdk5-mediated phosphorylation of MAP1B enhances its MT-binding affinity and so promotes axonal elongation [36]. In contrast, Cdk5-mediated phosphorylation of the MAP doublecortin at S297 reduces its affinity toward MTs in vitro, thereby reducing its effect on MT polymerization and neuronal migration (Fig. 2) [50] (Table 1). Accordingly, mutations in the doublecortin gene in humans result in similar cortical lamination defects in the developing brain as those seen in Cdk5-null mice. These mice exhibit severe abnormalities in lamination in the hippocampus, cerebral cortex, and cerebellum.
Likewise, Cdk5 phosphorylates tubulin polymerization-promoting protein (TPPP) at multiple sites in vitro and in vivo, which results in the loss of its MT-assembling activity [51] (Fig. 2). TPPP and tubulin are enriched in aggresomes and Lewy body in brain tissues from patients with synucleinopathies [52], suggesting that Cdk5-mediated phosphorylation of TPPP may prevent the formation of these inclusions in pathological states.
Of particular interest in neurodegenerative pathology, Cdk5-mediated phosphorylation of tau reduces its ability to bind to and stabilize MTs during axonal growth. This has important implications, as during brain development, tau is phosphorylated by the Cdk5-p39 complex at Ser-202 and Thr-205, but not by Cdk5-p35 [53]. Because p35 and p39 display differential expression patterns in the developing brain, Cdk5-mediated phosphorylation of tau promotes neuritic growth in a region-specific and developmentally regulated manner in developing axons. Consequently, at the period of brain development, phosphorylated tau is only present abundantly during strong neuritic outgrowth, whereas it diminishes to undetectable levels during neurite stabilization and synaptogenesis [54]. In the developing brain, the expression level of p39 is elevated in embryonic spinal cord, hind brain, and post-natal cerebral cortex, whereas p35 is highly abundant in cerebral cortex.
In a different mechanism, Cdk5 phosphorylates the MAP stathmin at Ser38, which results in axonal growth (Fig. 2 and Table 1) [55]. Stathmin sequesters free tubulin, thereby inhibiting MT growth. Cdk5-mediated phosphorylation of stathmin releases bound tubulin, thereby facilitating axonal growth and branching. A recent study demonstrated that stathmin-dependent changes in MT stability are crucial for synaptic function and memory formation [56]. Learning induces stathmin phosphorylation, which in turn regulates MT dynamics and long-term memory formation.
p35 can also act as a MAP; it binds to MTs through its N-terminus and promotes MT assembly and bundling of MT filaments [57]. Interestingly, the MT-binding and polymerizing activities of p35 are regulated both in a Cdk5-dependent and -independent manner. In the mechanism that is independent of Cdk5, p35, through its N-terminal domain, binds preferentially to calmodulin (CaM)-binding protein in the presence of Ca2+, and not to MTs, thereby inhibiting MT assembly. In the other mechanism, Cdk5 directly phosphorylates p35 at T138 and so inhibits its microtubule polymerization activity (Fig. 2) [58]. As T138 phosphorylation is highest in fetal brain and undetectable in adults, this suggests that Cdk5-mediated phosphorylation of p35 in developing brains may be required for the highly dynamic reorganization of the MT network by promoting both the rapid association and dissociation of MAPs, thereby allowing for a high plasticity during brain development.
Role of Cdk5 in Neuronal Migration
Although Cdk5 is known to have more than two dozen substrates that are associated with neuronal migration, we focus here on those substrates that promote migration predominantly through modulating MT organization.
The first step in neuronal migration is the extension of the neurite, which is followed by movement of the soma and the nucleus (nucleokinesis) into the leading process with simultaneous retraction of the trailing process. Cdk5 plays a key role in nucleokinesis by phosphorylating focal adhesion kinase (FAK) at S732 in vitro and in the developing brain, which results in its binding to specific MTs that originate from MT-organizing centers (MTOCs). Expression of phospho-resistant S732A FAK results in disorganization of MTs between the nucleus and the centrosome causing migration defects. This phenotype suggests that phosphorylation of FAK by Cdk5 at least in part promotes the proper organization of MTs that link the nucleus and the centrosome, thereby facilitating nucleokinesis and thus neuronal migration [59] (Fig. 3) (Table 1) .

Role of Cdk5 in neuronal migration. Cdk5-mediated phosphorylation of ErbB4 activates PI3K/Akt pathway leading to tangential migration of interneurons toward and within the cerebral cortex. Cdk5 promotes nucleokinesis by phosphorylating FAK at S732. Phosphorylation of FAK by Cdk5 is believed to promote the proper organization of MTs that link the nucleus and the centrosome, thereby facilitating nucleokinesis and thus neuronal migration. During neocortical development, Cdk5, through DISC1, attenuates the proliferation of progenitor cells, while concurrently directing them toward correct locations by increasing neuronal migration. Cdk5 phosphorylates DISC1 at Ser713, which increases its affinity toward BBS1 and BBS4, resulting in increased neuronal migration. Cdk5 also indirectly regulates DISC1-mediated neuronal migration by phosphorylating Dixdc1 at Ser250, which facilitates the formation of the DISC1-Dixdc1-Ndel1 complex that is essential for neuronal migration as it modulates the actin and MT cytoskeleton. In granular neurons, Cdk5 employs an indirect mechanism to activate Ndel1. Cdk5 activates Aurora A kinase through Cdk5-RAP2, which in turn phosphorylates Ndel1, resulting in MT remodeling and neuronal migration. In addition, Cdk5-mediated phosphorylation of Drebrin at Ser142 enables it to couple dynamic MTs to F-actin through MT-binding + TIP protein EB3 in growth cone resulting in increased neuronal migration
Another important substrate of Cdk5 is disrupted in schizophrenia 1 (DISC1), whose gene mutations are strong risk factors for several psychiatric disorders, such as autism, schizophrenia, depression, and bipolar disorder. In neurons, DISC1 is involved in multiple processes, including MT-mediated transport and regulation of dendritic spines [60]. During embryonic neocortical development, DISC1 regulates several steps of neurogenesis, including cell proliferation, cell migration, and axon and dendrite formation. In developing cerebral cortex, Cdk5 phosphorylates DISC1 at Ser713, which increases its affinity toward two centrosomal proteins, Bardet-Biedl syndrome (BBS)1 and BBS4, resulting in increased neuronal migration, while concurrently decreasing neuronal proliferation (Fig. 3) [61]. Thus, Cdk5-mediated phosphorylation of DISC1 acts as a developmentally regulated switch that, through increasing neuronal migration, attenuates the proliferation of progenitor cells and allows them to move toward their correct locations in the developing cerebral cortex (Table 1). Cdk5 also indirectly regulates DISC1-mediated neuronal migration through phosphorylating DIX domain containing-1 protein (Dixdc1) at Ser250, which facilitates the association of nudE nuclear distribution E homolog (Ndel1) to DISC1-Dixdc1 complex resulting in increased neuronal migration via modulating the actin and MT cytoskeleton (Fig. 3). Ndel1 is necessary for normal migration of neocortical projection neurons, and its ablation significantly diminishes this developmental process [62]. In the absence of Dixdc1 phosphorylation by Cdk5, Ndel1 does not bind to DISC1-Dixdc1 complex resulting in cell proliferation. Thus, the authors proposed that Cdk5-mediated phosphorylation of Dixdc1 is an alternate mechanism, which switches the involvement of DISC1 from progenitor cell proliferation to neuronal migration [63].
