
Abstract
Amiodarone (AM) is the most effective antiarrhythmic agent currently available. However, clinical application of AM is limited by its serious toxic adverse effects including optic neuropathy. The purpose of this study was to explore the effects of AM and to assess if insulin-like growth factor-1 (IGF-1) could protect retinal neuronal cells from AM-induced apoptosis, and to determine the molecular mechanisms underlying the effects. Accordingly, the phosphorylation/activation of Akt and FoxO3a were analyzed by Western blot while the possible pathways involved in the protection of IGF-1 were investigated by application of various pathway inhibitors. The full electroretinogram (FERG) was used to evaluate in vivo effect of AM and IGF-1 on rat retinal physiological functions. Our results showed that AM concentration dependently caused an apoptosis of RGC-5 cells, while IGF-1 protected RGC-5 cells against this effect by AM. The protective effect of IGF-1 was reversed by PI3K inhibitors LY294002 and wortmannin as well as the Akt inhibitor VIII. AM decreased p-Akt and p-FoxO3a while increased the nuclear localization of FoxO3a in the RGC-5 cells. IGF-1 reversed the effect of AM on the p-Akt and p-FoxO3a and the nuclear translocation of FoxO3a. Similar results were obtained in primary cultured retinal ganglia cells. Furthermore, FERG in vivo recording in rats showed that AM decreased a-wave and b-wave of FERG while IGF-1 reversed the effects of AM. These data show that AM induced apoptosis of retinal neuronal cells via inhibiting the PI3K/Akt/FoxO3a pathway while IGF-1 protected RGC-5 cells against AM-induced cell apoptosis by stimulating this pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amiodarone (AM) is one of the most frequently prescribed and efficacious drugs for both atrial fibrillation and ventricular tachyarrhythmias. Amiodarone is superior to other antiarrhythmic agents, including sotalol and propafenone, for the treatment of atrial fibrillation [1–3]. The clinical efficacy of amiodarone has been limited by its toxicity, with almost 50 % of long-term patients having to cease treatment with the drug. Amiodarone toxicity has high incidence rate and is clinically unpredictable; it affects the lungs, thyroid, nervous system, liver, skin, and eyes [4–6]. The toxic effects of amiodarone are related to the cumulative dose administered. The affected organs share a common feature: the accumulation of amiodarone and/or its toxic metabolite (i.e., desethylamiodarone) in the tissue. Both amiodarone and desethylamiodarone accumulate in the tissues due to their amphiphilic nature [7, 8].
Amiodarone-induced optic neuropathy is of great concern and is a leading cause of medication discontinuation. Many of the patients using amiodarone develop corneal microdeposits and related halo vision and suffer progressive visual loss from amiodarone-induced optic neuropathy, whereas some patients suffer from permanent blindness [9–15].
Several experimental studies have suggested that amiodarone induces neurotoxicity by blocking Na+, K+, or Ca2+ channels [16] and increases the levels of L-dopa and dopamine, shifting the ratio of neuroactive amino acids toward inhibitory transmitters [17]. The neurotoxicity is also related to the inhibition of protein kinase C (PKC) activity and calmodulin-regulated enzymes, modulating a variety of intracellular and extracellular signals across the neuronal membrane [18, 19]. Amiodarone is reported to cause apoptosis and necrosis in pulmonary epithelial cells. The apoptosis is accompanied with the activation of caspase-2, caspase-3, and caspase-8 and is reversed by caspase inhibitors [20]. However, the precise mechanism(s) of amiodarone-induced optic neuropathy is clearly not fully characterized [21–23].
RGC-5 cell is a clonal, retinal, neuronal cell line expressing characteristic retinal markers, including Thy-1, Brn-3c, and NMDA receptors [24–26]. The retinal ganglion cells (RGCs) are severely damaged in optic neuropathies, including glaucoma where a gradual but irreversible process of visual loss and blindness occurs in association with retinal ganglion degeneration. Retinal neuronal cells like RGCs are related to many eye diseases, and RGC-5 has been established as a model to study cellular and molecular mechanisms of glaucoma as a self-proliferative and morphologically undifferentiated in vitro system [24–26].
Insulin-like growth factor-1 (IGF-1) is a polypeptide growth factor similar to insulin in structure and function. IGF-1 plays roles in mitogenic, metabolic, and growth-stimulating activities in many cells and tissues, including the retina [27, 28]. IGF-1 is the major mediator of growth hormone activity in humans. We have previously reported that IGF-1 promotes the survival of various cell types in which cell death is initiated by serum deprivation. The cellular protective action of IGF-1 involves the PI3K/Akt signaling pathway, although the full characterization including upstream mediators and downstream effectors and targets have not been fully elucidated [29–33].
Several experimental studies have shown that IGF-1 protects RGCs from death and promotes the regeneration of axons after optic nerve injury in adult rats [34] and goldfish [35]. These data strongly indicate that IGF-1 is one of the most important molecules for regulating the regeneration of RGCs following damage to the optic nerve. It is possible that IGF-1 may also exert neuroprotective effects on the survival of RGCs. However, the cellular mechanism of amiodarone-induced retinal neuronal apoptosis and the impact of IGF-1 are unknown.
The biological actions of IGF-1 are mediated at least in part by type I IGF receptors. Binding of IGF-1 to this receptor initiates receptor autophosphorylation which in turn activates its intrinsic tyrosine kinase activity, leading to phosphorylation of several downstream intracellular substrates such as insulin receptor substrate-1 followed by activation of various signaling pathways, including the mitogen-activated protein (MAP) kinase (also called extracellular signal-regulated kinase (ERK)) and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway in various cell types [32, 33, 36–41]. The survival-promoting effects of IGF-1 are mediated, at least in part, through the activation of the PI3K/Akt pathway [33, 37].
Here, we report that amiodarone induced apoptosis of RGC-5 neuronal cells via inhibiting the PI3K/Akt/FoxO3a pathway while IGF-1 protected RGC-5 cells against amiodarone-induced apoptosis by stimulating this pathway.
