
Abstract
Phosphoinositide 3-kinase γ (PI3Kγ) is linked to neuroinflammation and phagocytosis. This study was conducted to elucidate conjectural differences of lipid kinase-dependent and kinase-independent functions of PI3Kγ in the evolvement of brain damage induced by focal cerebral ischemia/reperfusion. Therefore, PI3Kγ wild-type, knockout, and kinase-dead mice were subjected to middle cerebral artery occlusion followed by reperfusion. Tissue damage and cellular composition were assessed by immunohistochemical stainings. In addition, microglial cells derived from respective mouse genotypes were used for analysis of PI3Kγ effects on phagocytic activity, matrix metalloproteinase-9 release, and cAMP content under conditions of oxygen/glucose deprivation and recovery. Brain infarction was more pronounced in PI3Kγ-knockout mice compared to wild-type and kinase-dead mice 48 h after reperfusion. Immunohistochemical analyses revealed a reduced amount of galectin-3/MAC-2-positive microglial cells indicating that activated phagocytosis was reduced in ischemic brains of knockout mice. Cell culture studies disclosed enhanced metalloproteinase-9 secretion in supernatants derived from microglia of PI3Kγ-deficient mice after 2-h oxygen/glucose deprivation and 48-h recovery. Furthermore, PI3Kγ-deficient microglial cells showed a failed phagocytic activation throughout the observed recovery period. Lastly, PI3Kγ-deficient microglia exhibited strongly increased cAMP levels in comparison with wild-type microglia or cells expressing kinase-dead PI3Kγ after oxygen/glucose deprivation and recovery. Our data suggest PI3Kγ kinase activity-independent control of cAMP phosphodiesterase as a crucial mediator of microglial cAMP regulation, MMP-9 expression, and phagocytic activity following focal brain ischemia/recirculation. The suppressive effect of PI3Kγ on cAMP levels appears critical for the restriction of ischemia-induced immune cell functions and in turn tissue damage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brain ischemia causes both immediate and delayed cell death and is accompanied by a robust inflammatory response that can exacerbate injury during reperfusion. Recent evidence suggests that elements of the immune system are intimately involved in all stages of the ischemic cascade, from the acute intravascular events triggered by the interruption of the blood supply to the parenchymal processes leading to brain damage and the ensuing tissue repair [1]. Activation of ischemia-induced inflammation is mainly driven by “danger signals” released by dying and dead cells [2]. Whereas the progress of tissue damage after brain ischemia is forced by extracellular matrix metalloproteinase (MMP) activation [3, 4], phagocytic removal of dying cells and tissue debris appears to be important for resolution of inflammation and tissue repair [5, 6].
Microglial cells as the main cell population of resident immune-competent cells in the CNS are activated early after ischemia, preceding the invasion of blood-born immune cells [7]. Activation of microglia is normally strictly controlled by mechanisms involving neuronal-glial communication [8]. Furthermore, microglial activation in cortical tissue appears to be a general response to metabolic stress [9] associated with protein synthesis inhibition [10] at no detectable cortical damage [11]. Therefore, microglia activation is a key factor in the defense of neural parenchyma against various injuries, including brain ischemia [12]. Ischemic stroke has been shown as a powerful stimulus that disables the endogenous inhibitory signaling and triggers pronounced microglial activation [13]. Once activated, microglia exhibit a spectrum of phenotypes and functions to either exacerbate ischemic injury or to induce repair and regeneration—depending on different molecular signals received by the microglial receptors [14].
A wide variety of partly opposing responses have been described for activated microglia and associated with stroke pathophysiology; however, underlying signal transduction pathways are hitherto poorly characterized [15].