In granular neurons, Cdk5 employs another indirect mechanism to regulate Ndel1-mediated neuronal migration by acting through Aurora A kinase. Although the exact mechanism remains to be elucidated, it appears that cyclin-dependent kinase 5 regulatory-associated protein 2 Cdk5RAP2, a centrosomin family protein, mediates the interaction between Aurora A and Cdk5, which is critical for the correct centrosomal targeting of Aurora A. In turn, Aurora A phosphorylates Ndel1 at S251, resulting in MT remodeling and neuronal migration (Fig. 3) [64]. Depletion of Cdk5 reduced the centrosomal targeting of Cdk5RAP2, phosphorylation of Aurora A at T288, and that of Ndel1 at S251, which resulted in impaired neuronal movement. In addition, Cdk5RAP2 may have the role of inhibition of centrosomal Cdk5 during neurogenesis, perhaps taking part in a direct feedback loop with Cdk5 and may participate in regulation of brain size [65].
A recent study further revealed that Cdk5-mediated phosphorylation of receptor tyrosine-protein kinase erbB-4 (ErbB4) at Tyr1056, a docking site for PI3-kinase, is necessary for tangential migration of interneurons both toward and within the cerebral cortex [66] (Table 1). Importantly, phosphorylated ErbB4 reside in close proximity to MTs in adult interneurons, suggesting that Cdk5-directed migration of cortical interneurons likely occurs through MT cytoskeletal remodeling. Taken together, it is clear that Cdk5 utilizes several strategies to direct MT dynamics and neuronal migration.
Dynamic MTs Meet Actin Filaments in Growth Cones via Cdk5
Dendritic spines are highly dynamic membranous protrusions on post-synaptic dendrites. Structural changes in dendritic spines form the basis for learning and memory formation, making them central hubs for the processing and storage of information in the brain [67]. F-actin filaments are highly concentrated in the head, neck, and periphery regions of dendritic spines, whereas stable MTs are confined mainly to the axons and dendritic shafts. As a result, until recently, actin polymerization was deemed to be the underlying cause for the formation or enlargement of the spine during long-term potentiation (LTP), whereas actin depolymerization has been linked to spine shrinkage observed during long-term depression (LTD) [68, 69]. Recent studies, however, have revealed that dynamic MTs can polymerize directly into the dendritic spines, resulting in spine enlargement and increase in post-synaptic density protein 95 (PSD-95) levels [70–72]. As PSD-95 upregulation is strongly associated with enhanced synaptic strength, it suggests that MTs invasion directly promotes structural alterations in dendritic spines. More importantly, inhibition of MT dynamics alters spine morphology through the actin cytoskeleton, suggesting that the MT and actin cytoskeleton in fact act together in dendritic spines to regulate synaptic plasticity [73].
Of particular interest, Cdk5 appears to be responsible for coupling actin filaments to MTs through an F-actin-binding protein, drebrin, in primary cortical neurons [74] (Fig. 3). Drebrin is enriched in growth cones of developing neurons and promotes neuronal migration and neuritogenesis [75]. In mature neurons, it is highly expressed in dendritic spines and is essential for actin remodeling underlying memory [76]. Loss of drebrin in dendritic spines is causal to memory loss in mild cognitive impairment and AD [77]. In growth cones, drebrin was shown to couple dynamic MTs to actin through microtubule-binding + TIP protein EB3 [78]. Worth et al. showed that Cdk5-mediated phosphorylation of drebrin at Ser142 is responsible for this event. The authors proposed that drebrin phosphorylation by Cdk5 induces a conformational change that allows it to bind or bundle F-actin, which in turn interacts with EB3 located at the “plus” end of a MT invading the filopodium, and thus couple F-actin to MTs [74]. Furthermore, a recent study reported that Cdk5-p35-mediated phosphorylation of drebrin E at S142 and drebrin A at Ser142 and Ser342 promotes radial migration of neurons in embryonic cortex [79]. Drebrin E is embryonic form that is ubiquitously expressed, whereas drebrin A is adult brain-specific isoform. As neuronal migration requires highly coordinated regulation of actin and MT cytoskeleton, Cdk5 may regulate this process through dynamic reorganization of actin and MT network by phosphorylating drebrin (Fig. 3).
Cdk5 and Axonal Transport: Role in Brain Development
During development, various axonal proteins, organelles, and lipids are synthesized in the neuronal cell soma and delivered to growing axon terminals by anterograde axonal transport. Likewise, retrograde axonal transport is essential for transducing extracellular signals and recycling misfolded proteins from the nerve terminals to cell soma, thus avoiding the build-up of toxic aggregates. Axonal transport requires the cytoskeletal MT scaffold to serve as tracks, and the motor proteins kinesin and dynein that exert mechanical force to move cargoes anterogradely and retrogradely, respectively [80].