Materials and Methods
Materials
Amiodarone was from Sanofi Winthrop Industrie (Ambarès-et-Lagrave, France). IGF-1, phosphate-buffered saline solution, and dichlorofluorescein diacetate (DCFH-DA) kits were purchased from Sigma (St. Louis, MO, USA); LY294002, PD160316, JNK inhibitor, and PD98059 were from Calbiochem (La Jolla, CA, USA). Lipofectamine 2000 and all cell culture reagents were purchased from Invitrogen (Carlsbad, CA, USA). Anti-phospho-FoxO3a and anti-FoxO3a were obtained from Signalway (Pearland, TX) via Seajet Scientific (Beijing, China). Anti-phospho-AktThr473 and anti-phospho-ERK were from Cell Signaling (Beverly, MA, USA). All secondary antibodies conjugated with HRP were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Methyl thiazolyl tetrazolium (MTT), Hoechst 33342, and all other reagents were from Sigma Chemical (St. Louis, MO) or Fisher Scientific (Nepean, ON). The GFP-N1 and FoxO3a-GFP plasmids were kindly provided by Marten P. Smidt (Rudolf Magnus Institute of Neuroscience, Department of Pharmacology and Anatomy, University Medical Centre Utrecht, The Netherlands).
Cell Culture
The retinal neuronal cell line (RGC-5) was maintained in an RPMI-1640 medium (Gibco BRL, Grand Island, NY) supplemented with 10 % (v/v) fetal bovine serum (FBS), streptomycin (100 μg/ml), and penicillin (100 μg/ml) and incubated at 37 °C in a 5 % CO2 humidified atmosphere. Culture media were replaced twice a week with fresh medium. Stock cultures were routinely subcultured weekly at a 1:5 ratio. The RGC-5 cells used in the experiments were 2–3 passages postthawed to minimize the variability; the cells grew well and had normal morphology. The density of RGC-5 cells was approximately 80 % before the treatments.
Primary cultured RGCs were prepared according to the method described previously, with minor modifications [28, 29]. All of the procedures described in this study were performed in accordance with the Guide for the Care and Use of Laboratory Animals—Chinese Version (1996). Briefly, neonatal Sprague Dawley rats were killed by decapitation, and their eyes were rapidly removed and immersed in a calcium- and magnesium-free (CMF) salt solution (0.1 M Dulbecco’s phosphate-buffered saline (PBS); calcium- and magnesium-free; Gibco, Grand Island, NY). Approximately 30 eyes were harvested for each experiment. The retinas were rapidly isolated and incubated at 37 °C for 25 min in CMF containing 0.1 % trypsin. Then, the cells were incubated for 5 min with a mouse antimacrophage antibody. The cell suspensions were then incubated for 30 min on a petri dish coated with a goat anti-mouse immunoglobulin G (H + L chain) antibody. Suspensions containing cells that did not adhere to the petri dish were harvested and incubated for 1 h in a dish coated with an anti-Thy-1.1 antibody. The cells that adhered to the dish were then trypsinized (0.1 % trypsin for 10 min), after which they were diluted to 1 × 105 cells/ml and placed on dishes or glass coverslips that had previously been coated with 50 mg/ml poly-l-ornithine. Cells were incubated in a serum-free culture medium, which was prepared using a B27-supplemented Neurobasal medium. The cultures were maintained in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C, and the medium was changed every 3 days. Cells were used after 5 days.
Treatments
To study the effect of amiodarone on the viability of RGC-5 cells, cells were treated with various concentrations of amiodarone (0 to 10 μM). To evaluate toxicity of amiodarone, cell viability was measured by MTT assay while the phosphorylation of signaling proteins was determined by Western blotting. To investigate the protective effects of IGF-1, cells pretreated with IGF-1 were treated with amiodarone and then the cell viability and the phosphorylation of signaling proteins were determined. To examine the involvement of different signaling pathways, cells were treated with wortmannin (100 nM, 30 min), LY294002 (30 μM, 30 min), Akt inhibitor VIII (10 μM, 30 min), PD160316 (10 μM, 30 min), and PD98059 (25 μM, 30 min) before the treatment of amiodarone and IGF-1and then the cell viability and the phosphorylation of signaling proteins were determined. All experiments were carried out independently at least five times.
Cell Viability Assay
Before each experiment, cells were detached using 5 mM EDTA in PBS and seeded in 96-well plates (coated with 10 μg/ml poly-d-lysine) at a density of 4–8 × 105 cells/well in 1 % serum medium for 24 h. The culture medium was replaced with RPMI-1640 1 h before the experiments. Amiodarone was dissolved initially in dimethyl sulfoxide (DMSO) as a stock solution and then in culture media (final concentration of DMSO was 0.1 %, a concentration which has no effect in any assay). MTT assay or cell counting was performed 24 h after various treatments discussed above. For the MTT assay, cells were returned to the incubator for another 4-h period after replacement of the medium with 0.5 mg/ml MTT in RPMI-1640. Cells and MTT formazan crystals were then solubilized by trituration in a solution of isopropanol/HCl (0.1 N), and the survival profile of these cells was quantified by spectrophotometrically measuring the plate at 570 nM. Assays were repeated at least five times in quadruplicate. For cell counting, the number of surviving cells was estimated as the average of eight arbitrarily selected fields (0.3 mm2 each) by counting the cells under a light microscope. The mean of seven or eight culture dishes was determined.
Detection of Apoptotic Nuclei by Hoechst 33342 Staining
After various treatments, RGC-5 cells were fixed in 4 % paraformaldehyde in 0.1 M phosphate buffer (PB) for 20 min. Nonspecific binding was blocked using 5 % normal goat serum (Vector Laboratories, Burlingame, CA) in 0.01 M PBS containing 0.3 % Triton X-100 (PBS + T). Cells were washed twice with PBS and incubated with 5 μg/ml Hoechst 33342 in PBS for 10 min at room temperature. Chromatin staining pattern was then analyzed for individual cells by fluorescence microscopy.
FoxO3a Translocation Studies in RGC-5 Cells
RGC-5 cells were plated on 96-well cell culture plates coated with poly-d-lysine and fed with RPMI-1640 medium supplemented with 10 % FBS plus antibiotics in 5 % CO2 at 37 °C. The following day, cells were transfected with 0.8 μg of plasmid DNA (GFP-N1 or GFP-FoxO3a) with Lipofectamine 2000 for 4–6 h. Then, media were replaced with RPMI-1640 plus 5 % serum for 24 h. In the study of translocation, the culture medium was replaced with RPMI-1640 plus 1 % serum and treated with amiodarone (3 μM) or amiodarone (3 μM) plus 100 ng/ml IGF-1 for 30 to 60 min. Cells were fixed and stained with Hoechst 33342 to visualize the nucleus. The subcellular localization of FoxO3a was determined by fluorescence microscopy. For quantitative analysis of the subcellular localization of FoxO3a, the percentage of cells showing the majority of fluorescence in each compartment (nucleus or cytoplasm) of a cell was counted. A total of 400 cells were counted per well.