Phosphoinositide 3-kinase γ (PI3Kγ was originally characterized as a signaling protein mediating G protein-coupled receptor stimulation by its enzymatic activity to produce phosphatidylinositol 3,4,5-trisphosphate for downstream protein kinase B/Akt activation [16–18]. Subsequent studies of PI3Kγ-deficient mice revealed impaired respiratory burst and motility of peripheral leukocytes indicating a major regulatory function of this signaling protein in immune cells [19]. Unexpectedly, our investigations in heart revealed an intimate interplay of the PI3Kγ with the cAMP-signaling pathway. A first study revealed that PI3Kγ attenuates the cAMP/PKA pathway by working as an activator of cardiomyocytic phosphodiesterases, which hydrolyze cAMP to 5′-AMP [20]. Recently, we have disclosed the signaling protein PI3Kγ as a key mediator of microglial cAMP signaling. This pathway has been found responsible for LPS-induced microglial MMP-9 production with subsequent blood–brain barrier (BBB) deterioration and phagocytic control [21, 22]. A previous report revealed that PI3Kγ plays a significant role in ischemia/reperfusion-induced brain damage [23] without consideration of differences in its intracellular signaling performance.
Aim of this study was to elucidate conjectural differences of lipid kinase-dependent and kinase-independent functions of PI3Kγ in the evolvement of brain damage induced by focal cerebral ischemia/reperfusion. We hypothesized a specific role of PI3Kγ in microglial cAMP/PKA signaling on their inflammatory response in brain ischemia/reperfusion. Therefore, we used PI3Kγ knockout mice and derived microglial cell with complete loss of PI3Kγ and compared the respective functional and structural consequences after focal brain ischemia with mice and derived microglial cells with deficient PI3Kγ/AKT signaling by loss of lipid kinase activity induced by targeted point mutation [20] but preserved cAMP/PKA signaling. Our data indicate that PI3Kγ deficiency enhances the extent of brain damage during a later stage of acute recovery, likely mediated by aberrant microglial activity. Corroborating the in vivo analyses, investigations with primary microglial cells identified the suppressive effect of PI3Kγ on cAMP signaling on MMP-9 release and reinforced phagocytosis as essential for ameliorating neuroinflammatory responses and improved brain tissue survival.
Materials and Methods
Experiments were approved by the committee of the Thuringian State Government on Animal Research and performed according to the Protection of Animals Act of the Federal Republic of Germany. The animals were treated in accordance with the declaration of Helsinki and the guiding principles in the care and use of animals. Efforts were made to reduce the number of animals used and their suffering. All surgeries were performed under appropriate anesthesia (isoflurane).
Animals
Male 3-month-old PI3Kγ knockout mice (PI3Kγ−/−) [19] and PI3Kγ kinase-dead mice (PI3KγKD/KD; mice carrying a targeted point mutation in the PI3Kγ gene causing loss of lipid kinase activity) [20] were on the C57BL/6J background for more than ten generations. Consequently, age-matched C57BL/6 mice were used as controls. The animals were maintained with 12-h light and dark cycles with free access to food and water. Ambient temperature was 29 ± 1 °C during the whole experimental period.
Bone Marrow Transplantation
Six- to 8-week-old recipient mice (either wild-type or PI3Kγ−/− mice) underwent a lethal total body irradiation (1000 Rad). Freshly isolated total host bone marrow cells (derived from the appropriate genotype as indicated in the figure legends) were then injected into the lateral tail vein of syngeneic recipient mice (5 × 106 cells per mouse) 24 h after irradiation. Mice were maintained in an isolator, fed irradiation-sterilized chow, and allowed 4–6 weeks of recovery before surgery.
Induction of Transient Focal Brain Ischemia
Animals were anesthetized with 2.5 % isoflurane for induction and 1.5 % isoflurane for maintenance in 70/30 % nitrous oxide/oxygen administered by mask. Rectal temperature was maintained at 36.5 to 37 °C with a feedback-controlled heating blanket. Focal brain ischemia was induced by transient occlusion of the middle cerebral artery (MCA) using the intraluminal filament technique [24]. The right common carotid artery (CCA), the external carotid artery (ECA), and the internal carotid artery (ICA) were dissected from surrounding tissue. A 7–0 nylonmonofilament (70SPRe, Doccol Corp, USA) was inserted into the ICA (11 mm) for 45 min to occlude the MCA. Operation time per animal did not exceed 15 min. The intraluminal suture was left in situ for 45 min. Then, animals were reanesthetized, and the occluding monofilament was withdrawn to allow reperfusion. Sham animals underwent the same surgical procedure as the treatment group, except that occlusion of the middle cerebral artery was omitted. Animals were allowed to survive for 6 h (survival rate 100 % in all genotypes tested), 12 h (survival rate 100 % in all genotypes tested) or 48 h (survival rate of wild-type mice 10/12, PI3Kγ−/− mice 10/11, PI3KγKD/KD; 11/12), respectively. Immediately after recovery from anesthesia, and 24 and 48 h later, neurological deficits were scored in order to verify correct MCAO induction by a modified Bederson score [25] (scoring system: 0, no deficit; 1, forelimb flexion; 2, unidirectional circling; 3, longitudinal spinning; 4, no movement). Mice were excluded from analysis when subarachnoid hemorrhage was macroscopically observed during brain harvesting. No difference in exclusion rates between the groups was observed.
