
Abstract
In ischemic stroke, cell damage propagates from infarct core to surrounding tissue. To reveal proteins involved in neurodegeneration and neuroprotection, we explored the protein profile in penumbra surrounding the photothrombotic infarct core induced in rat cerebral cortex by local laser irradiation after Bengal Rose administration. Using antibody microarrays, we studied changes in expression of 224 signaling proteins 1, 4, or 24 h after photothrombotic infarct compared with untreated contralateral cortex. Changes in protein expression were greatest at 4 h after photothrombotic impact. These included over-expression of proteins initiating, regulating, or executing various apoptosis stages (caspases, SMAC/DIABLO, Bcl-10, phosphatidylserine receptor (PSR), prostate apoptosis response 4 (Par4), E2F1, p75, p38, JNK, p53, growth arrest and DNA damage inducible protein 153 (GADD153), glutamate decarboxylases (GAD65/67), NMDAR2a, c-myc) and antiapoptotic proteins (Bcl-x, p63, MDM2, p21WAF-1, ERK1/2, ERK5, MAP kinase-activated protein kinase-2 (MAKAPK2), PKCα, PKCβ, PKCμ, RAF1, protein phosphatases 1α and MAP kinase phosphatase-1 (MKP-1), neural precursor cell expressed, developmentally down-regulated 8 (NEDD8), estrogen and EGF receptors, calmodulin, CaMKIIα, CaMKIV, amyloid precursor protein (APP), nicastrin). Phospholipase Cγ1, S-100, and S-100β were down-regulated. Bidirectional changes in levels of adhesion and cytoskeleton proteins were related to destruction and/or remodeling of penumbra. Following proteins regulating actin cytoskeleton were over-expressed: cofilin, actopaxin, p120CTN, α-catenin, p35, myosin Va, and pFAK were up-regulated, whereas ezrin, tropomyosin, spectrin (α + β), βIV-tubulin and polyglutamated β-tubulin, and cytokeratins 7 and 19 were down-regulated. Down-regulation of syntaxin, AP2β/γ, and adaptin β1/2 indicated impairment of vesicular transport and synaptic processes. Down-regulation of cyclin-dependent kinase 6 (Cdk6), cell division cycle 7-related protein kinase (Cdc7 kinase), telomeric repeat-binding factor 1 (Trf1), and topoisomerase-1 showed proliferation suppression. Cytoprotection proteins AOP-1 and chaperons Hsp70 and Hsp90 were down-regulated. These data provide the integral view on penumbra response to photothrombotic infarct. Some of these proteins may be potential targets for antistroke therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is one of the leading causes of human disability and mortality. Acute ischemia caused by vascular occlusion very quickly, for few minutes, induces deficit of oxygen and glucose that is followed by necrosis and infarction of the nervous tissue. It is impossible to rescue these cells. However, the injurious processes propagate to the surrounding tissue beyond the infarction core. The tissue damage in this transition zone (penumbra) develops slower, for several hours, which provides time for neuroprotection therapy, which can potentially limit the brain injury [1, 2].
In order to find the antistroke neuroprotectors, various glutamate antagonists, blockers of Ca2+ channels, inhibitors of nitric oxide signaling, antioxidants, and ROS scavengers have been tested. However, efficient neuroprotective drugs for stroke pharmacotherapy have not been found yet [3]. Therefore, wider search based on comprehensive studies of molecular mechanisms of neurodegeneration and neuroprotection is needed [4, 5]. The cell response to acute injury is initially determined by proteins present in the cell. However, if their protective capability is insufficient, additional proteins are synthesized. The proteomic techniques have been used to characterize the cerebral ischemia-induced changes in expression of hundreds of proteins in the ischemic core and penumbra [6–11]. This information sheds light to some mechanisms of ischemic brain injury.
In the present work, we have studied the biochemical consequences of the focal photothrombotic infarction (PTI) in the rat cerebral cortex induced by the local photodynamic treatment. Photodynamic effect is based on dye photoexcitation in stained cells that is followed by generation of singlet oxygen and other ROS, oxidative stress, and finally, necrosis or apoptosis [12]. Cerebral PTI is induced by laser irradiation of the cerebral cortex after administration of Bengal Rose, a hydrophilic photosensitizer that does not penetrate cells and accumulates in the brain vessels. Following photoirradiation induces local oxidative damage of the vascular endothelium, aggregation of platelets, and occlusion of microvessels. This stroke model is little invasive. The injury location, size, and degree are easily controlled and reproducible [9, 10, 13–16].
Using less intensive but prolonged irradiation, we achieved a wider penumbra that was sufficient for biochemical analysis. Recently, we have studied changes in expression of 224 neuronal proteins in penumbra. We identified proteins whose expression increased or decreased after focal PTI [9, 10]. The aim of the present work was to study the changes in the expression of 224 signaling proteins in the penumbra around the PTI core in the rat cerebral cortex at different intervals after laser irradiation (1, 4, or 24 h) as compared with untreated contralateral cortex. We have revealed a number of up- or down-regulated proteins, which could be involved in the integral response of the penumbra tissue to photoinduced focal ischemia.
Materials and Methods
Chemicals
Cy3™ or Cy5™ monofunctional reactive dyes were supplied by GE Healthcare. The Panorama Ab Microarray Cell Signaling Kits (CSAA1, Sigma-Aldrich Co.) and other chemicals were obtained from Sigma-Aldrich-Rus (Moscow, Russia).
Animals
The experiments were performed on adult male Wistar rats (200–250 g). The animal holding room was maintained at a temperature of 22–25 °C, 12-h light/dark schedule, and an air exchange rate of 18 changes per hour. All experimental procedures were carried out in accordance with the European Union guidelines 86/609/ЕЕС for the use of experimental animals and local legislation for ethics of experiments on animals. The animal protocols were evaluated and approved by the Animal Care and Use Committee of the Southern Federal University (approval no. 02/2014).
Focal Photothrombotic Ischemia in the Rat Cerebral Cortex
The unilateral focal PTI in the rat somatosensory cortex was induced according to [9, 10, 13]. The rats were anesthetized with chloral hydrate (300 mg/kg, i.p.). After the longitudinal incision of the skull skin, the periosteum was removed. Bengal Rose (20 mg/kg) was injected in the vena subclavia. Then, the rats were fixed in the stereotactic holder. The unilateral irradiation of the somatosensory cortex was performed through the cranial bone using 532 nm diode laser (64 mW/cm2, Ø 3 mm, 30 min). Irradiation zone was located according to stereotactic coordinates in the sensorimotor cortex: 0.5 mm anterior to bregma, 0.5 mm lateral to the midline. The animals were euthanized with the chloral hydrate overdose (600 mg/kg, i.p.) and decapitated 1, 4, or 24 h after PTI. The symmetric cortical region in the contralateral hemisphere served as a control.
Histology and Immunohistochemistry
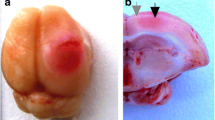
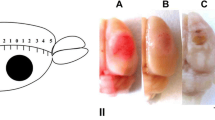
Twenty-four hours after light exposure, rats which brains should be stained by 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich Co.) were deeply anesthetized with chloral hydrate and then decapitated (n = 3). Brains were then rapidly removed, placed into a freezer (−70 °C) for 5–8 min, and sliced into 2-mm-thick coronal sections (Fig. 1). The hemisphere and infarction areas were traced and measured on each section using ImageJ analysis software (NIH). For histological study, the rats were perfused transcardially with 10 % buffered formalin (pH 7.2) under chloral hydrate anesthesia 4 h after laser irradiation. The extracted brains were post-fixed with formalin. The paraffin sections were stained with hematoxylin and eosin and imaged on the Eclipse FN1 microscope (Nikon, Japan; objective lens Plan Fluor ELWD, 40×/0.60) (Fig. 1a).