Western Blotting
Western blotting was performed according to protocols routinely used in our laboratory [42–44]. In brief, treated cells from different experimental conditions were rinsed once with ice-cold PBS and lysed in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1 % Igepal CA-630, 1 % sodium dodecyl sulfate (SDS), 50 mM NaF, 1 mM NaVO3, 5 mM phenylmethysulfonyl fluoride, 10 μg/ml leupeptin (Sigma), and 50 μg/ml aprotinin (Sigma)) or 2× sample buffer (final concentration of 62.5 mM Tris-HCl, pH 6.8, 2 % (w/v) SDS, 10 % glycerol, 50 mM dithiothreitol, and 0.1 % (w/v) bromophenol blue). Samples with equal amounts of protein were then separated by SDS polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membranes, then probed with specific antibodies, and visualized using enhanced chemiluminescence kit according to the manufacturer’s instructions. Image J was used as the image analyzer to carry out Western blot analysis.
FERG Test
Thirty Sprague Dawley rats were randomly assigned to three groups: group 1 was normal control group, group 2 was AM (1 μM) group, and group 3 was IGF-1 (100 ng/ml) + AM (1 μM) group; group 1 was vitreous injected with normal saline 5 μl, group 2 was vitreous injected with AM (1 μM) 2.5 μl + normal saline 2.5 μl, and group 3 was vitreous injected with IGF-1 (100 ng/ml) 2.5 μl + AM (1 μM) 2.5 μl, respectively. After above treatments for 24 h, full electroretinogram (FERG) studies were performed.
FERG was monitored by Roland RETI-port visual electric physiological system (Roland Consult, Germany). The experiment was performed under dim red illumination; FERG was conducted after 60 min of dark adaptation. Rats were anesthetized with 10 % chloral hydrate. The cornea was anesthetized with Pontocaine (0.5 %), and both pupils were dilated with compound tropicamide eye drops. A circle silver chloride electrode was placed on the center of the cornea. A reference electrode was placed in the mouth, and a grounding electrode was placed subcutaneous of the tail. Rats were kept warm during and after the procedure. All procedures were repeated for five times.
The average amplitudes of a- and b-waves were calculated. The amplitude of the a-wave was measured from the baseline to the bottom of the a-wave, and the amplitude of the b-wave was measured from the bottom of the a-wave to the peak of the b-wave.
Statistical Analysis
All statistical analysis was performed using the SPSS 13.0 statistical package. Data were presented as the mean ± SEM. Statistical analysis for multiple comparisons was performed by analysis of variance (one-way ANOVA) with Kruskal-Wallis test; the level of statistical significance is defined as p < 0.05.
Results
Amiodarone Caused Cell Death in RGC-5 Cells While IGF-1 Reversed the Effect of Amiodarone by Counter Regulation of the PI3K/Akt Pathway
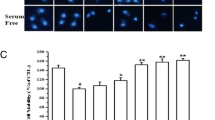
To establish a cellular model of amiodarone toxicity for mechanistic studies, we initially examined the effect of amiodarone on the viability of RGC-5 cells. Amiodarone induced cell death in a concentration-dependent manner in RGC-5 cells grown in a serum-free medium (Fig. 1a). Amiodarone induced cell death in RGC-5 cells at submicromolar concentrations and induced cellular apoptosis at concentrations as low as 0.5 μM, with approximately 50 % of the cells left viable at 3 μM. We thus selected this concentration of amiodarone (3 μM) for subsequent studies (Fig. 1a–c).

Amiodarone concentration dependently induced cell death in RGC-5 cells. a RGC-5 cells were treated with different concentrations of amiodarone for 24 h and the viability of cells was determined using the MTT assay. b Cellular apoptosis was evaluated using Hoechst 33342 staining at 24 h. c Representative photomicrographs (×100) of cells treated with amiodarone for 8 h. Nuclear condensation and fragmentation are seen at higher concentrations (arrows). The data are expressed as the mean ± SEM of results obtained from five independent experiments. *p < 0.05, **p < 0.01
We then examined the effect of IGF-1 in preventing amiodarone-induced cell death. IGF-1, in a concentration-dependent manner, protected RGC-5 cells against cell death induced by amiodarone (Fig. 2). The protective action of IGF-1 against the apoptotic action of amiodarone was observed at concentrations of 30 ng/ml with maximal protection at 100 ng/ml (Fig. 2). The protective effect of IGF-1 against amiodarone-induced cell death was further explored by Hoechst staining. Figure 3 showed that amiodarone triggered apoptotic-like cell death which was ameliorated by pretreatment of IGF-1.

Effects of IGF-1 on amiodarone-induced cell death in RGC-5 cells. RGC-5 cells were exposed to amiodarone (3 μM) for 24 h in the absence or presence of various concentration of IGF-1. Cell viability was determined using the MTT assay. Results are shown as the mean ± SEM and represent five independent experiments. *p < 0.05 compared with control, #p < 0.05 compared with amiodarone, ##p < 0.01 compared with amiodarone

Effects of IGF-1 on apoptosis of RGC-5 cells induced by amiodarone. Cells pretreated with or without IGF-1 were incubated with amiodarone. Apoptosis of cells was assessed by staining with Hoechst 33342. a Control. b Amiodarone (3 μM). c Amiodarone (3 μM) plus IGF-1 (100 ng/ml). Results are mean ± SEM and represent assays from at least five independent experiments. **p < 0.01 versus control, ##p < 0.01 versus amiodarone
We then used a pharmacological approach to investigate the role of various signaling pathways in mediating the protective effect of IGF-1 on amiodarone-induced toxicity (apoptosis). The protective effect of IGF-1 against cell death induced by amiodarone was specifically inhibited by pretreatment of the cells with LY294002 (30 μM), a PI3K inhibitor (Fig. 4). In contrast, PD98059 (25 μM), an upstream blocker of ERK1/2, and PD160316 (10 μM), a p38 MAPK inhibitor, had no effect (Fig. 4). These results suggested that the PI3K/Akt pathway is involved in the protective effect of IGF-1.