Assessment of Infarct Volume
Infarct volume was determined as described previously [26]. Briefly, brains were removed at various reperfusion times (6, 12, 48 h). Therefore, mice were deeply anesthetized and perfused with 4 % paraformaldehyde (PFA) in phosphate buffer after rinsing with PBS by cardiac puncture via the left ventricle. Brains were removed immediately after fixation and postfixed for 5 h in 4 % PFA at 4 °C. After cryoprotection in phosphate-buffered saline (PBS) containing 30 % sucrose, brains were frozen in methylbutane at −30 °C and stored at −80 °C. Whole brains were cut by coronal sections at 40 μm on a freezing microtome (Microm International GmbH, ThermoScientific, Germany). The slices were immunostained by MAP2 (see below) to visualize the infarctions. Sections were photographed with a digital Olympus DP50 camera. Planimetric measurements (ImageJ software, National Institutes of Health, Bethesda, MD) blinded to the treatment groups were used to calculate lesion volumes, which were corrected for brain edema accordingly.
Immunohistochemistry
Free-floating sections were treated with Tris-buffered saline containing 10 % normal donkey or goat serum, 1 % BSA, and 0.2 % Triton X-100. Sections were incubated with the desired primary antibody at 4 °C overnight, followed with the associated secondary antibody at 4 °C for 1 h, and visualized by fluorescence imaging. Control sections were incubated with the blocking solution in the absence of the respective primary antibody. The following primary antibodies were used: mouse anti-MAP2 (1:1000) antibody (Sigma-Aldrich Chemie Gmbh Munich, Germany) for MAP2 staining, goat polyclonal anti-Iba-1 (1:400) antibody (Abcam, Cambridge, UK) for Iba1 staining and rabbit polyclonal anti-MMP-9 (1:250) antibody (Cell Signaling Technology, Danvers, USA) for MMP-9, rabbit anti-mouse MAC-2/galectin-3 (1:750) antibody, and rabbit anti-mouse PMN (1:1000) antibody for neutrophil staining. For visualization, the secondary fluorescent isotype-specific antibodies Alexa Fluor® 488 and Alexa Fluor® 568 (Molecular Probes, Inc., Eugene, USA) were used. Cell numbers of respective immune-positive cells were counted from every five visual fields derived from sections adjacent to the both sections with the largest infarct size (according to the respective MAP2 staining) obtained from every three animals of each group. Counting was performed by one observer in a blinded fashion. For MAP2 visualization sections were processed by the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, USA) using a donkey anti-mouse biotinylated secondary antibody (Dianova, Hamburg, Germany). Finally, immunoreactivity was developed in 3,3′-diaminobenzidine tetrahydrochloride (DAB, Sigma-Aldrich Chemie Gmbh Munich, Germany).
Cell Culture
Primary microglial cells were isolated from neonatal mouse cerebral cortex of C57BL/6J wild-type (wt), PI3Kγ-knockout (PI3Kγ−/−), and PI3Kγ-kinase-dead (PI3KγKD/KD) mice at day 1 after birth (P1). Microglial cells were cultured at 37 °C and 5 % CO2 in DMEM with high glucose supplemented with 10 % FCS, 1 % penicillin/streptomycin, and 1 % amphotericin B. Co-cultures of microglial cells and astrocytes were incubated for 14 days at 37 °C and 5 % CO2. Adherent microglial cells were separated from co-cultures by addition of PBS/EDTA and careful shaking. Purity of microglia culture was >95 %. After harvesting, microglial cells were counted and seeded in 96-well plates (10,000 cells/well) and 6-well plates (300,000 cells/well) for oxygen-glucose-deprivation (OGD) experiments.