Cdk5 and Kinesins
Cdk5 orchestrates anterograde and retrograde axonal transport by regulating both kinesin and dynein, respectively. Cdk5 indirectly drives kinesin-induced axonal transport by inhibiting the activity of GSK-3β though PP1 (Fig. 4). GSK3β inhibits kinesin-mediated anterograde vesicle transport by releasing its cargo [47]. Cdk5 also directly phosphorylates the kinesin-3 family member 13B (KIF13B) at Thr506, which allows it to bind to transient receptor potential vanilloid 1 (TRPV1)-containing vesicles and deliver them to the membrane surface (Fig. 4). TRPV1 channels are important at the surface of primary sensory neurons as they regulate heat sensitivity. Accordingly, Cdk5-mediated increase in the surface localization of TRPV1s contributes to heat hyperalgesia [81]. In addition, Cdk5 also directly phosphorylates TRPV1 channel at T407, which too leads to its surface localization, causing inflammatory thermal hyperalgesia [82]. Furthermore, a recent study has revealed that Cdk5 directly phosphorylates kinesin-5 (also known as Eg5) at Thr926, which is essential for its association with MTs (Fig. 4) [83]. Kinesin-5 is a homotetrameric motor protein, which is ~100 times slower than dynein. As a result, it acts as a molecular “brake” that can effectively halt the movement of MTs by other motors, resulting in reduced neuronal growth [84]. Depletion or inhibition of kinesin-5 in neurons results in rapid but random axonal growth. Thus, the authors suggested that Cdk5 regulates the rate and directionality of neuronal growth and migration by phosphorylating kinesin-5 [83].

Cdk5-mediated regulation of axonal transport. Cdk5 orchestrates anterograde and retrograde axonal transport by regulating kinesin and dynein, respectively. Cdk5 drives kinesin-induced axonal transport by inhibiting GSK-3β activity via PP1 and facilitates dynein function by phosphorylating Ndel1, thereby strengthening its interaction with Lis1. Ndel1 and Lis1 both bind to dynein and stimulate its cargo transport capacity. Cdk5 also regulates the rate and directionality of neuronal growth and migration by inducing the association of kinesin-5 with MTs through phosphorylation of Thr926
Cdk5 and Dynein
Cdk5 also indirectly acts on dynein and promotes its function through phosphorylating Ndel1, a dynein-interacting protein [85]. Cdk5 phosphorylates Ndel1 at S197, T219, and S231, which enables it to bind to 14-3-3ε, an adaptor protein, which protects it from dephosphorylation as well as strengthens its interaction with Lis1, another dynein-binding factor (Fig. 4) [86]. Ndel1 and Lis1 are believed to regulate the processivity of dynein complex. Inhibition of Cdk5 has been shown to alter the localization of Ndel1 and its affinity toward the dynein complex, resulting in axonal swellings [87]. More recently, another study found that expression of a dominant-negative form of Cdk5 or a mutant of Ndel1 that cannot be phosphorylated by Cdk5 in adult axons not only completely abrogated retrograde transport but also severely reduced anterograde flux of acidic organelles [88], suggesting that there is a Cdk5-dependent switch that regulates Lis-Ndel1-Dynein-dependent transport in adult axons. In this model, Lis1 and unphosphorylated Ndel1 interact only weakly and although they bind to dynein inhibit its capacity to move cargo. Phosphorylation of Ndel1 by Cdk5 turns the switch “on,” triggering activation of the Lis1-Ndel1-dynein complex and stimulating the ability of dynein to cargo transport (Fig. 4). Therefore, Cdk5 appears to employ several direct and indirect strategies to recruit different motor proteins and direct MT dynamics, neuronal migration, and axonal transport.
Reduced Activity and Hyperactivity of Cdk5 Are Each Potentially Neurotoxic: Implications for Neurodevelopmental Disorders and Neurodegenerative Diseases
A variety of neurological disorders display profound cytoskeletal abnormalities, including AD, ALS, Huntington’s disease (HD), PD, and Down’s syndrome. In addition, several memory disorders and psychiatric illnesses such as mental retardation and schizophrenia also involve defects in the regulation of the MT cytoskeleton [89, 90]. Although many Cdk5 substrates are known to promote neurotoxicity, below, we mainly focus on the brain disorders that involves deregulation of the MT cytoskeleton caused either by reduced activity or hyperactivity of Cdk5 (Fig. 5).

Reduced activity and hyperactivity of Cdk5 are each potentially neurotoxic. In healthy cells, Cdk5 activity is exquisitely controlled. Increase in Cdk5 activity or loss of Cdk5 activity, both can give rise to neurodevelopmental or neurological disorders
Several neurological disorders originate due to loss or reduction in Cdk5 activity. In patients with NF1 microdeletion syndrome, lack of one copy of Cdk5R1 (p35) leads to severe mental retardation [91]. Similarly, in schizophrenia patients, p35 expression is reduced in specific brain regions. Importantly, mimicking an analogous reduction in p35 levels in heterozygous mice revealed similar cognitive deficits suggesting that impaired Cdk5 activity plays a causal role in schizophrenia [22]. p35 knockout mice have also been shown to exhibit spontaneous epileptic seizures [21]. Dredrup et al. further observed that p35 knockout mice display behavioral phenotypes reminiscent of ADHD [20]. In addition, as noted before, Cdk5 directs tangential migration of interneurons by activating ErbB4/PI3K pathway, dysfunction of which is repeated linked to neurodevelopmental disorders such as autism and schizophrenia [64]. The authors indeed show that loss in Cdk5 activity in p35 knockout mice causes permanent reduction in the final number of specific types of interneurons, which in turn may alter neuronal circuit formation, thereby increasing the risk of neurodevelopmental disorders.
In HD, most of the studies also favor a neuroprotective role of Cdk5. Reduced Cdk5 and p35 levels were reported in the brains of HD patients [92]; however, one study showed increased p25/p35 levels in HD patients [93]. Future studies are needed to unravel the mechanism of Cdk5 deregulation in this disease. Nevertheless, Cdk5 inhibits aggregation of Huntington (htt) by phosphorylating it at Ser434, which impairs its degradation by caspases [92]. Cdk5 also prevents aggregation of mutant htt by disrupting the MT network [94]. Intact MT cytoskeleton is required for aggregation of mutant htt [95]. Furthermore, DNA damage induced Cdk5 activation triggers the phosphorylation of htt at Ser1181 and Ser1201, which protects cultured striatal neurons from mutant htt-induced neurotoxicity [96]. Collectively, these findings underscore an essential role of normal Cdk5 activity for healthy neuronal functions.
By contrast, hyperactivation of Cdk5 is highly detrimental in several neurodegenerative diseases (Fig. 5). Mice that are transgenic for p25 show deregulated Cdk5 activity and display axonal swelling, cytoskeletal disorganization, and unusual clustering of lysosomes and mitochondria, all features that are consistent with the loss of a functioning MT network [97]. Disturbances in neuronal cytoskeletal organization observed were similar to those in several neurodegenerative diseases including AD [98]. Deregulation of Cdk5 in AD leads to loss of dendritic spines, with subsequent synaptic dysfunction and memory loss [99]. Similarly, it has been shown that hyperactivation of Cdk5 in the striatum of animal models reduces dendritic spine density and causes impaired motor coordination and decreased locomotor sensitization to cocaine [100].