LY294002, a PI3K inhibitor, attenuated the protective effect of IGF-1 on amiodarone-induced apoptosis of RGC-5 cells. RGC-5 cells were preincubated with PI3K inhibitor (LY294002, concentration shown in the figure), ERK pathway inhibitor (PD98059), or p38 MAP kinase inhibitor (PD160316) then treated with IGF-1 and amiodarone for 24 h as described in “Materials and Methods.” Cell viability was determined by the MTT assay. Data are expressed as a percentage of the corresponding control value which was set at 100 %. Results are shown as the mean ± SEM and represent assays from five independent experiments
To further investigate the role of the PI3K/Akt signaling pathway in IGF-1-mediated cellular protection, RGC-5 cells were pretreated with PI3K inhibitors LY294002 (30 μM) and wortmannin (100 nM) or the Akt kinase-specific inhibitor Akt inhibitor VIII (10 μM) and then the cells were exposed to amiodarone (3 μM) in the presence or absence of IGF-1 (100 ng/ml) for 40 min and cell viability was determined. Pretreatment of RGCs with LY294002, wortmannin, or Akt inhibitor VIII blocked the protective effects of IGF-1 (Fig. 5a). These results confirmed the role of PI3K and Akt, a key downstream target kinase of PI3K, in the protective effect of IGF-1. Importantly, the above results were also verified in primary cultured RGCs (Fig. 5b). Thus, in primary cultured RGC neurons, amiodarone decreased the cell viability while IGF-1 blocked the effect of amiodarone via the PI3K/Akt pathway.

Effects of PI3K and Akt inhibitors on the protective effect of IGF-1 on amiodarone-induced apoptosis of RGC-5 cells and primary cultured RGCs. a RGC-5 cells pretreated with the PI3K inhibitor LY294002 or wortmannin or the Akt inhibitor (Akt inhibitor VIII) were treated with IGF-1 and amiodarone for 24 h. Cell viability was determined by the MTT assay. b Primary cultured RGCs (see “Materials and Methods” for details) were pretreated with LY294002, wortmannin, or Akt inhibitor VIII, followed by treatment with IGF-1 and amiodarone for 24 h, cell viability was measured by the MTT assay. Data are expressed as a percentage of the corresponding control, which was set at 100 %. Results are shown as the mean ± SD and represent assays from five independent experiments. *p < 0.05 compared with amiodarone, #p < 0.05 compared with amiodarone plus IGF-1
IGF-1 Stimulated the Phosphorylation of Akt and FoxO3a in RGC-5 Cells and Reversed the Inhibitory Effects of Amiodarone on Phosphorylation of These Proteins
To investigate the role of PI3K/Akt/FoxO3a in modulating the effects of amiodarone and IGF-1 on the survival of RGC-5 cells, the activation/phosphorylation of Akt and FoxO3a in RGC-5 cell was studied by Western blotting after exposure to amiodarone and IGF-1. Amiodarone decreased the phosphorylation of FoxO3a and Akt in a concentration and time-dependent manner in RGC-5 cells (Fig. 6a–d). Inhibition of the phosphorylation of Akt and FoxO3a by amiodarone was apparent at an amiodarone 3 μM and reached maximal effect at 10 μM, the highest concentration tested (Fig. 6a, c, and d). Amiodarone had a biphasic time-dependent effect—at early time points (up to 3–6 h), phosphorylation was increased, followed by almost total elimination of the phosphorylation of Akt and FoxO3a after a 24-h treatment (Fig. 6b, e, and f). These data showed that amiodarone treatment at 3 μM for 24 h essentially abolished the phosphorylation of Akt and FoxO3a in RGCs.

The effects of amiodarone on the phosphorylation status of Akt and FoxO3a. RGC-5 cells were treated with various concentrations of amiodarone for 24 h (a) or treated with amiodarone (3 μM) for different times (b), and the phosphorylation of Akt and FoxO3a was analyzed by immunoblotting. c–f Quantification of the relative levels of p-Akt and p-FoxO3a versus β-actin in each sample was determined by densitometry of the blots. Densitometric analysis of the immunoblot was expressed as a percentage of control. Results are shown as the mean ± SEM and represent five independent experiments. *p < 0.05 compared with control
We then examined the effect of IGF-1 on the phosphorylation profile of Akt and FoxO3a with Western blotting in control and amiodarone-exposed RGC-5 cells. IGF-1concentration dependently increased the phosphorylated FoxO3a and Akt in RGC-5 cells, and the maximal effect was observed with 100 ng/ml (Fig. 7a, c, and d). Then, the cells were pretreated with amiodarone (3 μM) and then the actions of IGF-1 in these RGCs were determined. The dose-dependent stimulatory effect of IGF-1 on the phosphorylation of both FoxO3a and Akt was persevered in RGCs which had been pretreated with amiodarone (Fig. 7b, e, and f). Thus, amiodarone decreased the phosphorylation of Akt and FoxO3a in RGC-5 cells while IGF-1 treatment could overcome the effects of amiodarone.

Effects of IGF-1 on the inhibitory effect of amiodarone on the phosphorylation of Akt and FoxO3a. a RGC-5 cells were treated with IGF-1 in different concentrations and b RGC-5 cells were pretreated with various concentration of IGF-1 and then were incubated with amiodarone (3 μM) for 24 h. Expression level of phosphorylated Akt and FoxO3a was analyzed by immunoblotting. c–f Relative levels of p-FoxO3a and p-Akt versus β-actin in each sample were determined by densitometry of the blots, densitometric analysis of the immunoblot was expressed as a percentage of control. Results are shown as the mean ± SEM and represent five independent experiments. *p < 0.05 compared with control, #p < 0.05 compared with amiodarone
To further investigate the role of the PI3K/Akt pathway in the cell-protective effects of IGF-1 in amiodarone-treated RGCs, we again used pharmacological inhibitors of selected pathways. RGCs pretreated with LY294002 (the PI3K inhibitor) or PD98059 (an upstream blocker of ERK1/2) were treated with amiodarone with/without IGF-1, and the phosphorylation of Akt and ERK1/2 was determined. Amiodarone inhibited the phosphorylation of Akt, and this was reversed by the action of IGF-1 (Fig. 8a, b). The effect of IGF-1 was blocked by LY294002 but not by PD98059 (Fig. 8a, b). A similar response profile was observed for the effects of the above agents on the phosphorylation of FoxO3a (data not shown). These data suggest that the PI3K pathway and not the ERK1/2 pathway is mediating the cellular effects of IGF-1.