OGD
Primary microglia were seeded in single cell culture dishes (35 mm, 9.4 cm2, 300,000 cells/dish) or white 96-well plates and incubated over night at 37 °C and 5 % CO2 in DMEM high glucose containing 10 % FCS, 1 % penicillin/streptomycin, and 1 % amphotericin B. After becoming adherent, cells were starved 4 h in DMEM medium without FCS. After starvation, the medium was changed to DMEM without glucose, and cells were incubated under hypoxic conditions at 1 % O2 for 2 h. After OGD, the medium was replaced by DMEM high glucose, 1 % penicillin/streptomycin, and 1 % amphotericin B, and microglia were incubated at normoxic conditions at 37 °C and 5 % CO2 for 24 or 48 h (reoxygenation). Thereafter, microglial cells were used for protein lysis, phagocytosis assay, or cAMP assay.
Gel Zymography
As described previously [21], cells were seeded in 6-well plates and incubated at 37 °C (5 % CO2). Following adherence, cells were starved for 4 h and incubated under OGD conditions for the indicated times. Supernatants were collected and centrifuged (500g, 4 °C, 5 min). Thereafter, 500 μl of supernatant was concentrated using a 30-kDa Amicon Ultra 0.5-mL centrifugal filter (Millipore, Billerica, MA, USA). The whole concentrate was mixed with 5× nonreducing protein sample buffer (1.5 M Tris–HCl, pH 6.8, 2 % SDS, 50 % glycerol, 0.1 mg/ml bromophenol blue) and loaded to a 8 % polyacrylamide gel containing 0.6 g/l gelatin. Samples were separated at 15-mA constant current, the gels were washed in Triton X-100 (30 min) and in water (30 min), and then incubated in TAB buffer (100 mM Tris HCl, pH 7.8, 30 mM CaCl2, 0.01 % NaN3) for 20 h. Thereafter, the gels were stained with 0.25 % Coomassie blue G250 in de-/stain buffer (25 % methanol, 7 % acetic acid) [27]. Stained gels were scanned and analyzed by quantitative densitometry using FUJIFILM Multi Gauge Ver.3.0 software (Fuji Photo Film Co., Ltd., Tokyo, Japan).
Phagocytosis Assay
Estimation of microglial phagocytic activity has been described previously [22]. Briefly, microglial cells were seeded in single cell culture dishes (35 mm, 9.4 cm2, 300,000 cells/dish) and incubated under OGD conditions. At 23 or 47 h of reoxygenation, the phagocytosis assay was performed by using GFP-producing E. coli. For this purpose, 40 μl (optical density 10) of the suspended bacteria were added to the microglial cells and incubated for 1 h. Thereafter, cells were harvested, washed, and resuspended in PBS. The phagocytic activity of the cells was measured by flow cytometry using a FACS Canto instrument (BD, Heidelberg, Germany).
SDS Page and Western Blotting
Monoclonal anti-mouse p110γ antibody was produced in our facility in Jena. Other antibodies were obtained from Cell Signaling Technologies (Danvers, USA; Phospho-CREB, #9198) and Sigma-Aldrich Chemie Gmbh (Munich, Germany; β-Actin #A5441). For quantification of protein expression and phosphorylation under in vitro OGD conditions, microglial cells of equal cell numbers were seeded in single cell culture dishes (35 mm, 9.4 cm2) and incubated under hypoxic conditions for 2 h followed by reoxygenation at 37 °C (5 % CO2) for 24 or 48 h. Thereafter, cells were lysed in 200-μl RIPA lysis buffer composed of 50 mM Tris/HCl pH 8, 150 mM NaCl, 1 % (v/v) NP-40, 0.5 % (v/v) deoxycholate, 0.1 % (w/v) SDS, 100 μg/ml Pefa-Block, 1 μg/ml Pepstatin, 10 μM sodium orthovanadate, and 1 μg/ml leupeptin. Cell lysates were centrifuged at 13,000g at 4 °C for 20 min, and the supernatants were mixed with 5× protein sample buffer (5 % SDS, 33 % glycerol, 25 % β-mercaptoethanol) and heated to 95 °C for 5 min. Protein samples were separated by SDS-PAGE using 10 % PAGE-gels, transferred to a polyvinylidene fluoride membrane and developed with the indicated antibodies followed by enhanced chemiluminescence reaction.
cAMP Assay
Microglial cells were seeded in 96-well clear bottom plates and incubated under OGD conditions followed by reoxygenation. cAMP was measured following the manufactures protocol (Promega, cAMP GloAssayKit).