Morphological changes in the rat cerebral cortex after PTI. a Representative TTC staining images of the rat coronal brain sections at 24 h after light exposure. The red-colored areas represent normal areas, whereas the pale areas represent the infarct regions. Scale bar 3 mm. b Representative hematoxylin-eosin staining of the rat coronal brain sections at 4 h after light exposure. Coronal slice image with normal, infarct, and transition zones, labeled with white dotted line. Scale bar 500 μm. c, d The areas in the black rectangles in b. c Transition zone (penumbra) and normal tissue. d The tissues in the infarction core and transition zone (penumbra). Scale bar 50 μm
For immunohistochemical study, 5-μm sections of the paraffin-embedded experimental and control cortex tissues were used. The sections were demasked in Tris-EDTA buffer with 0.05 % Tween 20 (pH 9.0) in the programmed barocamera Pascal (Dako). Reactions were visualized using the REVEAL Polyvalent HRP-DAB Detection System (Spring Bioscience, SPD-060, USA) according to the manufacturer’s instructions. The primary antibodies against PAR4 (SAB4502078, Sigma-Aldrich, 1:3000) or cofilin (SAB4500148, Sigma-Aldrich, 1:3000) were used. The sections were counterstained with Mayer’s hematoxylin and imaged on the Eclipse FN1 microscope (Nikon, Japan; objective lens Plan Fluor ELWD, 40х/0.60). Omission of primary antibody was routinely used to certify the absence of nonspecific labeling. All observations were performed in lamina granularis externa, lamina pyramidalis, and lamina granularis interna. Twelve images were acquired per section and studied using the ImageJ software (NIH, USA) [19]. For quantification, the immunoreactivity coefficients, I = N ip / N t × 100 %, where N ip and N t are the numbers of immunopositive pixels and total number of pixels, respectively, were determined in each section in control (n = 3) and experimental (n = 3) samples. Statistical analysis was performed using Student’s t test for independent experiments. Quantitative data were presented as mean ± SEM.
Proteomic Study
The Panorama Antibody Ab Microarray Cell Signaling kit (CSAA1, Sigma-Aldrich Co.) contains two identical microarrays. In each of them, the nitrocellulose-coated glass slide contained 448 microdroplets with immobilized antibodies against 224 signaling proteins in duplicate. Nonlabeled bovine serum albumin was used as a negative control, and a single spot with Cy3 and Cy5-conjugated BSA was a positive control.
At 1, 4, or 24 h after photodynamic treatment, the rat cortex was extracted, and the PTI core region was excised using a Ø3-mm circular knife and discarded. After that, the 2-mm cortical ring around the PTI core (penumbra) was cut out by another Ø7-mm circular knife. A similar piece from the nonirradiated contralateral somatosensory cortex was used as control. These pieces were weighed, homogenized, and lyzed in the extraction/labeling buffer supplemented with protease and phosphatase inhibitor cocktails and nuclease bensonase (components of CSAA1). Then, the control and experimental lysates were centrifuged in the cooled centrifuge (10,000 rpm, 4 min, 4 °C). The supernatants were frozen in liquid nitrogen and stored at −80 °C for further analysis. After thawing, the protein contents in both experimental and control samples were determined using Bradford reagent. Then, the samples were diluted to 1 mg/ml protein content and incubated for 30 min in the dark at room temperature with Cy3 or Cy5, respectively. The unbound dye was removed by centrifugation (4000 rpm; 4 min) of the Sigma Spin Columns (CSAA1 components) filled with 200 μl of the labeled protein samples. The eluates were collected, and protein concentration was determined again. In another set, these samples were stained oppositely, by Cy5 and Cy3, respectively.
One microarray was incubated for 40 min at room temperature on a rocking shaker in 5 ml of the mixture of the control and experimental samples (10 μg/ml each) labeled with Cy3 and Cy5, respectively. Another microarray was incubated with the oppositely labeled samples: Cy5 and Cy3, respectively. Such swapped staining provided verification of results and compensation of a potential bias in binding of Cy3 or Cy5 dyes to protein samples. This ensures the double test and full control of the experiment. After following triple washing in the washing buffer (CSAA1 component) and triple washing in pure water, the microarray slides were air-dried overnight in the dark.
The microarrays were scanned using the GenePix 4100A Microarray Scanner (Molecular Devices, USA) at 532 and 635 nm (fluorescence maximums of Cy3 and Cy5, respectively). The integrated fluorescence intensity in each antibody spot was proportional to the quantity of the bound protein. The fluorescence images of the antibody microarrays were analyzed and normalized (ratio-based normalization) using the software GenePix Pro 6.0. Local fluorescence values in the rings around each spot were served as a background. The median fluorescence value that was determined over all spot pixels was used for estimation of the protein content in each spot, and the ratios of the experimental to control values characterized the difference in the protein level between photothrombotic and normal cortical tissue in the contralateral cortex. Two samples, labeled independently and reversely in duplicate, provided four experimental values for each protein. Each experiment was repeated four times for 1 h or 4 h reperfusion exposures, or two times for 24 h reperfusion exposure, and the experiment/control ratios were averaged. The standard statistical processing based on Student’s t test and 95 % significance level was used. Quantitative data in Table 1 are presented as means ± SD. Only proteins whose expression was more than 30 % different from the control are shown in Table 1 and are discussed. The reliability of the proteomic microarray data was proved by the self-control experiment using the swapped staining in the second microarray [9, 10].
Results
Morphological Changes in the Rat Cerebral Cortex after Photothrombotic Infarction
In the hematoxylin/eosin-stained cortical slices, the PTI core looked as a pale-stained area containing hypereosinophilic neurons with pyknotic nuclei (Fig. 1b, d). In the penumbra, the number of such abnormal neurons gradually decreased. Beyond the vacuolated area, the cortex retained a rather normal structure, with the neurons having the basophilic stippling of their cytoplasm (Fig. 1c). The mean width of the penumbra was 1.5–2 mm. At the periphery of penumbra, clusters of normal neurons were observed along with hypereosinophilic neurons having the pyknotic nuclei (Fig. 1c, d). The depth of the infarction zones was 2.7–3 mm (Fig. 1b). The infarct volume at 24 h after PTI l was 31 ± 2 mm3.
PTI-Induced Changes in Expression of Signaling Proteins in Penumbra
The histological (Fig. 1) and ultrastructural data [10] showed that the necrotic infarct core was surrounded by 1.5–2-mm-width penumbra. Morphological alterations decreased gradually across the penumbra from regions adjacent to the PTI core to the periphery.
The proteomic study of the expression of 224 signaling proteins in the penumbra as compared with the untreated contralateral cerebral cortex of the same rats showed >30 % up-regulation of 15, 51, and 37 proteins at 1, 4, or 24 h after PTI, respectively. At the same time, the levels of 14, 27, and 22 proteins, respectively, decreased (Table 1). The strongest changes were observed at 4 h after PTI (Table 1). The most characteristic processes in the post-stroke penumbra included simultaneous up-regulation of various pro and antiapoptotic proteins. We observed over-expression of proteins that are involved in execution of diverse apoptosis stages: SMAC/DIABLO (+75 %), Bcl-10 (+60 %), caspase 6 (+46 %), caspase 7 (+24 %), caspase 3 (+50 %) and its activated form (+32 %), and phosphatidylserine receptor (PSR) (+46 %). Some multifunctional proteins that can promote apoptosis in specific situations were also up-regulated. They included prostate apoptosis response 4 (Par4) (+94 %), E2F1 (+51 %); p75 (nerve growth factor receptor) (+54 %), MAP kinases p38 (+45 %) and JNK (+35 %), p53 (+51 %), growth arrest and DNA damage inducible protein 153, C/EBP homology protein 10 (GADD153/CHOP-10) (+50 %), glutamate decarboxylases (GAD65/67) (+49 %), glutamate receptor NMDAR2a (+46 %), inflammatory caspase 11 (+58 %), and c-myc (+56 %). The down-regulation of NF-κB activating kinase (NAK) (−54 %) also potentiates apoptosis.
At the same time, antiapoptotic neuroprotectors such as Bcl-x (+40 %), estrogen receptor (+62 %), ERK5 (+57 %), MAP kinase phosphatase-1 (MKP-1) (+57 %), and p53 antagonists p21WAF-1 (+50 %), MDM2 (+39 %), and p63 (+31 %) were up-regulated. The proinflammatory caspase 4 was 2.1-fold down-regulated.
The up-regulation of various signal transduction proteins was aimed to regulate diverse cell functions including survival. We observed over-expression of MAP kinase ERK1/2 (+68 %); MAP kinase-activated protein kinase-2 (MAKAPK2) (+62 %); EGF receptor (+53 %); protein kinases Cα, Cβ, and Cμ (or PKD) (+54, 36, and 32 %, respectively); protein phosphatase 1α (+36 %); RAF1 (+45 %); serum and glucocorticoid inducible kinase (SGK) (+41 %); calmodulin (+38 %); calcium/calmodulin-dependent protein kinases II and IV (CaM kinases IIα and IV) (+38 and 35 %, respectively); amyloid precursor protein (APP) (+44 %); and nicastrin (+40 %). Phospholipase Cγ1, S-100, and its β-chain were down-regulated by a factors of −1.98, −2.6, and −1.95, respectively.