Effects of LY294002 on the phosphorylation of Akt modulated by IGF-1 and amiodarone in RGC-5 cells. RGC-5 cells pretreated with LY294002 or PD98059 were treated with amiodarone or IGF-1 (a); the phosphorylation of Akt and ERK was analyzed by immunoblotting. b–c Densitometric analysis of the immunoblot was expressed as a percentage of control. Results are shown as the mean ± SEM and represent five independent experiments. **p < 0.01 compared with control, ##p < 0.01 compared with amiodarone
Amiodarone Modulated the Cellular Distribution of FoxO3a in RGC-5 Cells While IGF-1 Reversed Its Effect
The functions of the FoxO3a transcription factor are regulated by its phosphorylation status and cellular localization. Phosphorylation of FoxO3a induced by Akt leads to its localization in the cytosol and inhibition of its functions which include an apoptotic effect in neuronal cells. In contrast, dephosphorization of FoxO3a leads to the translocation to the nucleus and the initiation of an apoptotic response [47, 48]. Thus, we investigated the effects of amiodarone and IGF-1 on the subcellular distribution and localization of FoxO3a following treatments with these agents. RGC-5 cells were transfected with GFP-FoxO3a, and 24 h after transfection, the cells were treated with amiodarone alone or amiodarone and IGF-1 or amiodarone plus LY294002 and IGF-1. Amiodarone treatment induced a translocation of FoxO3a to the nucleus of RGCs (Fig. 9b compared to Fig. 9a and quantitated in Fig. 9d). In RGCs treated with both amiodarone and IGF-1, the action of amiodarone to stimulate translocation of FoxO3a to the nucleus (where it would cause apoptosis) was prevented by IGF-1 (Fig. 9c compared to Fig. 9b and quantitated in Fig. 9d).

The effect of amiodarone or IGF-1 on nuclear translocation of FoxO3a in RGC-5 cells. RGC-5 cells transfected with FoxO3a were cultured with 1 % FBS DMEM only (a) or treated with amiodarone (3 μM) (b) or treated with amiodarone (3 μM) and IGF-1 (100 ng/ml) (c). Subcellular localization of FoxO3a was determined as described in “Materials and Methods.” Densitometry analysis of the nuclear translocation of FoxO3a was expressed as a percentage of total cells. Results are shown as the mean ± SEM and represent five independent experiments
IGF-1 Showed a Protective Effect and Reversal of the Effects of Amiodarone on Entoretina Function Detected by FERG
To evaluate the retinal function among groups, FERG was performed after 24 h. Alterations in FERG were analyzed as shown in Fig. 10a–c. In AM (1 μM) group, b-wave amplitudes of max reaction in FERG were significantly decreased compared to the control group. There is also remarkable reduction in a-wave amplitudes of max reaction compared to the control group. Other reaction in FERG showed no statistical differences among groups. The a- and b-wave amplitudes of max reaction amplitude in FERG were all highly enhanced in the IGF-1 + AM treated group, compared to the AM group. This result demonstrated that IGF-1 had a protective effect on entoretina function after AM treated.

IGF-1 reversed the effects of amiodarone on entoretina functions detected by FERG. Rats were vitreous injected with normal saline 5 μl, AM (1 μM) 2.5 μl + normal saline 2.5 μl, and IGF-1 (100 ng/ml) 2.5 μl + AM (1 μM) 2.5 μl, respectively, and full electroretinogram (FERG) was used to investigate the effect of IGF-1 or amiodarone on rat eye entoretina functions as described in “Materials and Methods.” Representative of each group in max reaction (a) was shown. Results are shown as the mean ± SEM and represent five independent experiments. ##p < 0.01 compared with control, **p < 0.01 compared with amiodarone
Discussion
Amiodarone is an efficacious antiarrhythmic drug whose use is compromised by severe toxic side effects. The impairment of vision is one main cause of discontinuation in patients following long-term treatment of amiodarone [45], which usually begins with corneal deposit, microdeposits, and visual disturbance, which progress slowly to vision impairment level, optic neuritis, toxic optic neuropathy, and/or blindness [10, 12, 15, 46]. The mechanisms underlying amiodarone optic toxicity are still not clear. We used the RGC model to investigate the mechanisms of amiodarone toxicity and evaluated the potential for these effects to be prevented or reversed by a growth factor, IGF-1, which we have previously demonstrated to be cell protective in multiple cell types [47–49]. The major finding of the present study is that the drug, amiodarone, initiated apoptotic cell death in RGC-5 cells, whereas IGF-1 prevented cell death by stimulating the activation of the PI3K/Akt/FoxO3a pathway. These results indicated that IGF-1 could overcome the toxic effect of amiodarone.
We made the following observations: (1) amiodarone treatment of RGCs induced cell death in a concentration-dependent manner; (2) IGF-1 protected RGCs against amiodarone-induced cell death in a concentration-dependent manner; (3) the PI3K inhibitor, LY294002, and the Akt inhibitor VIII blocked the protective effects of IGF-1 against amiodarone-induced cell death; (4) amiodarone reduced the phosphorylation of Akt and FoxO3a, and these effects were reversed when cells were treated with IGF-1; (5) amiodarone induced the nuclear translocation of FoxO3a (which induces apoptosis), and this action was prevented by IGF-1 treatment. Thus, IGF-1 can activate the PI3K/Akt/FoxO3a pathway to oppose the apoptotic action of amiodarone in RGCs.
Amiodarone was recently reported to cause neurotoxicity in brain cell culture [50]. Amiodarone activates both apoptotic and necrotic pathways to induce cytotoxicity in transformed human peripheral lung epithelial cells [51]. Molecular investigations revealed that amiodarone downregulated activator protein-1 (AP-1) and nuclear factor kappa-B (NF-kappaB) DNA-binding activities in activated human T cells also inhibited DNA-binding and transcriptional activities of both AP-1 and NF-kappa B in Jurkat cells [52]. Amiodarone caused cell death in Hepa1c1c7 cells, while TNF cotreatment potentiated its toxicity. Activation of caspases 9 and 3/7 was observed in AM/TNF-cotreated cells, and caspase inhibitors provided protection from cytotoxicity [53]. In vivo study of the apoptotic effects of amiodarone on rat testes with immunohistochemical staining showed an evidence of apoptosis induced by amiodarone which included increased expression of caspase-3, caspase-9, and Bax and increased DNA fragmentation detected via a terminal deoxynucleotidyl transferase dUTP nick end labeling assay [54]. Amiodarone also triggers apoptosis through an iodine-independent mechanism which is not mediated by p53 [55, 56].