Statistical Analysis
Data are reported as means ± SD, if not otherwise indicated. Comparisons between groups were made with one-way or two-way analysis of variance or Mann–Whitney rank sum test, if appropriate. Post hoc comparisons were made with the Holm–Sidak test. Differences were considered significant when p < 0.05.
Results
PI3Kγ Deficiency Provokes Maturation of Enlarged Brain Infarction
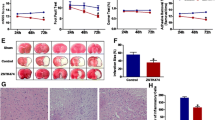
Evaluation of infarct size after temporary focal brain ischemia by MCAO revealed more pronounced enlargement of brain infarction and neurologic deficit score in PI3Kγ−/− mice compared to wild-type mice 48 h after reperfusion (p < 0.05, Fig. 1, Table 1). Early infarct demarcation (6 and 12 h after reperfusion) was similar in both genotypes (Fig. 2a, b). In order to assess the causal impact of specific PI3Kγ signaling reactions on this phenotype, we also studied the response of knock-in mice expressing lipid kinase-dead PI3Kγ (PI3KγKD/KD). Infarct size after temporary MCAO and 48-h recirculation was similar in PI3KγKD/KD and wild-type mice indicating that PI3Kγ inhibits enlargement of infarct size not through its catalytic but through its scaffolding function. In addition, differences shown in infarct volume were not caused by differences in vascular anatomy between wild-type mice and mutants under consideration (Fig. 1 Suppl., Fig. 2 Suppl.).

PI3Kγ deficiency (PI3Kγ KO) enhances infarct volume 48 h after temporary (45 min) MCAO (values are means + SD, n = 10–12 per group; *p < 0.05, *significant difference to wild-type mice; triangles indicate the individual values of infarct size; one-way ANOVA, followed by Holm–Sidak test for post hoc multiple comparisons)

Time-dependent increase of infarct volume 6 (a) and 12 h (b) after temporary (45 min) MCAO. Note that there was no dependency of PI3Kγ on the extent of infarct volume. c Enhanced infarct volume in chimeras of PI3Kγ−/−/wild-type bone marrow chimeras in comparison to wild-type/PI3Kγ−/−-bone marrow (BM) 48 h after temporary (45 min) MCAO (values are means + SD, *p < 0.05, *significant difference between both groups; Mann–Whitney rank sum test; n = 9–10 per group; triangles indicate the individual values of infarct size)
Since PI3Kγ controls multiple functions of hematopoetic cells including cell migration [28] and invasion of blood-born leukocytes into the injured brain tissue, we checked for polymorphonuclear leukocytes (PMN) infiltration 48 h after focal brain ischemia. We found a marked increase of invaded PMN in the infarct core regions, compared to corresponding peri-infarct areas (Table 2). Importantly, PI3Kγ−/− mice exhibited a reduced amount of PMN invasion as compared to wild-type mice, making the contribution of PMN to the increased infarction in PI3Kγ−/− mice unlikely. PI3KγKD/KD mice exhibited similar PMN accumulation in the infarct core as seen in PI3Kγ−/− mice.
To further elucidate the potential contribution of PI3Kγ in invading leukocytes to infarct progression, we determined infarct size 48 h after MCAO in the bone marrow chimeras. Intriguingly, PI3Kγ−/− mice transplanted with bone marrow harvested from wild-type mice developed enlarged brain infarctions compared to wild-type mice transplanted with PI3Kγ−/− bone marrow (Fig. 2c). These data indicate that the increased damage seen in PI3Kγ−/− mice following MCAO injury occurred independently of its role in bone-marrow-derived hematopoietic cells.