Bidirectional changes in the levels of proteins that are involved in the intercellular interactions, adhesion, and cytoskeleton were associated with destruction and remodeling of the penumbra tissue. Some proteins that regulate the actin cytoskeleton were up-regulated; they included cofilin (+72 %), actopaxin (+37 %), and focal adhesion kinase, phosphorylated at tyrosine 577 (FAK-pTyr577) (+39 %). However, the phosphorylation of another focal adhesion kinase Pyk2 decreased by 2.3 times. Catenin p120CTN and α-catenin that link cadherins with the actin cytoskeleton were also up-regulated (+50 and +77 %, respectively). The protein p35 associated with the β-catenin/N-cadherin complex was up-regulated by 61 %. At the same time, ezrin that links the actin cytoskeleton to the plasma membrane was down-regulated (−76 %). The level of myosin Va increased by 42 %. Other cytoskeleton components such as tropomyosin (−3-fold), spectrin (α + β) (−2.5-fold), βIV-tubulin and polyglutamated β-tubulin (−5.7- and −2.8-fold), and cytokeratins 7 and 19 (−3- and −1.9-fold) were down-regulated.
Down-regulation of syntaxin (−3-fold), adaptor proteins AP2β and AP2γ (−67 and −98 %, respectively), adaptin (β1 + β2) (−79 %), and general receptor for phosphoinositides-1 (ARF nucleotide binding site opener, also called cytohesin-3) (GRP1(ARNO3 cytohesin-3)) (−66 %) indicated impairment of Golgi apparatus, vesicular transport, and transport of synaptic vesicles. Nevertheless, synaptosome-associated protein of 25 kDa (SNAP25) (+55 %) and coat protein β-COP (+34 %) were up-regulated. Nuclear transport factor 2 (NTF2) was 2-fold down-regulated.
The down-regulation of cyclin-dependent kinase 6 (Cdk6), Cdc7 kinase (cell division cycle 7-related protein kinase), Trf1 (telomeric repeat-binding factor 1), and topoisomerase-1 (−2-, −2-, −1.8-, and 1.5-fold, respectively) indicated the suppression of proliferation. At the same time, the level of cyclin B1 that regulates proliferation was up-regulated (+33 %).
Some cytoprotection mechanisms were suppressed: the levels of the mitochondrial antioxidant protein AOP-1 and chaperons Hsp70 and Hsp90 decreased by −50 %, −80 %, and −2.8-fold, respectively. Up-regulation of ubiquitin-like protein neural precursor cell expressed, developmentally down-regulated 8 (NEDD8) (+38 %) indicated activation of proteolysis. At the same time, the level of cystatin A, inhibitor of lysosomal proteases, decreased (−57 %).
Smaller changes were observed at 1 or 24 h after PTI (Table 1). Over-expression of such proteins as Par4, p120CTN, SMAC/DIABLO, cofilin, ERK1, MAKAPK2, p35, Bcl-10, ERK5, MKP-1, caspase 3, and MDM2 and down-regulation of βIV-tubulin, β-tubulin polyglutamylated, cytokeratin, syntaxin, Hsp70 and Hsp90, S100 and S100β, spectrin (α + β), phosphorylated Pyk2, caspase 4, phospholipase Cγ1, Trf1, and adaptin (β1 + β2) occurred throughout the entire reperfusion interval from 1 to 24 h after PTI. These proteins are likely to play the major role in the response of penumbra tissue to PTI. We did not observe up-regulation of proteins only at 1 h after PTI, at the early stage of the response of the cortical tissue. At the late reperfusion stage, 24 h after PTI, changes in the protein expression were weaker than at 4 h: 37 proteins were up-regulated and 22 were down-regulated. The expression of apoptosis-inducing factor 2 (AIF), caspase 7, Cdk4, and activating transcription factor 2 (ATF2) increased by 31–41 % only at this stage, whereas AP2α was down-regulated (−44 %).
Generally, the integral response of the penumbra tissue to PTI included multiple changes in the expression of diverse proteins involved in various cellular subsystems and processes.
Immunohistochemical Study of the Expression of Par4 and Cofilin in Penumbra
Immunohistochemical analysis showed significant increase in Par4 expression in the penumbra at 1, 4, and 24 h after PTI as compared with the contralateral cerebral cortex of the same animals (+51, +74, and +48 %, p < 0.05, respectively) (Fig. 2). These changes were observed mainly in the vicinity of the infarct core, but not at the periphery of the penumbra, where the Par4 level was low.

Expression of Par4 in the untreated contralateral (CL) rat brain cortex (A) and in the penumbra 1, 4, or 24 h after PTI (b–d, respectively). e The index of Par4 immunoreactivity in the control (CL) cortex areas and in the penumbra 1, 4, and 24 h after PTI. Mean ± standard error. (Student’s t test for independent groups; *р < 0.05; n = 3). Scale bar 50 μm
The expression of cofilin in the penumbra (Fig. 3) also increased significantly 1, 4, and 24 h after PTI (+60, 75, and +50 %, p < 0.05, respectively).

Expression of cofilin in the untreated contralateral (CL) rat brain cortex (A) and in the penumbra 1, 4, or 24 h after PTI (b–d, respectively). e The index of cofilin immunoreactivity in the control (CL) cortex areas and in the penumbra 1, 4, and 24 h after PTI. Mean ± standard error. (Student’s t test for independent groups; *р < 0.05; n = 3). Scale bar 50 μm
Discussion
The early change in the protein profile in the penumbra that was observed 1 h after PTI enhanced at 4 h and involved the larger group of proteins. The penumbra response 24 h after the impact included fewer proteins (Table 1). These proteins perform diverse functions, and changes in their levels represent the integral response of the penumbra (Fig. 4), which included the coordinated reactions of various cellular subsystems leading either to survival and recovery or to cell death. These data are summarized on Fig. 4.

The summary scheme of changes in the expression of signaling proteins in the penumbra 4 h after focal photothrombotic infarct in the rat cerebral cortex
Proteins that Regulate Cell Death and Survival
One of the most significant effects observed in the penumbra after PTI was the concerted up-regulation of diverse proteins that can initiate, mediate, or regulate apoptosis. These included apoptosis execution proteins such as SMAC/DIABLO; AIF; Bcl-10; execution caspases 3, 6, and 7; proapoptotic signaling protein p75; MAP kinases p38 and JNK; transcription factors that induce expression of apoptosis-regulating genes p53, c-myc, E2F1, and GADD153/CHOP-10; and multifunctional proteins such as Par4, glutamate decarboxylase, glutamate receptor NMDAR2a, caspase 11, and PSR that, except other functions, can promote apoptosis in some situations (Table 1).
The transcription factor E2F1 is one of the key players that determine the cell fate. It induces up-regulation of a variety of genes involved in the regulation of DNA synthesis and repair, cell cycle, and apoptosis [17–19]. Its expression is controlled by MAP kinase p38 and transcription factor c-Myc [20]. E2F1 can regulate apoptosis either in a p53-dependent or in a p53-independent manner [17, 18]. It induces up-regulation of different proapoptotic proteins such as SMAC/DIABLO, Apaf-1, BH3-only proapoptotic proteins of Bcl-2 family, caspase 3, caspase 7, caspase 8, caspase 9, p53, and p73 [17, 19]. We observed over-expression of E2F1, p53, c-myc, p38, SMAC/DIABLO, Bcl-x, caspase 3, caspase 6, and caspase 7 in penumbra after PTI (Table 1). This corresponds to the data on up-regulation of E2F1 [21] and p53 [22] after transient ischemic stroke in rats. Cerebral ischemia has been shown to induce significant up-regulation of caspase 3, caspase 6, and caspase 7 [23]. On the other hand, inhibition of the p53/E2F1 pathway prevented neuronal apoptosis [24]. One can suggest that the up-regulation of caspase 3, caspase 7, SMAC/DIABLO, Bcl-x, Bcl-10, p21/Waf1, and p53 in the penumbra was associated with the over-expression of E2F1.
p53 is known to stimulate expression of caspase 6 [25], p21/Waf1, and MDM2. It could be responsible for the up-regulation of these proteins in the penumbra after PTI. MDM2 negatively regulates p53. It inhibits the transcription activity of p53 and stimulates its proteolysis. Over-expression of MDM2 after transient middle cerebral artery occlusion (MCAO) in rats could mediate the recovery of injured neurons [26]. p21WAF-1 is a target gene of p53. DNA damage up-regulates р21Waf-1, which then inhibits p53-mediated apoptosis and arrests the cell cycle. This enables the repair of the damaged DNA [27, 28]. The observed up-regulation of p21WAF-1 in the penumbra after PTI was in agreement with the over-expression of p21WAF-1 mRNA in neurons of hippocampus, cortex, and striatum after transient forebrain ischemia in rats [29]. Since DNA is not damaged in the penumbra, and cell proliferation is limited, the inhibition of apoptosis could be the major effect of p21WAF-1. The protein p63, an antagonist of p53, was also over-expressed in the penumbra. Under experimental ischemia, it is rapidly induced and inhibits apoptosis of cortical neurons [30]. According to [31], survival, death, or senescence of neuronal cells depends on the balance between activities of p53, p63, and p73.