IGF-1, a key mediator of growth hormone action in mammals, plays an important role in the regulation of cellular and tissue functions. IGF-1 can protect cells from damage induced by hypoxia and hyperglycemia. IGF-1 and Akt (also known as protein kinase B (PKB)) proteins have been reported to exhibit gastroprotective effects by reducing water immersion and restraint stress (WRS)-induced gastric mucosal cellular apoptosis. The IGF-1/PTEN/Akt/FoxO signaling pathway is effective in protecting against gastric ulcers [57, 58]. In line with these studies, in the present in vitro experiments, we have revealed that IGF-1 rescues RGC-5 cells from the amiodarone-induced toxic effects via the PI3K/AKT/FoxO3a pathway. The protective effect of IGF-1 was completely abolished in the presence of the PI3K inhibitor LY294002 but was unaffected by treatment with the MEK inhibitor PD98059 or the p38 inhibitor PD160316, indicating that IGF-1 elicits its protective effects via PI3K signaling but not via the MAP kinase ERK1/2 or p38 pathway in these cells. In addition, treatment of RGC-5 cells with IGF-1 leads to the rapid generation of phosphorylated Akt. Lastly, this effect on Akt phosphorylation is blunted in the presence of LY294002. Together, these data suggest that IGF-1 promotes RGC cell survival and protects them from the toxic effects of amiodarone by specifically activating the prosurvival PI3K/Akt pathway.
The FoxO3a transcription factor is an important molecule in the cell cycle as its nuclear localization results in apoptosis. FoxO3a is a transcription factor and a proapoptotic protein, when it is dephosphorylated, it stays in the nucleus and binds to the promoter of its target genes like Fas, Bim, and CC3 to induce apoptosis. When FoxO3a is phosphorylated, it translocates to the cytoplasm, leaving its target genes, leading to cell survival [59, 60]. We have previously reported that IGF-1 and neurotrophins can induce the phosphorylation of FoxO3a in primary cultured neurons, leading to cell survival [43, 44]. IGF-I activates the FoxO kinase Akt/PKB, which phosphorylates FoxO and causes its rapid translocation to the cytosol, whereas inhibition of Akt has an opposite effect. In a two-state mathematical model for FoxO1 nuclear-cytoplasmic redistribution, FoxO phosphorylation/dephosphorylation is assumed to be fast compared with nuclear influx and efflux [44, 61, 62]. Since FoxO3a is a direct downstream target of Akt, we examined the potential role of FoxO3a in the effect of IGF-1 to promote cell survival. Intriguingly, amiodarone inhibited basal levels of phosphorylated FoxO3a, in contrast to IGF-1 which increased these levels. IGF-1 was capable of returning the levels of phosphorylated FoxO3a to the basal level in the presence of amiodarone. These results are similar to the effects seen on phosphorylated Akt. Furthermore, amiodarone caused a marked increase in nuclear translocation of FoxO3a, consistent with an apoptotic phenotype. In contrast, IGF-1 prevented this effect and promoted nuclear exclusion of FoxO3a, consistent with an Akt-mediated antiapoptotic phenotype. Together, these data indicate that IGF-1 may mediate the survival and protection from amiodarone-induced RGC-5 cell death via stimulation of the PI3K/Akt/FoxO3a signaling pathway. The protective effect of IGF-1 was further confirmed by the FERG analysis showing a reversal of the effects of amiodarone on entoretina function.
Our finding is significant as clinical application of amiodarone is limited by its toxicity, and understanding the underlying molecular mechanism is important for the prevention and treatment of toxic side effect of amiodarone [11, 13].
In summary, this study highlights the role of PI3K/Akt/FoxO3a in the protective action of IGF-1 in RGC-5 cells against cell death induced by amiodarone (Fig. 11). The present study also demonstrated that the FoxO3a transcription factor participates in the protective effect of IGF-1 against amiodarone. Therefore, IGF-1 analogs and stimulators of PI3K/Akt pathway may be potential therapeutic agents for amiodarone-related eye diseases.

Possible mechanisms underlying the effect of IGF-1 and amiodarone in retinal neuronal cells. Amiodarone decreased the p-Akt and p-FoxO3a which leads to the nuclear localization of FoxO3a and apoptosis of RGC-5 cells. IGF-1 stimulated the activation of the PI3K/Akt pathway. Active Akt phosphorylated FoxO3a and caused its cytosolic localization, antagonized the effect of AM, and caused cell survival [59, 63, 64]
Abbreviations
- AM:
-
Amiodarone
- IGF-1:
-
Insulin-like growth factor-1
- RGCs:
-
Retinal ganglion cells
- MAP kinase:
-
Mitogen-activated protein kinase
- PI3K:
-
Phosphatidylinositol 3-kinase
References
Simopoulos V et al (2014) Ranolazine enhances the antiarrhythmic activity of amiodarone by accelerating conversion of new-onset atrial fibrillation after cardiac surgery. Angiology 65:294–297
Wurdeman RL, Mooss AN, Mohiuddin SM, Lenz TL (2002) Amiodarone vs. sotalol as prophylaxis against atrial fibrillation/flutter after heart surgery: a meta-analysis. Chest 121:1203–1210
Wirth KJ, Knobloch K (2001) Differential effects of dofetilide, amiodarone, and class lc drugs on left and right atrial refractoriness and left atrial vulnerability in pigs. Naunyn Schmiedeberg's Arch Pharmacol 363:166–174
Mosher MC (2011) Amiodarone-induced hypothyroidism and other adverse effects. Dimens Crit Care Nurs 30:87–93
Vereckei A et al (1991) The role of oxidative stress, caused by amiodarone, in the side effects of the drug. Orv Hetil 132(483–484):487–488
Murphy JA, Fitzsimons BM, Meute MA, Wilkinson WC, Luck JC, Wiley SW (1990) Amiodarone: a postmarketing evaluation of monitoring for drug-induced toxicity. DICP 24:1001–1006