We therefore sought for another cellular basis and potential molecular mediators of the destructive processes involved in the discovered PI3Kγ-dependent ischemic brain damage. We considered the contribution of brain-resident microglial cells because recent own findings indicated PI3Kγ as a crucial mediator of microglial cell activation, MMP expression, and subsequent BBB deterioration in the development of sepsis-induced brain damage [21]. Indeed, IHC evaluation of brain slices revealed a distinct immunoreactivity for MMP-9 in Iba-1-positive cells (a marker of microglial cells), suggesting expression of this proteinase in activated microglial cells within the ischemic core (Fig. 3).

Detectable cells co-labeling for Iba1 (a marker of microglial cells in red, left panel) and MMP-9 (middle panel, in green) in brain slices derived from wild-type (upper part), PI3Kγ-deficient (middle part) and PI3Kγ kinase-dead (lower part) mice (right panel, merged pictures, bar 50 μm) (color figure online)
PI3Kγ Deficiency Enhances Microglial cAMP Level After OGD/Reoxygenation Challenge
Our recent reports identified negative control of cAMP signaling as a prominent lipid kinase-independent activity of PI3Kγ in resting and activated microglial cells [21, 22]. We used oxygen-glucose deprivation (OGD) to mimic in vitro conditions in the infarcted tissue. As shown in Fig. 4a, PI3Kγ-deficient microglia exhibited strongly increased cAMP in comparison with wild-type microglia or cells expressing PI3KγKD/KD. This effect was further enhanced early after OGD/reoxygenation challenge. The missing response of intracellular cAMP content in wild type as well as PI3KγKD/KD microglial cells owing to OGD/reoxygenation challenge was accompanied by PI3Kγ upregulation (Fig. 4b) indicating a PI3Kγ-related negative control of cAMP signaling to prevent its excessive activation. A similar relation was observed if cAMP levels in these microglial cells were indirectly measured by assaying phosphorylation of the cAMP response element-binding transcription factor CREB (Fig. 4b). These data confirmed the role of PI3Kγ as a critical mediator of cAMP signaling in microglia in response to OGD/reoxygenation challenge, presumably through a scaffold-dependent stimulation of cAMP phosphodiesterase.

Effects of PI3Kγ on cAMP level in microglia. a Increased cAMP levels in primary microglial cells derived from knockout (KO, PI3Kγ−/−, open columns) mice compared to those from wild-type (Wt, black columns) and PI3 Kγ-kinase-dead (KD, PI3KγKD/KD, hatched columns) mice. Cells were seeded in 6-well plates, starved overnight, administered to oxygen/glucose deficiency (OGD) for 2 h followed by 24 h and 48 h recovery under normoxic/normoglycemic conditions. cAMP levels were evaluated using Promega cAMP GloAssayKit. b cAMP-dependent CREB phosphorylation in Wt, KO, and KD primary microglia. CREB phosphorylation has been assayed by Western blotting using specific pCREB antibodies. A representative blot is shown. Values are mean + SEM, n = 10–12. *§ p < 0.05; §significant difference to normoxia (two-way ANOVA, treatment effect), *significant difference between Wt and KO within the respective treatment group (one-way ANOVA followed by Holm–Sidak test for post hoc multiple comparisons)
PI3Kγ Mediates Microglial MMP-9 Release
Prompted by the reported in vivo data, further investigations were focused on regulatory functions of PI3Kγ in microglial cells with potential relevance for infarct enlargement in PI3Kγ-deficient mice. As shown in Fig. 4b, challenge with OGD induced a marked increase of PI3Kγ production in primary microglial cells derived from wild-type mice, consistent with an involvement in the control of neuroinflammatory reactions of these immune cells. Next, we asked for the role of PI3Kγ in microglial MMP expression. We determined the activity of microglial MMP-9 release into the supernatant by gelatin zymography. As shown in Fig. 5, 2-h OGD and 48-h recovery caused an enhanced MMP-9 secretion, with the highest activity in supernatants derived from microglia of PI3Kγ-deficient mice. Wild type as well as PI3KγKD/KD microglia exhibited only low levels of liberated MMP-9 activity after OGD challenge. This observation indicates that the PI3Kγ scaffold function—and presumably related effects on cAMP signaling—is important for negative control of OGD-induced MMP-9 production.