Another transcription factor c-myc was also over-expressed in the penumbra after PTI. This multifunctional protein regulates energy metabolism, protein synthesis, and cell cycle. It can potentiate apoptosis [19, 32]. Its up-regulation also occurred after transient global or focal cerebral ischemia in the rodent brain [33]. Ferrer et al. (2003) showed the increased phosphorylation of c-myc in the penumbra 4 h after focal ischemia in rats [34]. It was suggested that c-myc contributes to the delayed neuronal death after transient global ischemia [33]. The observed over-expression of transcription factor GADD153/CHOP-10 in the penumbra could also stimulate apoptosis. It is known to inhibit proliferation and induce apoptosis via down-regulation of Bcl-2, depletion of glutathione, and increased ROS production. Its level is normally low, but increases under DNA damage, ER stress, or cerebral ischemia [21, 35].
The activation of MAP kinases p38 and JNK can induce apoptosis. Both of them were up-regulated in the penumbra after PTI. JNK phosphorylates c-Jun, a component of the transcription factor AP-1, which regulates the expression of some proteins that regulate apoptosis. It also enhances the proapoptotic activity of p53 [36]. The p38-mediated proapoptotic pathway involves E2F1 [37]. Phosphorylation of ERK, JNK, p38, CREB, Elk-1, c-Myc, ATF-2, and c-Jun, which are associated with cell death or survival, was observed in the penumbra at 4 h after MCAO in rats [34]. Under stress conditions, the p38 mitogen-activated protein kinase MAKAPK2 phosphorylates chaperones HSP25/HSP27, which stabilize the protein structure and prevent apoptosis. Its up-regulation after ischemia/reperfusion injury can be involved in brain protection [38].
The nerve growth factor receptor p75 is known to mediate neuronal apoptosis [39]. In our study, it was up-regulated in the penumbra 4–24 h after PTI. Its expression was also observed in the penumbra after cortical ischemia [40].
Not only apoptotic proteins but also some multifunctional proteins that among other functions regulate apoptosis were over-expressed in the penumbra after PTI. In our experiments, the level of Par4 significantly increased in the penumbra during 1–24 h of reperfusion. This is in agreement with the up-regulation of Par4 observed under neurodegeneration, stroke, and excitotoxicity [41]. Its proapoptotic effect was associated with the suppression of NF-B activity and following down-regulation of Bcl-2, IAP, and calbindin [40]. The early induction of Par4, p53 and Bcl-2 protein family, which mediate mitochondrial dysfunction and subsequent release of proapoptotic proteins, has been suggested to be the leading mechanism of apoptosis in ischemia [42].
The phagocytic removal of apoptotic neurons occurs after exposure of phosphatidylserine, the “eat-me” signal. The PTI-induced up-regulation of PSR that recognizes apoptotic cells and stimulates their phagocytic elimination was observed in our experiments. Similarly, its over-expression was observed in microglial cells after focal brain ischemia [43]. We also observed the up-regulation of inflammatory caspase 11, which mediates apoptosis of astrocytes and microglia following inflammation [44]. Therefore, degeneration of glial cells could contribute to the overall apoptosis in the penumbra. Another inflammatory caspase 4 was down-regulated. Under ER stress, it is known to mediate activation of caspase 3 and apoptosis [45]. Possibly, ER stress-mediated apoptosis did not play a significant role in death of penumbra cells.
Upon glutamate binding, glutamate receptor NMDAR2a provides influx of Na+ and Ca2+ into neurons and release of K+, which depolarizes adjacent cells and mediates propagation of excitotoxicity. The Ca2+-dependent excitotoxic response leads to apoptosis [46]. Over-expression of glutamate decarboxylase (GAD65/67) observed in the penumbra after PTI was another proapoptotic effect associated with glutamate metabolism. This effect is in agreement with the observation that the embryonic splice variant of GAD67 induces apoptosis in the adult brain under stroke [47].
The down-regulation of cytoprotective proteins such as mitochondrial antioxidant protein AOP-1 and molecular chaperons HSP-70 and HSP-90 could reduce the cell viability in penumbra.
Not only proapoptotic but also antiapoptotic processes occurred in the penumbra after PTI. We observed the up-regulation of diverse signaling proteins with the protective, antiapoptotic activity such as protein kinases ERK 1/2, ERK5, MAKAPK2, PKCα, PKCβ, PKCμ (PKD), and RAF1; protein phosphatases 1α and MKP-1; estrogen and EGF receptors; NEDD8; and antiapoptotic proteins Bcl-x, p63, MDM2, and p21WAF-1. The observed changes in their expression are generally in agreement with the literature data. The prosurvival extracellular signal-regulated kinases ERK1/2 are known to phosphorylate various transcription factors and cytoskeleton proteins and thereby regulate proliferation, cell mobility, and survival. For example, Kitagawa et al. (1999) have reported the over-expression of ERK1/2 at 3–8 h after MCAO. At 24 h, its level was elevated only in the penumbra, but decreased in the ischemic core [48]. Ferrer et al. (2003) observed the phosphorylation of ERK in the penumbra following reperfusion [34]. ERK5 is known to be activated by neurotrophic factors or oxidative stress and to participate in neuroprotection and ischemic tolerance due to its antiapoptotic activity [49]. Activation of ERK by reactive oxygen species is mediated by the Raf1-1-dependent signaling pathway [50]. The observed over-expression of Raf1 in penumbra after PTI was in agreement with the simultaneous up-regulation of ERK1/2 and ERK5. We also observed the over-expression of the EGF receptor, which stimulates the downstream pathways Raf1/ERK1/2 and PI 3-kinase/Akt under MCAO-induced ischemia. The cross-talk of these EGF receptor-associated signaling pathways either protects brain cells or potentiates their damage [51].
Protein kinase C isoforms α, β, and μ control multiple cell functions such as proliferation, neurotransmission, apoptosis, and brain response to stroke [52]. PKCα and PKCβ were also up-regulated after incomplete global cerebral ischemia [53]. PKCμ (also named as PKD) plays the neuroprotective role in cerebral ischemia: it phosphorylates chaperon HSP27, which then suppresses ASK1/JNK-mediated neuron apoptosis [54].
Protein phosphatases 1α and MKP-1 dephosphorylate and inactivate numerous phosphorylated proteins. Over-expression of MKP-1 under hypoxia/reoxygenation suppressed neuronal death via inhibition of JNK [55], whereas its inhibition increased injury [56]. Up-regulation of these protein phosphatases in the penumbra after PTI indicated mobilization of such neuroprotective mechanism.
Estrogen protects the brain from ischemic stroke through transcriptional activation of the antiapoptotic proteins Bcl-2 and Bcl-xL [57]. The observed up-regulation of the estrogen receptor in the penumbra after PTI was apparently aimed to neuroprotection.
Different stressful impacts such as reperfusion 1–4 h after transient global cerebral ischemia [58] or photothrombotic infarct, in our work, increased the expression of serum/glucocorticoid-regulated kinase SGK1, which regulates ion channels, cell volume, cell cycle, and apoptosis. It links cell hydration and metabolism and possibly plays a protective role in oxidative stress situations [59].
Phospholipase Cγ releases diacylglycerol, which stimulates protein kinase C, and inositol 1,4,5-trisphosphate, which stimulates Ca2+ release from endoplasmic reticulum. PLCγ1 is involved in necrosis and apoptosis of glial cells [60] and death of neurons [61]. Its down-regulation in the penumbra 1–24 h after PTI could play the neuroprotective role. The Ca2+/calmodulin-dependent signaling pathways regulate production and secretion of neurotransmitters, cytoskeleton remodeling, axonal transport, gene expression, cell survival, learning, and memory. Calmodulin, CaMKIIα, and CaMKIV are abundant in the mammalian brain. Nevertheless, their expression simultaneously increased in the penumbra after PTI. The neuroprotective role of CaMKII can be associated with phosphorylation and inhibition of some proapoptotic proteins such as NO synthase and GSK-3. Nonetheless, the excessive CaMKII can enhance excitotoxic neuronal death through phosphorylation of NMDA and AMPA receptors and Ca2+ influx. Application of CaMKII inhibitors has protected the brain from ischemic damage [62], whereas CaMKIV can inhibit excitotoxicity through disruption of the NMDAR/PSD95 complex, activation of the antiapoptotic PI3K/Akt pathway, and phosphorylation of the transcription factor CREB, which stimulates expression of some antiapoptotic genes [63].