Rodrigues M, Alves G, Ferreira A, Queiroz J, Falcao A (2013) A rapid HPLC method for the simultaneous determination of amiodarone and its major metabolite in rat plasma and tissues: a useful tool for pharmacokinetic studies. J Chromatogr Sci 51:361–370
Deng P, You T, Chen X, Yuan T, Huang H, Zhong D (2011) Identification of amiodarone metabolites in human bile by ultraperformance liquid chromatography/quadrupole time-of-flight mass spectrometry. Drug Metab Dispos 39:1058–1069
Van Elmbt G, Andris C, Collignon N (2007) Amiodarone associated optic neuropathies—two cases reports. Bull Soc Belge Ophtalmol: 75–80
Mindel JS (2014) Absence of amiodarone-associated optic neuropathy. Ophthalmology 121:2074–2075
Mindel JS (2008) Amiodarone and optic neuropathy. Am Heart J 156:411–413
Martinez-LoPez-Portillo MA, Martinez-Gamero BO, Mohamed-Noriega J, Cavazos-Adame MH, Mohamed-Hamsho MJ (2014) Behaviour of disc oedema during and after amiodarone optic neuropathy: case report. J Clin Diagn Res 8:D4–D5
Kervinen M, Falck A, Hurskainen M, Hautala N (2013) Bilateral optic neuropathy and permanent loss of vision after treatment with amiodarone. J Cardiovasc Pharmacol 62:394–396
Kerrison JB (2004) Optic neuropathies caused by toxins and adverse drug reactions. Ophthalmol Clin N Am 17:481–488
Chassang B, Bonnin N, Moisset X, Citron B, Clavelou P, Chiambaretta F (2014) Two cases of bilateral amiodarone-associated optic neuropathy. J Fr Ophtalmol 37:231–236
Nicolas J, Hendriksen PJ, de Haan LH, Koning R, Rietjens IM, Bovee TF (2015) In vitro detection of cardiotoxins or neurotoxins affecting ion channels or pumps using beating cardiomyocytes as alternative for animal testing. Toxicol in Vitro 29:281–288
Turovaya AY et al (2005) Effects of verapamil and amiodarone on sympathoadrenal system and balance of excitatory and inhibitory amino acids in rat medulla oblongata. Bull Exp Biol Med 139:665–667
Vig PJ, Desaiah D (1991) Modulation of protein kinase C activity by amiodarone and desethylamiodarone. Neurotoxicology 12:595–601
Silver PJ, Connell MJ, Dillon KM, Cumiskey WR, Volberg WA, Ezrin AM (1989) Inhibition of calmodulin and protein kinase C by amiodarone and other class III antiarrhythmic agents. Cardiovasc Drugs Ther 3:675–682
Yano T, Itoh Y, Yamada M, Egashira N, Oishi R (2008) Combined treatment with L-carnitine and a pan-caspase inhibitor effectively reverses amiodarone-induced injury in cultured human lung epithelial cells. Apoptosis 13:543–552
Miguel A, Henriques F, Azevedo LF, Pereira AC (2014) Ophthalmic adverse drug reactions to systemic drugs: a systematic review. Pharmacoepidemiol Drug Saf 23:221–233
Li J, Tripathi RC, Tripathi BJ (2008) Drug-induced ocular disorders. Drug Saf 31:127–141
Bratulescu M, Zemba M, Gheorghieva V, Andrei S, Cucu B, Dobrescu N (2005) Ocular manifestation in amiodarone toxicity—case report. Oftalmologia 49:18–23
Wang S, Li K (2014) MicroRNA-96 regulates RGC-5 cell growth through caspase-dependent apoptosis. Int J Clin Exp Med 7:3694–3702
Zhou X et al (2014) Neuroprotective effects of methyl 3,4-dihydroxybenzoate against H(2)O(2)-induced apoptosis in RGC-5 cells. J Pharmacol Sci 125:51–58
Jiang SH et al (2014) The effect and underlying mechanism of Timosaponin B-II on RGC-5 necroptosis induced by hydrogen peroxide. BMC Complement Altern Med 14:459
Bu SY, Yu GH, Xu GX (2013) Expression of insulin-like growth factor 1 receptor in rat retina following optic nerve injury. Acta Ophthalmol 91:e427–e431
Biswas SK, Zhao Y, Nagalingam A, Gardner TW, Sandirasegarane L (2008) PDGF- and insulin/IGF-1-specific distinct modes of class IA PI 3-kinase activation in normal rat retinas and RGC-5 retinal ganglion cells. Invest Ophthalmol Vis Sci 49:3687–3698
Yang X, Wei A, Liu Y, He G, Zhou Z, Yu Z (2013) IGF-1 protects retinal ganglion cells from hypoxia-induced apoptosis by activating the Erk-1/2 and Akt pathways. Mol Vis 19:1901–1912
Wang H, et al. (2014) IGF-1 signaling via the PI3K/Akt pathway confers neuroprotection in human retinal pigment epithelial cells exposed to sodium nitroprusside insult. J Mol Neurosci
Sun C, Meng Q, Zhang L, Wang H, Quirion R, Zheng W (2012) Glutamate attenuates IGF-1 receptor tyrosine phosphorylation in mouse brain: possible significance in ischemic brain damage. Neurosci Res 74:290–297
Zheng WH, Quirion R (2006) Insulin-like growth factor-1 (IGF-1) induces the activation/phosphorylation of Akt kinase and cAMP response element-binding protein (CREB) by activating different signaling pathways in PC12 cells. BMC Neurosci 7:51
Zheng WH, Kar S, Quirion R (2002) Insulin-like growth factor-1-induced phosphorylation of transcription factor FKHRL1 is mediated by phosphatidylinositol 3-kinase/Akt kinase and role of this pathway in insulin-like growth factor-1-induced survival of cultured hippocampal neurons. Mol Pharmacol 62:225–233
Homma K, Koriyama Y, Mawatari K, Higuchi Y, Kosaka J, Kato S (2007) Early downregulation of IGF-I decides the fate of rat retinal ganglion cells after optic nerve injury. Neurochem Int 50:741–748
Koriyama Y et al (2007) Upregulation of IGF-I in the goldfish retinal ganglion cells during the early stage of optic nerve regeneration. Neurochem Int 50:749–756
Wang H et al (2012) Insulin-like growth factor-1 induces the phosphorylation of PRAS40 via the PI3K/Akt signaling pathway in PC12 cells. Neurosci Lett 516:105–109
Nitta A, Zheng WH, Quirion R (2004) Insulin-like growth factor 1 prevents neuronal cell death induced by corticosterone through activation of the PI3k/Akt pathway. J Neurosci Res 76:98–103
Olianas MC, Dedoni S, Onali P (2011) delta-Opioid receptors stimulate GLUT1-mediated glucose uptake through Src- and IGF-1 receptor-dependent activation of PI3-kinase signalling in CHO cells. Br J Pharmacol 163:624–637