Increased extracellular MMP-9 activity of primary microglial cell-derived knockout (KO, open columns) mice after OGD/reoxygenation challenge, analyzed by zymography of cell culture supernatants (black columns, wild-type (Wt); hatched columns, PI3Kγ kinase-dead (KD) microglial cells). Cells were seeded in 6-well plates, starved overnight and used as well (Normoxia) or stimulated with OGD for 2 h and incubated in normoxia for 24 h (2 h OGD 24 h Reox) or incubated in normoxia for 48 h (2 h OGD 48 h Reox). Five hundred microliters of cell culture supernatant was concentrated (30 kDa size exclusion) and used for gelantin zymography. Values are mean + SD, n = 5 independent experiments derived from five different microglia isolations. *§ p < 0.05, *significant difference vs. wt within the respective state, §significant difference vs. normoxia (two-way ANOVA, followed by Holm–Sidak test for post hoc multiple comparisons)
PI3Kγ Deficiency Inhibits Microglial Phagocytic Activity
Considering that microglial phagocytic activity is important for removal of dying cells and debris after focal brain ischemia and recent own data revealing PI3Kγ-dependent suppression of cAMP signaling as a critical regulatory element of microglial phagocytosis [22], we asked for specific signaling functions of PI3Kγ involved in brain ischemia/recirculation-induced microglial phagocytosis.
To assess phagocytic activity of microglial cells within the brain infarction after MCAO and 48 h recovery, an IHC analysis of galectin-3/MAC-2 immunoreactivity [29] in Iba-1 positive cells was performed. Intriguingly, a markedly reduced number of Iba1- and galectin-3/MAC-2 positive cells were found within the peri-infarct regions of PI3Kγ-deficient mice compared with wild-type and kinase-dead PI3Kγ knockin-mice (Fig. 6).

Representative pictures (a) of cells co-labeling for Iba1 (a marker of microglial cells in red, left panel) and galectin-3/MAC-2 (middle panel, in green) in brain slices derived from wild-type (upper part), PI3Kγ-deficient (middle part), and PI3Kγ kinase-dead (lower part) mice (right panel, merged pictures, bar 50 μm). (b) Number of Iba1- and MAC-2/Galectin-3- immunoreactive cells per tissue section for different groups of mice (wild-type, black columns; PI3Kγ-/-, open columns; PI3KγKD/KD, hatched columns). Values are given as means + SD, *,# P < 0.05, * significant difference to wild-type (Wt) mice, # significant difference to PI3KγKD/KD mice (MAC-2: MAC-2/Galectin-3).
Subsequent studies on primary microglial cells subjected to OGD/reoxygenation challenge were consistent with the IHC findings. OGD/reoxygenation caused a marked increase of the phagocytic activity in microglial cells derived from wild-type mice, whereas PI3Kγ-deficient microglial cells showed a markedly diminished phagocytic activity already under control conditions and a failed phagocytic activation throughout the observed recovery period following OGD (Fig. 7). Microglial cells derived from PI3KγKD/KD mice showed a comparable phagocytic activity as seen in wild-type microglial cells. Furthermore, the OGD/reoxygenation challenge induced a similar phagocytic activation in PI3KγKD/KD microglial cells, compared with wild-type microglial cells. These data clearly indicate a microglial deficiency of phagocytosis activation in the absence of PI3Kγ, which depends on PI3Kγ scaffold functions but not on its lipid kinase activity.