Role of Cdk5 in Neurofibrillary Tangles
At the molecular level, Cdk5 deregulation not only results in hyperphosphorylation of several of its physiological substrates but also in the phosphorylation of non-physiological targets that cause neurotoxicity. For example, during development, tau is phosphorylated by Cdk5-p39 but not by Cdk5-p35 in axons, resulting in axonal growth in a region-specific manner [53]. However, upon neurotoxic insults, such as excitotoxicity or β-amyloid stimulation, formation of p25 from p35, hyperactivates Cdk5, which in turn hyperphosphorylates tau, which aggregates to form the neurofibrillary tangles observed in AD. Neurofibrillary tangles (NFTs) contribute to axonal degeneration by disrupting mitochondrial transport in AD [101]. Likewise, under physiological conditions, Cdk5 phosphorylates neurofilament heavy chain (NF-H) at KSP motifs in its tail domain, which results in axonal support and neurite outgrowth although it reduces axonal transport [102]. However, in several neurological diseases, NFs are hyperphosphorylated and aggregate in cell bodies. Many kinases, including deregulated GSK3β, PKA, MAPK, CaMK2, or Cdk5-p25, are known to substantially contribute to aberrant phosphorylation of NFs in diseased neurons [101]. Similarly, under physiological conditions, Cdk5-mediated direct or indirect regulation of CRMP2 is involved in axonal elongation or growth cone collapse. However, in AD, hyperphosphorylation of CRMP2 causes its aggregation and so contributes to NFT formation. Therefore, owing to hyperactivation and mislocalization, the Cdk5-p25 complex hyperphosphorylates several of its physiological targets, thereby leading to neuronal toxicity. Interestingly, hyperphosphorylation of CRMP2 in AD patients is observed prior to formation of amyloid plaques and NFTs, suggesting it might be early event in AD pathogenesis [103].
In the Niemann–Pick type C (NPC) mouse model, deregulated Cdk5-p25 hyperphosphorylates several neuronal cytoskeletal proteins, including neurofilaments, tau, and MAP2, at 4 weeks of age, the earliest time point studied here, resulting in the formation of axonal spheroids, suggesting a causal role of Cdk5 in NPC pathogenesis. The severity of these cytoskeletal abnormalities rapidly spread to the other parts of the brain resulting in neurodegeneration in NPC [104]. An environmental trigger for late-life Cdk5-mediated tau hyperphosphorylation may be early-life exposure to heavy metals, such as lead (Pb). Developmental (post-natal days 1–20) exposure to Pb in drinking water resulted at 24 months of age in greater levels of Cdk5 and tau protein and mRNA and of tau hyperphosphorylation in mouse brains. These changes did not occur in animals only exposed later in life to Pb (7–9 months age) [105], which is consistent with the previously proposed LEARn model for late-life neuropsychiatric disorders [106].
Conditions Caused by Axonal Transport Defects upon Cdk5 Deregulation
Failure of axonal transport is one of the crucial triggers in the onset and progression of several neurodegenerative disorders, including injury and motor neuron diseases [107, 108]. Defects in transportation often result in the accumulation of abnormal organelles (such as damaged mitochondria) or protein aggregates that cause swollen axons or spheroids [109]. In AD, hyperphosphorylation of tau and CRMP2 by Cdk5 not only lead to the formation of NFTs but also significantly impair axonal transport, causing neuronal death [110]. Similarly, deregulation of Cdk5 by ectopic expression of p25 results in increased pausing of mitochondria in neurons [111]. The resulting mitochondrial “traffic jam” causes a drop in ATP levels, resulting in synaptic dysfunction and ultimately neuronal death [112].
A recent study reported that Aβ inhibits kinesin-5 and so causes a reduced transport of neurotrophin and of neurotransmitter receptors to the cell surface, which results in neuronal death; however, a potential role of Cdk5 was not examined in this work [113]. As Cdk5 is known to phosphorylate kinesin-5 at Thr926, which enhances its MT-binding ability [83], it is likely that Cdk5 hyperactivation in Aβ-exposed neurons could result in hyperphosphorylation of kinesin-5, thereby completely halting axonal transport. Future studies are needed to unravel the underlying mechanism. In summary, while physiological Cdk5 activity is crucial for promoting axonal transport, deregulated Cdk5 elicits defects in axonal transport through the hyperphosphorylation of several of its substrates that are involved in axonal transport.
Conclusions
Research over the past two decades has uncovered numerous physiological and pathological roles of Cdk5 in various tissue types. Under physiological conditions, Cdk5-p35 acts as a major orchestrator of neuronal cytoskeletal dynamics. As a result, Cdk5 activity is essential for normal brain development during embryogenesis and, in adults, it is important for all aspects of synaptic signaling as well as for drug addiction, pain signaling, and long-term behavior changes. At the other end of the spectrum, deregulation of Cdk5 triggers a number of degradative pathways, which result in profound remodeling of neuronal cytoskeleton, loss of synapses, and ultimately neurodegeneration. In this context, it is worth noting that hyperactivation of Cdk5 not only results in the hyperphosphorylation of many of its physiological targets that are associated with the cytoskeleton but also in its mislocalization, which allows it to access and phosphorylate multiple non-physiological, cytoplasmic, and nuclear targets, thus causing further toxicity.
Consequently, Cdk5 is an attractive drug target for multiple neurological disorders. Although there are many known Cdk5 inhibitors that exhibit high potency, these suffer from poor selectivity and inhibit other Cdks with equal or higher potency. Therefore, despite their potential, Cdk5 inhibitors have thus far not been successful in clinical trials due to serious side effects. Given the pivotal contribution of Cdk5 in the brain, it would be beneficial to develop specific Cdk5 inhibitors or to engineer inhibitors that selectively abrogate the interactions of Cdk5 with p25, but not with p35. Such an approach would allow targeting the deleterious effects of Cdk5, while retaining its physiological and beneficial functions in the brain.