The observed up-regulation of calreticulin in the penumbra 4 h after PTI could play a protective role. Calreticulin is a Ca2+-binding endoplasmic reticulum chaperone, which regulates Ca2+ homeostasis and controls protein folding. It regulates apoptosis via modulation of p53 expression and functional activity. Its level correlates with the rate of apoptosis [64]. During the early stage of ischemic stroke, calreticulin inhibited Fas/FasL-mediated apoptosis of neuronal cells [65].
Ca2+-binding protein S-100 and its β-chain (S-100β) were down-regulated in the penumbra after PTI. S-100β is specific for the nervous tissue, in particular, for glial cells. In the nanomolar range, it demonstrates neuroprotective properties, whereas at micromolar concentrations, it can induce apoptosis. Serum S-100 and S-100β are markers of brain injury such as acute stroke, neurotrauma, brain tumors, and neurodegenerative disorders [66, 67].
The observed up-regulation of proteins that positively control the cell cycle such as cyclin B1, cdk4, E2F1, c-myc, and EGF receptor in the penumbra after PTI indicated the proliferation processes. This corresponded to the stroke-induced proliferation of microvessels (angiogenesis) [68], astrocytes and microglia [69], and neural progenitor cells [70] in the penumbra. At the same time, other regulators of the cell cycle such as Cdk6 that controls G1/S transition, Cdc7 kinase that is involved in the initiation of DNA replication, topoisomerase-1 that mediates DNA replication, and Trf1 that controls the telomere length were down-regulated. These data indicated the suppression of proliferation. This discrepancy could be associated with different regulation of proliferation in various penumbra components, glial cells, or endothelium and in different locations, close to the infarction core or at the periphery of the penumbra.
The over-expression of APP in the penumbra after PTI was evidently associated with the up-regulation of nicastrin, a component of the γ-secretase protein complex that releases the intracellular C-terminal domain of APP (AICD). After translocation into the nucleus, AICD regulates transcription of GSK-3β, p53, and APP itself. APP regulates Ca2+ homeostasis, cell adhesion and migration, axonal outgrowth and restoration after injury, cell survival, and apoptosis [71]. Our results were in agreement with the up-regulation of APP after cerebral ischemia [72] that is considered as an adaptive and neuroprotective response [71, 72].
Cytoskeleton
Bidirectional changes in the expression of various cytoskeleton elements such as actin network, microtubules, and intermediate fibers were associated either with destruction or with remodeling of the penumbra tissue. The tissue integrity is provided by integrin- and cadherin-mediated cell adhesion to the extracellular matrix and to other cells, respectively. Inside the cell, the actin fibers are attached to the protein scaffold formed at the cytoplasmic tails of integrins or cadherins. These scaffolds consist of different structural and signaling proteins. The actin cytoskeleton is responsible for maintaining the cell shape, motility, and intracellular movements. Diverse proteins associated with actin cytoskeleton such as α-catenin, catenin p120CTN, p35, cofilin, actopaxin, and myosin Va were up-regulated in the penumbra after PTI. Catenin p120CTN and α-catenin that link E- and N-cadherins with the actin fibers and intracellular signaling proteins regulate the architecture and dynamics of the actin cytoskeleton. These proteins play critical roles in protection against cerebral infarct and brain recovery by remodeling of the neurovascular unit and cytoskeleton [73]. Actopaxin links integrins to the intracellular actin fibers. It recruits integrin-linked kinase and focal adhesion kinase, which signals about the integrin-dependent reorganization of the actin cytoskeleton [74]. FAK regulates the cell shape, motility, and some stages of apoptosis. Loss of the adhesion contacts leads to FAK-mediated rounding of the cell, membrane blebbing, chromatin condensation, and fragmentation of the nucleus. However, excessive FAK can inhibit apoptosis. Myosin Vα mediates the organelle transport along actin fibers in neuronal and glial cells and participates in the synaptic transmission. The up-regulation of these proteins in the penumbra could maintain the integrity of the brain parenchyma after cerebral ischemia.
At the same time, the changes in expression of some cytoskeleton-associated proteins in the penumbra after PTI could be associated with remodeling or death of the cell. Cofilin depolymerizes and reorganizes fibrillar actin and thereby changes the cell shape and motility. Its over-expression may be associated either with apoptosis or with the cell remodeling caused by impaired adhesion [75]. Under ischemic stroke, cofilin rapidly forms the rod-shaped actin bundles, which suppress the axonal transport and contribute to synaptic deficit [76]. Oxidative stress induces cofilin translocation into mitochondria and release of cytochrome c, which promotes apoptosis [77]. The siRNA-mediated down-regulation of cofilin increased viability of cultured cortical neurons, thereby indicating the involvement of cofilin in neurodegeneration and apoptosis [75, 76].
Spectrin (α and β isoforms) lines the intracellular side of the plasma membrane, maintains its integrity, and forms a scaffold for the actin cytoskeleton. Under brain injury, it is cleaved by calpain (in the case of necrosis) or caspases (in the case of apoptosis). This destroys the cytoskeleton and causes membrane blebbing [78]. Like in our study, the breakdown of spectrin was observed in the ischemic core and, to a lesser extent, in the penumbra 2–3.5 h after MCAO [79]. Tropomyosin, oppositely, stabilizes fibrillar actin and mediates formation of the complex actin network. It controls the actin filament dynamics and the cell shape during migration, morphogenesis, and cytokinesis. Its down-regulation in the penumbra after PTI indicated tissue destruction [80]. The level of ezrin, which links the plasma membrane to the actin cytoskeleton and controls the cell shape and motility, also decreased in the penumbra. This could be a result of PTI-induced tissue destruction. The focal adhesion kinase Pyk2, specific for CNS, activates ERK, JNK, and p38 after formation of the adhesive contacts and modulates the cytoskeleton architecture, cell morphology, ion channels, neuronal activity, and cell death. The decline in its phosphorylation could be associated with destruction of the penumbra tissue.
The microtubular cytoskeleton was also impaired 1–24 h after PTI. We have observed the decrease in the levels of βIV-tubulin and polyglutamated β-tubulin, which regulate the assembly of microtubules and their interactions with motor proteins and other microtubule-associated proteins [81]. The over-expression of p35 in the penumbra could be associated with cell damage and death. p35 binds to protein kinase Cdk5, and this complex controls the dynamics of the microtubular cytoskeleton. It hyperphosphorylates tau protein and regulates the microtubule remodeling [82]. Cerebral ischemia has been shown to up-regulate p35 and Cdk5 in the peri-infarct neurons and blood vessels and to induce cell death [73]. Over-activation of Cdk5 is a crucial prodeath signal in neurons, astrocytes, and endothelium, whereas its inhibition provides neuroprotection. It decreases the infarct size and induces functional recovery [73]. The down-regulation of cytokeratins 7 and 19, components of intermediate fibers, could be due to destruction of the penumbra tissue after PTI.
Vesicular Transport
PTI also impaired the vesicular transport in the penumbra. At 4 h after PTI, we observed down-regulation of the adaptor proteins AP2α, AP2β, and AP2γ as well as adaptins β1 and β2, which mediate formation of clathrin vesicles and endocytosis. Calpain-mediated suppression of the clathrin-dependent endocytosis and vesicular transport is known to sensitize cells to glutamate-mediated excitotoxicity and neurodegeneration [83], whereas β-COP that mediates the vesicular transport between endoplasmic reticulum and Golgi stacks was up-regulated. The similar up-regulation of β-COP in hippocampal neurons after transient ischemia indicated the activation of biosynthetic processes and vesicle transport [84]. The down-regulation of GRP1(ARNO3, cytohesin-3), which controls Golgi structure and regulates protein sorting and membrane trafficking, could be a result of destruction of the Golgi apparatus. The down-regulation of cystatin A, an inhibitor of lysosomal proteases, was possibly associated with stimulation of the lysosomal activity in the penumbra.
Vesicle transport plays the central role in synaptic transmission. Bidirectional changes in the expression of synaptic proteins were observed in the penumbra after PTI. On the one hand, tyrosine hydroxylase that synthesizes catecholamines (l-DOPA, dopamine, epinephrine, norepinephrine) and syntaxin, which is involved in the docking and fusion of synaptic vesicles with the presynaptic membrane and in neurotransmitter release, was down-regulated. On the other hand, SNAP25 that also mediates fusion of synaptic vesicles and release of neurotransmitters from presynaptic nerve terminals was up-regulated. This discrepancy remained unclear because both SNAP-25 and syntaxin are involved in the same synaptic processes [85].