Xu L, Chen WF, Wong MS (2009) Ginsenoside Rg1 protects dopaminergic neurons in a rat model of Parkinson’s disease through the IGF-I receptor signalling pathway. Br J Pharmacol 158:738–748
De Luca A, Pierno S, Liantonio A, Camerino C, Conte CD (1998) Phosphorylation and IGF-1-mediated dephosphorylation pathways control the activity and the pharmacological properties of skeletal muscle chloride channels. Br J Pharmacol 125:477–482
de Cupis A, Noonan D, Pirani P, Ferrera A, Clerico L, Favoni RE (1995) Comparison between novel steroid-like and conventional nonsteroidal antioestrogens in inhibiting oestradiol- and IGF-I-induced proliferation of human breast cancer-derived cells. Br J Pharmacol 116:2391–2400
Wang R et al (2014) Stereoselective reduction of 1-o-isopropyloxygenipin enhances its neuroprotective activity in neuronal cells from apoptosis induced by sodium nitroprusside. ChemMedChem 9:1397–1401
Wang H et al (2013) FoxO3a negatively regulates nerve growth factor-induced neuronal differentiation through inhibiting the expression of neurochondrin in PC12 cells. Mol Neurobiol 47:24–36
Wen Q, Wang H, Little PJ, Quirion R, Zheng W (2012) Forkhead family transcription factor FoxO and neural differentiation. Neurogenetics 13:105–113
Nantsupawat T, Nugent K, Phrommintikul A (2013) Atrial fibrillation in the elderly. Drugs Aging 30:593–601
Passman RS et al (2012) Amiodarone-associated optic neuropathy: a critical review. Am J Med 125:447–453
Wang H et al (2015) IGF-1 signaling via the PI3K/Akt pathway confers neuroprotection in human retinal pigment epithelial cells exposed to sodium nitroprusside insult. J Mol Neurosci 55:931–940
Ma J et al (2015) Transplantation of human neural progenitor cells expressing IGF-1 enhances retinal ganglion cell survival. PLoS One 10:e125695
Chen C, Xu Y, Song Y (2014) IGF-1 gene-modified muscle-derived stem cells are resistant to oxidative stress via enhanced activation of IGF-1R/PI3K/AKT signaling and secretion of VEGF. Mol Cell Biochem 386:167–175
Pomponio G et al (2015) Amiodarone biokinetics, the formation of its major oxidative metabolite and neurotoxicity after acute and repeated exposure of brain cell cultures. Toxicol in Vitro 30:192–202
Mulder JE, Brien JF, Racz WJ, Takahashi T, Massey TE (2011) Mechanisms of amiodarone and desethylamiodarone cytotoxicity in nontransformed human peripheral lung epithelial cells. J Pharmacol Exp Ther 336:551–559
Cheng SM et al (2015) Modulation of both activator protein-1 and nuclear factor-kappa B signal transduction of human T cells by amiodarone. Exp Biol Med (Maywood) 240:99–108
Lu J, Miyakawa K, Roth RA, Ganey PE (2013) Tumor necrosis factor-alpha potentiates the cytotoxicity of amiodarone in Hepa1c1c7 cells: roles of caspase activation and oxidative stress. Toxicol Sci 131:164–178
Ozkaya AK, Dilber E, Gurgen SG, Kutlu O, Cansu A, Gedik Y (2016) Effects of chronic amiodarone treatment on rat testis. Acta Histochem 118:271–277
Di Matola T et al (2000) Amiodarone induces cytochrome c release and apoptosis through an iodine-independent mechanism. J Clin Endocrinol Metab 85:4323–4330
Parks A, Marceau F (2016) Lysosomotropic cationic drugs induce cytostatic and cytotoxic effects: role of liposolubility and autophagic flux and antagonism by cholesterol ablation. Toxicol Appl Pharmacol 305:55–65
Huang P et al (2012) Effect of the IGF-1/PTEN/Akt/FoxO signaling pathway on the development and healing of water immersion and restraint stress-induced gastric ulcers in rats. Int J Mol Med 30:650–658
Huang P, Zhou ZR, Zheng MQ, Shi FX (2012) Effect of the IGF-1/PTEN/Akt/FoxO signaling pathway in the duodenal mucosa of rats subjected to water immersion and restraint stress. Genet Mol Res 11:4775–4788
Li D et al (2015) Involvement of the JNK/FOXO3a/Bim pathway in neuronal apoptosis after hypoxic-ischemic brain damage in neonatal rats. PLoS One 10:e132998
Ahn JS, Li J, Chen E, Kent DG, Park HJ, Green AR (2016) JAK2V617F mediates resistance to DNA damage-induced apoptosis by modulating FOXO3A localization and Bcl-xL deamidation. Oncogene 35:2235–2246
Wang H et al (2013) The role of Akt/FoxO3a in the protective effect of venlafaxine against corticosterone-induced cell death in PC12 cells. Psychopharmacology 228:129–141
Wen Q et al (2011) Characterization of intracellular translocation of Forkhead transcription factor O (FoxO) members induced by NGF in PC12 cells. Neurosci Lett 498:31–36
Shrestha A et al (2016) Critical role of AMPK/FoxO3A Axis in globular Adiponectin-induced cell cycle arrest and apoptosis in cancer cells. J Cell Physiol 231:357–369
Li Y et al (2016) MiR-182-5p protects inner ear hair cells from cisplatin-induced apoptosis by inhibiting FOXO3a. Cell Death Dis 7:e2362
Acknowledgments
This research was financially supported by the Guangdong Provincial Project of Science and Technology (2011B050200005), SRG2015-00004-FHS and MYRG2016-00052-FHS from the University of Macau, the Science and Technology Development Fund (FDCT) of Macau (FDCT 021/2015/A1), Guangdong natural science foundation (No.2014A030310039), and the National Natural Science Foundation of China (31371088).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
ᅟ
All of the procedures described in this study were performed in accordance with the Guide for the Care and Use of Laboratory Animals—Chinese Version (1996).
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Rifang Liao, Fengxia Yan, and Zhuanping Zeng contributed equally to this work.
Rights and permissions
About this article

Cite this article
Liao, R., Yan, F., Zeng, Z. et al. Amiodarone-Induced Retinal Neuronal Cell Apoptosis Attenuated by IGF-1 via Counter Regulation of the PI3k/Akt/FoxO3a Pathway. Mol Neurobiol 54, 6931–6943 (2017). https://doi.org/10.1007/s12035-016-0211-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0211-x