References
Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2(10):749–59. doi:10.1038/35096019
Shah K, Lahiri DK (2014) Cdk5 activity in the brain—multiple paths of regulation. J Cell Sci 127(Pt 11):2391–400. doi:10.1242/jcs.147553
Chang KH, Vincent F, Shah K (2012) Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J Cell Sci 125(Pt 21):5124–37. doi:10.1242/jcs.108183
Tsai LH, Delalle I, Caviness VS Jr, Chae T, Harlow E (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. 371(6496):419–23. doi:10.1038/371419a0
Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH. (1995) An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem. (45), 26897–903. doi:10.1074/jbc.270.45.26897
Brinkkoetter PT, Pippin JW, Shankland SJ (2010) Cyclin I-Cdk5 governs survival in post-mitotic cells. Cell Cycle 9(9):1729–31. doi:10.4161/cc.9.9.11471
Sun KH, Chang KH, Clawson S, Ghosh S, Mirzaei H, Regnier F, Shah K (2011) Glutathione-S-transferase P1 is a critical regulator of Cdk5 kinase activity. J Neurochem 118(5):902–14. doi:10.1111/j.1471-4159.2011.07343.x
Modi PK, Komaravelli N, Singh N, Sharma P (2012) Interplay between MEK-ERK signaling, cyclin D1, and cyclin-dependent kinase 5 regulates cell cycle reentry and apoptosis of neurons. Mol Biol Cell 23(18):3722–30. doi:10.1091/mbc.E12-02-0125
Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, Geng Y, Gawrzak S et al (2011) Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell 21(4):655–68. doi:10.1016/j.devcel.2011.08.009
Fu WY, Chen Y, Sahin M, Zhao XS, Shi L, Bikoff JB, Lai KO, Yung WH et al (2007) Cdk5 regulates EphA4-mediated dendritic spine retraction through an ephexin1-dependent mechanism. Nat Neurosci 10(1):67–76. doi:10.1038/nn1811
Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM et al (2000) Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 26(3):633–46. doi:10.1016/S0896-6273(00)81200-3
Kobayashi H, Saito T, Sato K, Furusawa K, Hosokawa T, Tsutsumi K, Asada A, Kamada S et al (2014) Phosphorylation of cyclin-dependent kinase 5 (Cdk5) at Tyr-15 is inhibited by Cdk5 activators and does not contribute to the activation of Cdk5. J Biol Chem 289(28):19627–36. doi:10.1074/jbc.M113.501148
Asada A, Yamamoto N, Gohda M, Saito T, Hayashi N, Hisanaga S (2008) Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cyclin-dependent kinase 5 complexes. J Neurochem 106(3):1325–36. doi:10.1111/j.1471-4159.2008.05500.x
McLinden KA, Trunova S, Giniger E (2012) At the fulcrum in health and disease: Cdk5 and the balancing acts of neuronal structure and physiology. Brain Disord Ther 2012(Suppl 1):001. doi:10.4172/2168-975X
Bibb JA, Chen J, Taylor JR, Svenningsson P, Nishi A, Snyder GL, Yan Z, Sagawa ZK et al (2001) Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature 410(6826):376–80. doi:10.4172/2168-975X
Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J (2002) Cyclin-dependent kinase 5 is required for associative learning. J Neurosci 22(9):3700–7. doi:10.3389/fnbeh.2013.00216
Takahashi S, Ohshima T, Cho A, Sreenath T, Iadarola MJ, Pant HC, Kim Y, Nairn AC et al (2005) Increased activity of cyclin-dependent kinase 5 leads to attenuation of cocaine-mediated dopamine signaling. Proc Natl Acad Sci U S A 102(5):1737–42. doi:10.1073/pnas.0409456102
Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM et al (2007) Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci 10(7):880–6. doi:10.1038/nn1914
Hisanaga S, Endo R (2010) Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J Neurochem 115(6):1309–21. doi:10.1111/j.1471-4159.2010.07050.x
Drerup JM, Hayashi K, Cui H, Mettlach GL, Long MA, Marvin M, Sun X, Goldberg MS et al (2010) Attention-deficit/hyperactivity phenotype in mice lacking the cyclin-dependent kinase 5 cofactor p35. Biol Psychiatry 68(12):1163–71. doi:10.1016/j.biopsych.2010.07.016
Patel LS, Wenzel HJ, Schwartzkroin PA (2004) Physiological and morphological characterization of dentate granule cells in the p35 knock-out mouse hippocampus: evidence for an epileptic circuit. J Neurosci 24(41):9005–14. doi:10.1523/JNEUROSCI.2943-04.2004
Engmann O, Hortobágyi T, Pidsley R, Troakes C, Bernstein HG, Kreutz MR, Mill J, Nikolic M et al (2011) Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain 134(Pt 8):2408–21. doi:10.1093/brain/awr155
Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH (2005) Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron 48(5):825–38. doi:10.1016/j.neuron.2005.10.033
Su SC, Tsai LH (2011) Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol 27:465–91. doi:10.1146/annurev-cellbio-092910-154023
Shukla V, Skuntz S, Pant HC (2012) Deregulated Cdk5 activity is involved in inducing Alzheimer's disease. Arch Med Res 43(8):655–62. doi:10.1016/j.arcmed.2012.10.015
Meyer DA, Torres-Altoro MI, Tan Z, Tozzi A, Di Filippo M, DiNapoli V, Plattner F, Kansy JW et al (2014) Ischemic stroke injury is mediated by aberrant Cdk5. J Neurosci 34(24):8259–67. doi:10.1523/JNEUROSCI.4368-13.2014
Sun KH, de Pablo Y, Vincent F, Johnson EO, Chavers AK, Shah K (2008) Novel genetic tools reveal Cdk5's major role in Golgi fragmentation in Alzheimer's disease. Mol Biol Cell 19(7):3052–69. doi:10.1091/mbc.E07-11-1106
Sun KH, de Pablo Y, Vincent F, Shah K (2008) Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J Neurochem 10:265–278. doi:10.1111/j.1471-4159.2008.05616.x
Sun KH, Lee HG, Smith MA, Shah K (2009) Direct and indirect roles of Cdk5 as an upstream regulator in the JNK cascade: relevance to neurotoxic insults in Alzheimer’s disease. Mol Biol Cell 20(21):4611–9. doi:10.1091/mbc.E09-05-0433
Chang KH, Pablo Y, Lee H, Lee H, Smith M, Shah K (2010) Cdk5 is a major regulator of p38 cascade: relevance to neurotoxicity in Alzheimer’s disease. J Neurochem 113(5):1221–9. doi:10.1111/j.1471-4159.2010.06687.x