The up-regulation of the ubiquitin-like protein NEDD8 is another hallmark of cell injury in the penumbra. Neddylation regulates proteasomal degradation of proteins. It is involved in the maintenance of synapses [86]. NEDD8 expression can serve as a biomarker of ischemic brain injury that may be helpful for the early detection and prognosis of ischemic stroke [87].
Thus, the expression of diverse proteins involved in signal transduction, regulation of apoptosis, remodeling of the cytoskeleton, adhesion, vesicular transport, and the cell cycle changed positively or negatively in the penumbra surrounding the infarct core during 1, or 4, or 24 h reperfusion. The early changes in the protein profile were observed 1 h after the PTI. After 4 h, the changes in the protein profile in the penumbra increased and included the larger set of proteins. In the infarction core, most of cells die rapidly from necrosis, whereas in the penumbra apoptosis plays a significant role [88]. As commonly assumed, apoptosis is executed by existing proteins in the cell, which consecutively activate each other. On the other hand, apoptosis is known to require protein synthesis [89]. Which proteins are synthesized? The present results on the concerted up-regulation of various proapoptotic and signaling proteins in the penumbra suggest that the existing protein pool is insufficient, and additional proteins are necessary. This raises the question on transcription factors and signaling pathways responsible for the expression of different proteins. Not all of them are still known.
According to the current paradigm, in the ischemic locus, the oxygen and glucose deficit induces ATP depletion, generation of reactive oxygen species, oxidative membrane injury, loss of ionic gradients, Ca2+ influx, K+ release, oxidative stress, and tissue edema. Ca2+ activates hydrolytic enzymes and induces necrosis or apoptosis. K+-mediated depolarization assists the opening of NMDA channels in neighboring neurons, additional Ca2+ influx, and release of K+ and glutamate. Such self-developing excitotoxic process results in the injury propagation outside the infarction core [1, 2]. [Ca2+]i increase is, therefore, the key event that promotes cell death. How do Ca2+ ions regulate the gene transcription? Massive Ca2+ influx into ischemic cells can occur through either synaptic or extrasynaptic NMDA receptors. Correspondingly, it can induce either neuronal survival via transcription of prosurvival genes and suppression of apoptotic cascades or neuronal death [90, 91]. The first effect is mediated by the Ca2+-dependent stimulation of Ras/Raf/ERK or CaMKI and CaMKIV pathways that stimulate the CREB-mediated transcription of prosurvival genes. CREB also mediates the expression of the mitochondrial Ca2+ uniporter that limits the Ca2+ overload [92]. Ca2+ entry through the extrasynaptic NMDA receptors inactivates the ERK pathway and suppresses the CREB-mediated transcription. At the same time, it can activate the FOXO-mediated transcription of various proapoptotic genes [89, 90]. Transcription repressor DREM is another Ca2+-dependent regulator of gene expression. It represses transcription by direct binding to DNA, but its affinity for DNA is reduced upon Ca2+ binding [93]. Ca2+-dependent phosphatase calcineurin dephosphorylates nuclear factor of activated T-cells (NFAT), which then translocates to the nucleus and regulates the transcription of α-actin and calponin, which are involved in formation and remodeling of the actin cytoskeleton [94]. Ca2+ also controls gene expression indirectly through the Ca2+-dependent interaction between calmodulin and S-100 with the basic helix-loop-helix transcription factors that prevents the binding of these factors to DNA [95].
It is still unclear, which cellular components of the penumbra—neurons, glial cells, or blood vessels—contribute more into the PTI-induced changes in the protein profile in the cerebral cortex. Another important reason that influences the PTI-induced tissue injury is the damage degree, which decreases gradually from the PTI core to the penumbra periphery. Both neurodegeneration and neuroprotection processes occur simultaneously in the penumbra, but neurodegeneration dominates in the area adjacent to the infarction core, whereas recovery processes prevail at the penumbra periphery. The aim of the neuroprotective therapy is to prevent the expansion of the death/survival boundary and reduce the degeneration zone.
The proteomic data provide a list of proteins with the changed expression in the penumbra at different time intervals after photothrombotic infarct. These results, like genomic data, do not disclose the reasons of these changes because signaling pathways and transcription factors, which control the expression of these proteins, remain unknown. In some cases, one can suggest the key functions of such transcription factors as E2F1 or p53. Further mechanistic studies will answer these questions. Some of these proteins are novel in the context of the stroke research, and the detailed investigation of the reasons and consequences of changes in their levels will discover the novel mechanism of the penumbra response to ischemia. Some over-expressed proteins may be the potential therapeutic targets for stroke therapy.
References
Iadecola C, Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14:1363–1368. doi:10.1038/nn.2953
Meisel A, Prass K, Wolf T, Dirnagl U (2004) Stroke. In: Bähr M (ed) Neuroprotection: models, mechanisms and therapies. Wiley, New York, pp. 9–43
Ginsberg MD (2008) Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology 55:363–389. doi:10.1016/j.neuropharm.2007.12.007
Mehta SL, Manhas N, Raghubir R (2007) Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 54:34–66
Puyal J, Ginet V, Clarke PG (2013) Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol 105:24–48. doi:10.1016/j.pneurobio.2013.03.002
Bu X, Zhang N, Yang X, Liu Y, Du J, Liang J, Xu Q, Li J (2011) Proteomic analysis of PKCβII-interacting proteins involved in HPC-induced neuroprotection against cerebral ischemia of mice. J Neurochem 117:346–356. doi:10.1111/j.1471-4159.2011.07209.x
Datta A, Park JE, Li X, Zhang H, Ho ZS, Heese K, Lim SK, Tam JP et al (2010) Phenotyping of an in vitro model of ischemic penumbra by iTRAQ-based shotgun quantitative proteomics. J Proteome Res 9:472–484. doi:10.1021/pr900829h
Dayon L, Turck N, Garcí-Berrocoso T, Walter N, Burkhard PR, Vilalta A, Sahuquillo J, Montaner J et al (2011) Brain extracellular fluid protein changes in acute stroke patients. J Proteome Res 10:1043–1051. doi:10.1021/pr101123t
Demyanenko SV, Panchenko SN, Uzdensky AB (2015) Expression of neuronal and signaling proteins in penumbra around a photothrombotic infarction core in rat cerebral cortex. Biochem Mosc 80:790–799. doi:10.1134/S0006297915060152
Uzdensky A, Demyanenko S, Fedorenko G, Lapteva T, Fedorenko A (2016) Photothrombotic infarct in the rat brain cortex: protein profile and morphological changes in penumbra. Mol Neurobiol 53:1–17. doi:10.1007/s12035-016-9964-5
Villa RF, Gorini A, Ferrari F, Hoyer S (2013) Energy metabolism of cerebral mitochondria during aging, ischemia and post-ischemic recovery assessed by functional proteomics of enzymes. Neurochem Int 63:765–781. doi:10.1016/j.neuint.2013.10.004
Uzdensky AB (2010) Cellular and molecular mechanisms of photodynamic therapy. Nauka, Sankt Petersburg in Russian
Dietrich WD, Watson BD, Busto R, Ginsberg MD, Bethea JR (1987) Photochemically induced cerebral infarction. I. Early microvascular alterations. Acta Neuropathol 72:315–325
Pevsner PH, Eichenbaum JW, Miller DC, Pivawer G, Eichenbaum KD, Stern A, Zakian KL, Koutcher JA (2001) A photothrombotic model of small early ischemic infarcts in the rat brain with histologic and MRI correlation. J Pharmacol Toxicol Methods 45:227–233
Schmidt A, Hoppen M, Strecker JK, Diederich K, Schäbitz WR, Schilling M, Minnerup J (2012) Photochemically induced ischemic stroke in rats. Exp Transl Stroke Med 4:13. doi:10.1186/2040-7378-4-13