Chang KH, Multani PS, Sun KH, Vincent F, de Pablo Y, Ghosh S, Gupta R, Lee HP et al (2011) Nuclear envelope dispersion triggered by deregulated Cdk5 precedes neuronal death. Mol Biol Cell 22(9):1452–62. doi:10.1091/mbc.E10-07-0654
Alvarez A, Muñoz JP, Maccioni RB (2001) A Cdk5-p35 stable complex is involved in the beta-amyloid-induced deregulation of Cdk5 activity in hippocampal neurons. Exp Cell Res 264(2):266–74. doi:10.1006/excr.2001.5152
Luo L (2002) Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol 18:601–35. doi:10.1146/annurev.cellbio.18.031802.150501
Poulain FE, Sobel A (2010) The microtubule network and neuronal morphogenesis. Dynamic and coordinated orchestration through multiple players. Mol Cell Neurosci 43:15–32. doi:10.1016/j.mcn.2009.07.012
Yuan A, Rao MV, Nixon RA, Veeranna (2012) Neurofilaments at a glance. J Cell Sci 125(Pt 14):3257–63. doi:10.1242/jcs.104729
Paglini G, Pigino G, Kunda P, Morfini G, Maccioni R, Quiroga S, Ferreira A, Cáceres A (1998) Evidence for the participation of the neuron-specific CDK5 activator P35 during laminin-enhanced axonal growth. J Neurosci 18(23):9858–69. doi:10.1016/S0960-9822(00)00487-5
Connell-Crowley L, Le Gall M, Vo DJ, Giniger E (2000) The cyclin-dependent kinase Cdk5 controls multiple aspects of axon patterning in vivo. Curr Biol 10(10):599–602. doi:10.1016/S0960-9822(00)00487-5
Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P (2003) HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J 22(5):1168–79. doi:10.1093/emboj/cdg115
North BJ, Marshall BL, Borra MT, Denu JM, Verdin E (2003) The human Sir2 ortholog, SIRT2, is an NAD + −dependent tubulin deacetylase. Mol Cell 11(2):437–44. doi:10.1016/S1097-2765(03)00038-8
Ip JP, Shi L, Chen Y, Itoh Y, Fu WY, Betz A, Yung WH, Gotoh Y et al (2011) α2-chimaerin controls neuronal migration and functioning of the cerebral cortex through CRMP-2. Nat Neurosci 15(1):39–47. doi:10.1038/nn.2972
Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K et al (2005) Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells 10(2):165–79. doi:10.1111/j.1365-2443.2005.00827.x
Yamashita N, Ohshima T, Nakamura F, Kolattukudy P, Honnorat J, Mikoshiba K, Goshima Y (2012) Phosphorylation of CRMP2 (collapsin response mediator protein 2) is involved in proper dendritic field organization. J Neurosci 32(4):1360–5. doi:10.1523/JNEUROSCI.5563-11.2012
Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R (2009) An atypical role for ollapsing response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem 284(45):31375–90. doi:10.1074/jbc.M109.009951
Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T et al (2004) Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J Neurosci 24(41):8994–9004. doi:10.1523/JNEUROSCI.3184-04.2004
Niisato E, Nagai J, Yamashita N, Nakamura F, Goshima Y, Ohshima T (2013) Phosphorylation of CRMP2 is involved in proper bifurcation of the apical dendrite of hippocampal CA1 pyramidal neurons. Dev Neurobiol 73(2):142–51. doi:10.1002/dneu.22048
Fang WQ, Ip JP, Li R, Ng YP, Lin SC, Chen Y, Fu AK, Ip NY (2011) Cdk5-mediated phosphorylation of Axin directs axon formation during cerebral cortex development. J Neurosci 31(38):13613–24. doi:10.1523/JNEUROSCI.3120-11.2011
Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST (2004) A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J 23(11):2235–45. doi:10.1038/sj.emboj.7600237
Jiang H, Guo W, Liang X, Rao Y (2005) Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. (1), 123–35. DOI 10.1016/j.cell.2004.12.033.
Chow HM, Guo D, Zhou JC, Zhang GY, Li HF, Herrup K, Zhang J (2014) CDK5 activator protein p25 preferentially binds and activates GSK3β. Proc Natl Acad Sci U S A 111(45):E4887–95. doi:10.1073/pnas.1402627111
Tanaka T, Serneo FF, Tseng HC, Kulkarni AB, Tsai LH, Gleeson JG (2004) Cdk5 phosphorylation of doublecortin ser297 regulates its effect on neuronal migration. Neuron 41(2):215–27. doi:10.1016/S0896-6273(03)00852-3
Hlavanda E, Klement E, Kókai E, Kovács J, Vincze O, Tökési N, Orosz F, Medzihradszky KF et al (2007) Phosphorylation blocks the activity of tubulin polymerization-promoting protein (TPPP): identification of sites targeted by different kinases. J Biol Chem 282(40):29531–9. doi:10.1074/jbc.M703466200
Kovács GG, László L, Kovács J, Jensen PH, Lindersson E, Botond G, Molnár T, Perczel A et al (2004) Natively unfolded tubulin polymerization promoting protein TPPP/p25 is a common marker of alpha-synucleinopathies. Neurobiol Dis 17(2):155–62. doi:10.1016/j.nbd.2004.06.006
Takahashi S, Saito T, Hisanaga S, Pant HC, Kulkarni AB (2003) Tau phosphorylation by cyclin-dependent kinase 5/p39 during brain development reduces its affinity for microtubules. J Biol Chem 278(12):10506–15. doi:10.1074/jbc.M211964200
Brion JP, Octave JN, Couck AM (1994) Distribution of the phosphorylated microtubule-associated protein tau in developing cortical neurons. Neuroscience 63(3):895–909. doi:10.1016/0306-4522(94)90533-9
Hayashi K, Pan Y, Shu H, Ohshima T, Kansy JW, White CL 3rd, Tamminga CA, Sobel A et al (2006) Phosphorylation of the tubulin-binding protein, stathmin, by Cdk5 and MAP kinases in the brain. J Neurochem 99(1):237–50. doi:10.1111/j.1471-4159.2006.04113.x
Uchida S, Martel G, Pavlowsky A, Takizawa S, Hevi C, Watanabe Y, Kandel ER, Alarcon JM et al (2014) Learning-induced and stathmin-dependent changes in microtubule stability are critical for memory and disrupted in ageing. Nat Commun 5:4389. doi:10.1038/ncomms5389