Shanina EV, Redecker C, Reinecke S, Schallert T, Witte OW (2005) Long-term effects of sequential cortical infarcts on scar size, brain volume and cognitive function. Behav Brain Res 158:69–77
Engelmann D, Pützer BM (2010) Translating DNA damage into cancer cell death—a roadmap for E2F1 apoptotic signalling and opportunities for new drug combinations to overcome chemoresistance. Drug Resist Updat 13:119–131. doi:10.1016/j.drup.2010.06.001
Udayakumar T, Shareef MM, Diaz DA, Ahmed MM, Pollack A (2010) The E2F1/Rb and p53/MDM2 pathways in DNA repair and apoptosis: understanding the crosstalk to develop novel strategies for prostate cancer radiotherapy. Semin Radiat Oncol 20:258–266. doi:10.1016/j.semradonc.2010.05.007
Meng P, Ghosh R (2014) Transcription addiction: can we garner the Yin and Yang functions of E2F1 for cancer therapy? Cell Death Dis 5:e1360. doi:10.1038/cddis.2014.326
Bretones G, Delgado MD, León J (2015) Myc and cell cycle control. Biochim Biophys Acta 1849:506–516. doi:10.1016/j.bbagrm.2014.03.013
Jin K, Mao XO, Eshoo MW, Nagayama T, Minami M, Simon RP, Greenberg DA (2001) Microarray analysis of hippocampal gene expression in global cerebral ischemia. Ann Neurol 50:93–103
Li Y, Chopp M, Zhang ZG, Zaloga C, Niewenhuis L, Gautam S (1994) p53-immunoreactive protein and p53 mRNA expression after transient middle cerebral artery occlusion in rats. Stroke 25:849–855
Prunell GF, Arboleda VA, Troy CM (2005) Caspase function in neuronal death: delineation of the role of caspases in ischemia. Curr Drug Targets CNS Neurol Disord 4:51–61
Camins A, Verdaguer E, Folch J, Beas-Zarate C, Canudas AM, Pallàs M (2007) Inhibition of ataxia telangiectasia-p53-E2F-1 pathway in neurons as a target for the prevention of neuronal apoptosis. Curr Drug Metab 8:709–715
MacLachlan TK, El-Deiry WS (2002) Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc Natl Acad Sci U S A 99:9492–9497
Tu Y, Hou ST, Huang Z, Robertson GS, MacManus JP (1998) Increased Mdm2 expression in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab 18:658–669
Cox LS (1997) Multiple pathways control cell growth and transformation: overlapping and independent activities of p53 and p21Cip1/WAF1/Sdi1. J Pathol 183:134–140
Cmielová J, Rezáčová M (2011) p21Cip1/Waf1 protein and its function based on a subcellular localization. J Cell Biochem 112:3502–3506. doi:10.1002/jcb.23296
van Lookeren CM, Gill R (1998) Increased expression of cyclin G1 and p21WAF1/CIP1 in neurons following transient forebrain ischemia: comparison with early DNA damage. J Neurosci Res 53:279–296
Bui T, Sequeira J, Wen TC, Sola A, Higashi Y, Kondoh H, Genetta T (2009) ZEB1 links p63 and p73 in a novel neuronal survival pathway rapidly induced in response to cortical ischemia. PLoS One 4:e4373. doi:10.1371/journal.pone.0004373
Fatt MP, Cancino GI, Miller FD, Kaplan DR (2014) p63 and p73 coordinate p53 function to determine the balance between survival, cell death, and senescence in adult neural precursor cells. Cell Death Differ 21:1546–1559. doi:10.1038/cdd.2014.61
Dang CV (2012) MYC on the path to cancer. Cell 149:22–35. doi:10.1016/j.cell.2012.03.003
McGahan L, Hakim AM, Robertson GS (1998) Hippocampal Myc and p53 expression following transient global ischemia. Brain Res Mol Brain Res 56:133–145
Ferrer I, Friguls B, Dalfó E, Planas AM (2003) Early modifications in the expression of mitogen-activated protein kinase (MAPK/ERK), stress-activated kinases SAPK/JNK and p38, and their phosphorylated substrates following focal cerebral ischemia. Acta Neuropathol 105:425–437
Onoue S, Kumon Y, Igase K, Ohnishi T, Sakanaka M (2005) Growth arrest and DNA damage-inducible gene 153 increases transiently in the thalamus following focal cerebral infarction. Brain Res Mol Brain Res 134:189–197
Gao Y, Signore AP, Yin W, Cao G, Yin X, Sun F, Luo Y, Graham SH et al (2005) Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab 25:694–712
Hou ST, Xie X, Baggley A, Park DS, Chen G, Walker T (2002) Activation of the Rb/E2F1 pathway by the nonproliferative p38 MAPK during Fas (APO1/CD95)-mediated neuronal apoptosis. J Biol Chem 277:48764–48770
Piao CS, Kim SW, Kim JB, Lee JK (2005) Co-induction of alphaB-crystallin and MAPKAPK-2 in astrocytes in the penumbra after transient focal cerebral ischemia. Exp Brain Res 163:421–429
Roux PP, Barker PA (2002) Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67:203–233
Angelo MF, Aviles-Reyes RX, Villarreal A, Barker P, Reines AG, Ramos AJ (2009) p75 NTR expression is induced in isolated neurons of the penumbra after ischemia by cortical devascularization. J Neurosci Res 87:1892–1903. doi:10.1002/jnr.21993
Culmsee C, Zhu Y, Krieglstein J, Mattson MP (2001) Evidence for the involvement of Par-4 in ischemic neuron cell death. J Cereb Blood Flow Metab 21:334–343
Culmsee C, Landshamer S (2006) Molecular insights into mechanisms of the cell death program: role in the progression of neurodegenerative disorders. Curr Alzheimer Res 3:269–283
Neher JJ, Emmrich JV, Fricker M, Mander PK, Théry C, Brown GC (2013) Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A 110:E4098–E4107. doi:10.1073/pnas.1308679110
Suk K (2005) Role of caspases in activation-induced cell death of neuroglial. Curr Enzyme Inhibition 1:43–50
Kim SJ, Zhang Z, Hitomi E, Lee YC, Mukherjee AB (2006) Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum Mol Genet 15:1826–1834
Zhou X, Ding Q, Chen Z, Yun H, Wang H (2013) Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J Biol Chem 288:24151–24159. doi:10.1074/jbc.M113.482000
Jaenisch N, Popp A, Guenther M, Schnabel J, Witte OW, Frahm C (2014) Pro-apoptotic function of GABA-related transcripts following stroke. Neurobiol Dis 70:237–244. doi:10.1016/j.nbd.2014.06.015
Kitagawa H, Warita H, Sasaki C, Zhang WR, Sakai K, Shiro Y, Mitsumoto Y, Mori T et al (1999) Immunoreactive Akt, PI3-K and ERK protein kinase expression in ischemic rat brain. Neurosci Lett 274:45–48
Su C, Sun F, Cunningham RL, Rybalchenko N, Singh M (2014) ERK5/KLF4 signaling as a common mediator of the neuroprotective effects of both nerve growth factor and hydrogen peroxide preconditioning. Age (Dordr) 36:9685. doi:10.1007/s11357-014-9685-5
Wu HW, Li HF, Wu XY, Zhao J, Guo J (2008) Reactive oxygen species mediate ERK activation through different Raf-1-dependent signaling pathways following cerebral ischemia. Neurosci Lett 432:83–87. doi:10.1016/j.neulet.2007.11.073
Zhou J, Du T, Li B, Rong Y, Verkhratsky A, Peng L (2015) Crosstalk between MAPK/ERK and PI3K/AKT signal pathways during brain ischemia/reperfusion. ASN Neuro 7 . doi:10.1177/1759091415602463pii: 1759091415602463
Bright R (2005) The role of protein kinase C in cerebral ischemic stroke. Stroke 36:2781–2790
Fleegal MA, Hom S, Borg LK, Davis TP (2005) Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol 289:H2012–H2019
Stetler RA, Gao Y, Zhang L, Weng Z, Zhang F, Hu X, Wang S, Vosler P et al (2012) Phosphorylation of HSP27 by protein kinase D is essential for mediating neuroprotection against ischemic neuronal injury. J Neurosci 32:2667–2682. doi:10.1523/JNEUROSCI.5169-11.2012
Koga S, Kojima S, Kishimoto T, Kuwabara S, Yamaguchi A (2012) Over-expression of map kinase phosphatase-1 (MKP-1) suppresses neuronal death through regulating JNK signaling in hypoxia/re-oxygenation. Brain Res 1436:137–146. doi:10.1016/j.brainres.2011.12.004
Liu L, Doran S, Xu Y, Manwani B, Ritzel R, Benashski S, McCullough L, Li J (2014) Inhibition of mitogen-activated protein kinase phosphatase-1 (MKP-1) increases experimental stroke injury. Exp Neurol 261:404–411. doi:10.1016/j.expneurol.2014.05.009
Hurn PD, Macrae IM (2000) Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab 20:631–652
Nishida Y, Nagata T, Takahashi Y, Sugahara-Kobayashi M, Murata A, Asai S (2004) Alteration of serum/glucocorticoid regulated kinase-1 (sgk-1) gene expression in rat hippocampus after transient global ischemia. Brain Res Mol Brain Res 123:121–125
Schoenebeck B, Bader V, Zhu XR, Schmitz B, Lübbert H, Stichel CC (2005) Sgk1, a cell survival response in neurodegenerative diseases. Mol Cell Neurosci 30:249–264
Uzdenskii AB, Kolosov MS, Lobanov AV (2008) Neuron and gliocyte death induced by photodynamic treatment: signal processes and neuron-glial interactions. Neurosci Behav Physiol 38:727–735. doi:10.1007/s11055-008-9042-1
Matrone C, Marolda R, Ciafrè S, Ciotti MT, Mercanti D, Calissano P (2009) Tyrosine kinase nerve growth factor receptor switches from prosurvival to proapoptotic activity via Abeta-mediated phosphorylation. Proc Natl Acad Sci U S A 106:11358–11363. doi:10.1073/pnas.0904998106
Coultrap SJ, Vest RS, Ashpole NM, Hudmon A, Bayer KU (2011) CaMKII in cerebral ischemia. Acta Pharmacol Sin 32:861–872. doi:10.1038/aps.2011.68
Bell KF, Bent RJ, Meese-Tamuri S, Ali A, Forder JP, Aarts MM (2013) Calmodulin kinase IV-dependent CREB activation is required for neuroprotection via NMDA receptor-PSD95 disruption. J Neurochem 126:274–287. doi:10.1111/jnc.12176
Mesaeli N, Phillipson C (2004) Impaired p53 expression, function, and nuclear localization in calreticulin-deficient cells. Mol Biol Cell 15:1862–1870
Chen B, Wu Z, Xu J, Xu Y (2015) Calreticulin binds to Fas ligand and inhibits neuronal cell apoptosis induced by ischemia-reperfusion injury. Biomed Res Int 2015:895284. doi:10.1155/2015/895284
Korfias S, Stranjalis G, Papadimitriou A, Psachoulia C, Daskalakis G, Antsaklis A, Sakas DE (2006) Serum S-100B protein as a biochemical marker of brain injury: a review of current concepts. Curr Med Chem 13:3719–3731
Kumar H, Lakhotia M, Pahadiya H, Singh J (2015) To study the correlation of serum S-100 protein level with the severity of stroke and its prognostic implication. J Neurosci Rural Pract 6:326–330. doi:10.4103/0976-3147.158751
Szpak GM, Lechowicz W, Lewandowska E, Bertrand E, Wierzba-Bobrowicz T, Dymecki J (1999) Border zone neovascularization in cerebral ischemic infarct. Folia Neuropathol 37:264–268
Barreto GE, Sun X, Xu L, Giffard RG (2011) Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS One 6:e27881. doi:10.1371/journal.pone.0027881
Jin K, Wang X, Xie L, Mao XO, Zhu W, Wang Y, Shen J, Mao Y et al (2006) Evidence for stroke-induced neurogenesis in the human brain. Proc Natl Acad Sci U S A 103:13198–13202
Nalivaeva NN, Turner AJ (2013) The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett 587:2046–2054. doi:10.1016/j.febslet.2013.05.010
Clarke J, Thornell A, Corbett D, Soininen H, Hiltunen M, Jolkkonen J (2007) Overexpression of APP provides neuroprotection in the absence of functional benefit following middle cerebral artery occlusion in rats. Eur J Neurosci 26:1845–1852
Posada-Duque RA, Barreto GE, Cardona-Gomez GP (2014) Protection after stroke: cellular effectors of neurovascular unit integrity. Front Cell Neurosci 8:231. doi:10.3389/fncel.2014.00231
Clarke DM, Brown MC, LaLonde DP, Turner CE (2004) Phosphorylation of actopaxin regulates cell spreading and migration. J Cell Biol 166:901–912
Madineni A, Alhadidi Q, Shah ZA (2016) Cofilin inhibition restores neuronal cell death in oxygen-glucose deprivation model of ischemia. Mol Neurobiol 53:867–878. doi:10.1007/s12035-014-9056-3
Maloney MT, Bamburg JR (2007) Cofilin-mediated neurodegeneration in Alzheimer's disease and other amyloidopathies. Mol Neurobiol 35:21–44
Klamt F, Zdanov S, Levine RL, Pariser A, Zhang Y, Zhang B, Yu LR, Veenstra TD et al (2009) Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat Cell Biol 11:1241–1246. doi:10.1038/ncb1968
Sun M, Zhao Y, Gu Y, Xu C (2010) Neuroprotective actions of aminoguanidine involve reduced the activation of calpain and caspase-3 in a rat model of stroke. Neurochem Int 56:634–641. doi:10.1016/j.neuint.2010.01.009
Yao H, Ginsberg MD, Eveleth DD, LaManna JC, Watson BD, Alonso OF, Loor JY, Foreman JH et al (1995) Local cerebral glucose utilization and cytoskeletal proteolysis as indices of evolving focal ischemic injury in core and penumbra. J Cereb Blood Flow Metab 15:398–408
Schevzov G, Curthoys NM, Gunning PW, Fath T (2012) Functional diversity of actin cytoskeleton in neurons and its regulation bytropomyosin. Int Rev Cell Mol Biol 298:33–94. doi:10.1016/B978-0-12-394309-5.00002-X
Fukushima N, Furuta D, Hidaka Y, Moriyama R, Tsujiuchi T (2009) Post-translational modifications of tubulin in the nervous system. J Neurochem 109:683–693. doi:10.1111/j.1471-4159.2009.06013.x
Shah K, Lahiri DK (2016) A tale of the good and bad: remodeling of the microtubule network in the brain by Cdk5. Mol Neurobiol. doi:10.1007/s12035-016-9792-7
Rudinskiy N, Grishchuk Y, Vaslin A, Puyal J, Delacourte A, Hirling H, Clarke PG, Luthi-Carter R (2009) Calpain hydrolysis of alpha- and beta2-adaptins decreases clathrin-dependent endocytosis and may promote neurodegeneration. J Biol Chem 284:12447–12458. doi:10.1074/jbc.M804740200
Katayama T, Imaizumi K, Tsuda M, Mori Y, Takagi T, Tohyama M (1998) Expression of an ADP-ribosylation factor like gene, ARF4L, is induced after transient forebrain ischemia in the gerbil. Brain Res Mol Brain Res 56:66–75
Xu J, Luo F, Zhang Z, Xue L, Wu XS, Chiang HC, Shin W, Wu LG (2013) SNARE proteins synaptobrevin, SNAP-25, and syntaxin are involved in rapid and slow endocytosis at synapses. Cell Rep 3:1414–1421. doi:10.1016/j.celrep.2013.03.010
Scudder SL, Patrick GN (2015) Synaptic structure and function are altered by the neddylation inhibitor MLN4924. Mol Cell Neurosci 65:52–57. doi:10.1016/j.mcn.2015.02.010
Wong YH, Wu CC, Lai HY, Jheng BR, Weng HY, Chang TH, Chen BS (2015) Identification of network-based biomarkers of cardioembolic stroke using a systems biology approach with time series data. BMC Syst Biol 9(Suppl 6):S4. doi:10.1186/1752-0509-9-S6-S4
Love S (2003) Apoptosis and brain ischaemia. Prog Neuro-Psychopharmacol Biol Psychiatry 27:267–282
Lockshin RA, Zakeri Z (2001) Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol 2:545–550
Hagenston AM, Bading H (2011) Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb Perspect Biol 3:a004564. doi:10.1101/cshperspect.a004564
Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696. doi:10.1038/nrn2911
Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, Hines KJ, Ragheb J et al (2015) Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal 8:ra23. doi:10.1126/scisignal.2005673
Carrión AM, Link WA, Ledo F, Mellström B, Naranjo JR (1999) DREAM is a Ca2 + −regulated transcriptional repressor. Nature 398:80–84
Barlow CA, Rose P, Pulver-Kaste RA, Lounsbury KM (2006) Excitation-transcription coupling in smooth muscle. J Physiol 570:59–64
Mellström B, Naranjo JR (2001) Ca2+-dependent transcriptional repression and derepression: DREAM, a direct effector. Semin Cell Dev Biol 12:59–63
Acknowledgments
The work was supported by the Russian Science Foundation (grant 14-15-00068). A. Uzdensky’s work was supported by the Ministry of Education and Science of Russian Federation (grant “Science organization” no. 790). The authors used the equipment of the Center for Collective Use of Southern Federal University “High Technology” supported by the Ministry of Education and Science of the Russian Federation (project RFME_FI59414X0002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experimental procedures were carried out in accordance with the European Union guidelines 86/609/ЕЕС for the use of experimental animals and local legislation for ethics of experiments on animals. The animal protocols were evaluated and approved by the Animal Care and Use Committee of the Southern Federal University (approval no. 02/2014).
Competing Interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Demyanenko, S., Uzdensky, A. Profiling of Signaling Proteins in Penumbra After Focal Photothrombotic Infarct in the Rat Brain Cortex. Mol Neurobiol 54, 6839–6856 (2017). https://doi.org/10.1007/s12035-016-0191-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0191-x