Hou Z, Li Q, He L, Lim HY, Fu X, Cheung NS, Qi DX, Qi RZ (2007) Microtubule association of the neuronal p35 activator of Cdk5. J Biol Chem 282(26):18666–70. doi:10.1074/jbc.C700052200
He L, Hou Z, Qi RZ (2008) Calmodulin binding and Cdk5 phosphorylation of p35 regulate its effect on microtubules. Biol Chem 283(19):13252–6. doi:10.1074/jbc.M706937200
Xie Z, Sanada K, Samuels BA, Shih H, Tsai LH (2003) Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell 114(4):469–82. doi:10.1016/S0092-8674(03)00605-6
Wang Q, Brandon NJ (2011) Regulation of the cytoskeleton by disrupted-in-schizophrenia 1 (DISC1). Mol Cell Neurosci 48(4):359–64. doi:10.1016/j.mcn.2011.06.004
Ishizuka K, Kamiya A, Oh EC, Kanki H, Seshadri S, Robinson JF, Murdoch H, Dunlop AJ et al (2011) DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 473(7345):92–6. doi:10.1038/nature09859
Sasaki S, Mori D, Toyo-oka K, Chen A, Garrett-Beal L, Muramatsu M, Miyagawa S, Hiraiwa N et al (2005) Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Mol Cell Biol 25:7812–7827. doi:10.1128/MCB.25.17.7812-7827.2005
Singh KK, Ge X, Mao Y, Drane L, Meletis K, Samuels BA, Tsai LH (2010) Dixdc1 is a critical regulator of DISC1 and embryonic cortical development. Neuron 67(1):33–48. doi:10.1016/j.neuron.2010.06.002
Takitoh T, Kumamoto K, Wang CC, Sato M, Toba S, Wynshaw-Boris A, Hirotsune S (2012) Activation of Aurora-A is essential for neuronal migration via modulation of microtubule organization. J Neurosci 32(32):11050–66. doi:10.1523/JNEUROSCI.5664-11.2012
Bond J, Woods CG (2006) Cytoskeletal genes regulating brain size. Curr Opin Cell Biol 18(1):95–101. doi:10.1016/j.ceb.2005.11.004
Rakic S, Kanatani S, Hunt D, Faux C, Cariboni A, Chiara F, Khan S, Wansbury O et al (2015) Cdk5 phosphorylation of ErbB4 is required for tangential migration of cortical interneurons. Cereb Cortex 25(4):991–1003. doi:10.1093/cercor/bht290
Bourne JN, Harris KM (2008) Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31:47–67. doi:10.1146/annurev.neuro.31.060407.125646
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. doi:10.1038/nature02617
Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional Plasticity. Nat Neurosci 7:1104–1112. doi:10.1038/nn1311
Mitsuyama F, Niimi G, Kato K, Hirosawa K, Mikoshiba K, Okuya M, Karagiozov K, Kato Y et al (2008) Redistribution of microtubules in dendrites of hippocampal CA1 neurons after tetanic stimulation during long-term potentiation. Ital J Anat Embryol 113:17–27. doi:10.1523/JNEUROSCI.2583-12.2013
Jaworski J, Kapitein LC, Gouveia SM, Dortland BR, Wulf PS, Grigoriev I, Camera P, Spangler SA et al (2009) Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61:85–100. doi:10.1016/j.neuron.2008.11.013
Hu X, Ballo L, Pietila L, Viesselmann C, Ballweg J, Lumbard D, Stevenson M, Merriam E et al (2011) BDNF-induced increase of PSD-95 in dendritic spines requires dynamic microtubule invasions. J Neurosci 31(43):15597–603. doi:10.1523/JNEUROSCI.2445-11.2011
Merriam EB, Millette M, Lumbard DC, Saengsawang W, Fothergill T, Hu X, Ferhat L, Dent EW (2013) Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J Neurosci 33(42):16471–82. doi:10.1523/JNEUROSCI.0661-13.2013
Worth DC, Daly CN, Geraldo S, Oozeer F, Gordon-Weeks PR (2013) Drebrin contains a cryptic F-actin-bundling activity regulated by Cdk5 phosphorylation. J Cell Biol 202(5):793–806. doi:10.1083/jcb.201303005
Dun XP, Bandeira de Lima T, Allen J, Geraldo S, Gordon-Weeks P, Chilton JK (2012) Drebrin controls neuronal migration through the formation and alignment of the leading process. Mol Cell Neurosci 49(3):341–50. doi:10.1016/j.mcn.2012.01.006
Ivanov A, Esclapez M, Ferhat L (2009) Role of drebrin A in dendritic spine plasticity and synaptic function: implications in neurological disorders. Commun Integr Biol 2(3):268–70. doi:10.1155/2012/474351
Shim KS, Lubec G (2002) Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer's disease and Down syndrome. Neurosci Lett 324(3):209–12. doi:10.1016/S0304-3940(02)00210-0
Geraldo S, Khanzada UK, Parsons M, Chilton JK, Gordon-Weeks PR (2008) Targeting of the F-actin-binding protein drebrin by the microtubule plus-tip protein EB3 is required for neuritogenesis. Nat Cell Biol 10(10):1181–9. doi:10.1038/ncb1778
Tanabe K, Yamazaki H, Inaguma Y, Asada A, Kimura T, Takahashi J, Taoka M, Ohshima T et al (2014) Phosphorylation of drebrin by cyclin-dependent kinase 5 and its role in neuronal migration. PLoS One 9(3):e92291. doi:10.1371/journal.pone.0092291
Hirokawa N, Niwa S, Tanaka Y (2010) Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68(4):610–38. doi:10.1016/j.neuron.2010.09.039
Xing BM, Yang YR, Du JX, Chen HJ, Qi C, Huang ZH, Zhang Y, Wang Y (2012) Cyclin-dependent kinase 5 controls TRPV1 membrane trafficking and the heat sensitivity of nociceptors through KIF13B. J Neurosci 32(42):14709–21. doi:10.1523/JNEUROSCI.1634-12.2012
Liu J, Du J, Yang Y, Wang Y. (2015) Phosphorylation of TRPV1 by cyclin-dependent kinase 5 promotes TRPV1 surface localization, leading to inflammatory thermal hyperalgesia. Exp Neurol. 273, 253--62. doi:10.1016/j.expneurol.2015.09.005
Kahn OI, Sharma V, González-Billault C, Baas PW (2015) Effects of kinesin-5 inhibition on dendritic architecture and microtubule organization. Mol Biol Cell 226(1):66–77. doi:10.1091/mbc.E14-08-1313
Myers KA, Baas PW (2007) Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J Cell Biol 178(6):1081–91. doi:10.1083/jcb.200702074
Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S (2000) A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 28(3):681–96. doi:10.1016/S0896-6273(00)00146-X
Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH et al (2003) 14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet 34(3):274–85. doi:10.1038/ng1169
Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH (2000) NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28(3):697–711. doi:10.1016/S0896-6273(00)00147-1
Pandey JP, Smith DS (2011) A Cdk5-dependent switch regulates Lis1/Ndel1/dynein-driven organelle transport in adult axons. J Neurosci 31(47):17207–19. doi:10.1523/JNEUROSCI.4108-11.2011
Benitez-King G, Ramírez-Rodríguez G, Ortíz L, Meza I (2004) The neuronal cytoskeleton as a potential therapeutical target in neurodegenerative diseases and schizophrenia. Curr Drug Targets CNS Neurol Disord 3(6):515–33. doi:10.2174/1568007043336761
Newey SE, Velamoor V, Govek EE, Van Aelst L (2005) Rho GTPases, dendritic structure, and mental retardation. J Neurobiol 64(1):58–74. doi:10.1002/neu.20153
Venturin M, Guarnieri P, Natacci F, Stabile M, Tenconi R, Clementi M, Hernandez C, Thompson P et al (2004) Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J Med Genet 41:35–41. doi:10.1111/j.1529-8817.2005.00203.x
Luo S, Vacher C, Davies JE, Rubinsztein DC (2005) Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol 169(4):647–56. doi:10.1083/jcb.200412071
Paoletti P, Vila I, Rifé M, Lizcano JM, Alberch J, Ginés S (2008) Dopaminergic and glutamatergic signaling crosstalk in Huntington's disease neurodegeneration: the role of p25/cyclin-dependent kinase 5. J Neurosci 28(40):10090–101. doi:10.1523/JNEUROSCI.3237-08.2008
Kaminosono S, Saito T, Oyama F, Ohshima T, Asada A, Nagai Y, Nukina N, Hisanaga S (2008) Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J Neurosci 28(35):8747–55. doi:10.1523/JNEUROSCI.0973-08.2008
Muchowski PJ, Ning K, D'Souza-Schorey C, Fields S (2002) Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proc Natl Acad Sci U S A 99(2):727–32. doi:10.1073/pnas.022628699
Anne SL, Saudou F, Humbert S (2007) Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J Neurosci 27(27):7318–28. doi:10.1523/JNEUROSCI.1831-07.2007
Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40(3):471–83. doi:10.1016/S0896-6273(03)00627-5
Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A et al (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A 97(6):2910–5. doi:10.1073/pnas.040577797
Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA (2011) S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A 108(34):14330–5. doi:10.1073/pnas.1105172108
Meyer DA, Richer E, Benkovic SA, Hayashi K, Kansy JW, Hale CF, Moy LY, Kim Y et al (2008) Striatal dysregulation of Cdk5 alters locomotor responses to cocaine, motor learning, and dendritic morphology. Proc Natl Acad Sci U S A 105(47):18561–6. doi:10.1073/pnas.0806078105
Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M et al (2012) Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci 32(7):2430–41. doi:10.1523/JNEUROSCI.5927-11.2012
Shea TB, Yabe JT, Ortiz D, Pimenta A, Loomis P, Goldman RD, Amin N, Pant HC (2004) Cdk5 regulates axonal transport and phosphorylation of neurofilaments in cultured neurons. J Cell Sci 117(Pt 6):933–41. doi:10.1242/jcs.00785
Cole AR, Noble W, van Aalten L, Plattner F, Meimaridou R, Hogan D, Taylor M, LaFrancois J et al (2007) Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J Neurochem 103(3):1132–44. doi:10.1111/j.1471-4159.2007.04829.x
Bu B, Li J, Davies P, Vincent I (2002) Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci 22(15):6515–25. doi:10.1007/s11596-009-0312-0
Bihaqi SW, Bahmani A, Adem A, Zawia NH (2014) Infantile postnatal exposure to lead (Pb) enhances tau expression in the cerebral cortex of aged mice: relevance to AD. Neurotoxicology 44:114–20. doi:10.1016/j.neuro.2014.06.008
Lahiri DK, Maloney B, Zawia NH (2009) The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases. Mol Psychiatry 14(11):992–1003. doi:10.1038/mp.2009.82
El-Kadi AM, Soura V, Hafezparast M (2007) Defective axonal transport in motor neuron disease. J Neurosci Res 85(12):2557–66. doi:10.1002/jnr.21188
Vicario-Orri E, Opazo CM, Muñoz FJ (2015) The pathophysiology of axonal transport in Alzheimer's disease. J Alzheimers Dis 43(4):1097–113. doi:10.3233/JAD-141080
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P et al (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307(5713):1282–8. doi:10.1093/hmg/ddt313
Hensley K, Venkova K, Christov A, Gunning W, Park J (2011) Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol 43(3):180–91. doi:10.1007/s12035-011-8166-4
Morel M, Authelet M, Dedecker R, Brion JP (2010) Glycogen synthase kinase-3beta and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience 167(4):1044–56. doi:10.1016/j.neuroscience.2010.02.077
Whiteman IT, Gervasio OL, Cullen KM, Guillemin GJ, Jeong EV, Witting PK, Antao ST, Minamide LS et al (2009) Activated actin-depolymerizing factor/cofilin sequesters phosphorylated microtubule-associated protein during the assembly of alzheimer-like neuritic cytoskeletal striations. J Neurosci 29(41):12994–3005. doi:10.1523/JNEUROSCI.3531-09.2009
Ari C, Borysov SI, Wu J, Padmanabhan J, Potter H (2014) Alzheimer amyloid beta inhibition of Eg5/kinesin 5 reduces neurotrophin and/or transmitter receptor function. Neurobiol Aging 35:1839–49. doi:10.1016/j.neurobiolaging.2014.02.006
Acknowledgments
This work was supported by grant from the National Institutes of Health (NIAR21AG 47447) to KS and DKL. The authors sincerely thank Bryan Maloney (IUPUI) for his critical reading and helpful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shah, K., Lahiri, D.K. A Tale of the Good and Bad: Remodeling of the Microtubule Network in the Brain by Cdk5. Mol Neurobiol 54, 2255–2268 (2017). https://doi.org/10.1007/s12035-016-9792-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9792-7