Reduced phagocytic activity in PI3Kγ-deficient microglial cells (open columns) with missing activation after OGD/reoxygenation challenge (black columns, wild type; hatched columns, PI3Kγ kinase-dead microglial cells). Cells were seeded in 6-well plates, starved for 4 h and used as well (Normoxia) or stimulated with OGD for 2 h and incubated in normoxia for 24 h (2 h OGD 24 h Reox) or incubated in normoxia for 48 h (2 h OGD 48 h Reox). To assay phagocytic activity, a suspension of GFP-producing E. coli cells (40 μl) was added and incubated together for the last 60 min, respectively. Values are mean + SD, n = 4 independent experiments derived from four different microglia isolations. *$§ p < 0.05, *significant difference vs. wt within the respective state, $significant difference vs. KD within the respective state, §significant difference vs. normoxia (two-way ANOVA, followed by Holm–Sidak test for post hoc multiple comparisons)
Discussion
Our comparative investigation of infarct expansion after temporary focal brain ischemia revealed an enhanced time-dependent progression in PI3Kγ-deficient mice, compared with wild-type mice or mice harboring a lipid kinase-dead version of PI3Kγ. The time-course of infarct expansion and experiments with bone-marrow-transplanted mice suggests that the genotype-related differences of resulting brain damage are likely related to brain intraparenchymal processes. As underlying reasons, we identified distinct differences in neuroinflammatory control mechanisms presumably leading to altered removal of dying cell and debris as well as to alterations of extracellular proteolysis.
Compliant infarct sizes in wild-type and PI3Kγ-deficient mice early (6 and 12 h) after recirculation confirm our findings that differences in infarct volume were not caused by differences in vascular anatomy between WT and KO mice (Figs. 1,2 Suppl.) and verified previous results [23]. Furthermore, we document an enhanced enlargement in infarct size 48 h after recirculation. The latter finding contradicts published results from Jin et al. who showed reduced ischemia/reperfusion-induced BBB disruption and brain damage in PI3Kγ-deficient mice [23]. Conclusions for the reasons of discrepancy between both studies cannot be drawn from this study. However, in the previous report, the documented enhanced BBB disturbance and associated brain damage in wild-type mice resulted from a longer-lasting period of focal ischemia (1 h) and occurred already earlier (24 h) after recirculation. In contrast, our data showed that the enhanced enlargement of infarct size in KO mice was detectable clearly only at a later stage (48 h) of infarct maturation suggesting different underlying mechanisms. Indeed, the authors of the previous study claimed a markedly enhanced PMN infiltration into the ischemic area of wild-type mice as the main source for increased MMP-9 secretion and ROS production and as the relevant executors for enlarged brain tissue damage. Our data clearly show that after a shorter period of brain ischemia with probably reduced BBB disturbance [30] an anticipated markedly increased PMN accumulation occurred in wild-type mice within the ischemic core but was associated with smaller infarct size. In contrast, KO mice with enlarged infarct size exhibited a reduced PMN accumulation (Table 2). These findings suggest that PMN invasion is unlikely to play a decisive role for PI3Kγ-dependent differences in infarct maturation during later stages of recirculation after focal brain ischemia. This viewpoint is supported by our findings in chimeras harboring PMN (and other leukocytes) with opposing genotype in respect to the host organism including residing microglial cells with corresponding alterations in infarct size 48 h after recirculation (Fig. 2c).
Consequently, we sought for alternative cellular components which may be causally involved in PI3Kγ-dependent modification of infarct maturation. Our recent studies revealed that major inflammatory functions of activated microglial cells are markedly controlled by PI3Kγ. We were able to identify PI3Kγ as a key mediator of microglial cAMP signaling responsible for LPS-induced microglial MMP-9 production with subsequent BBB deterioration and phagocytic control [21, 22]. There is likewise compelling evidence that cAMP serves as a second messenger for a variety of immunoregulatory and inflammatory responses in microglial cells after transient brain ischemia by a wide range of external stimuli including adrenergic activation of microglial cells via β-adrenergic stimulation [8]. Transient brain ischemia is accompanied by pronounced extracellular norepinephrine release [31, 32] suggesting that adrenergic pathways may contribute to microglial activation early after brain ischemia via cAMP-mediated signaling. Sustained modulation of microglial cAMP signaling during formation of secondary ischemic brain injury is mainly driven by ATP released from damaged cells, being a universal “danger” signal as well as its degrading products formed by numerous endonucletidases also acting as signaling molecules [33]. This is accompanied by pronounced upregulation of different G protein-coupled purinergic receptors in activated microglia with stimulating as well as inhibitory effects on membrane-bound adenylate cyclase activity affecting intracellular cAMP content [34, 35]. However, intracellular control of cAMP signaling in activated microglial cells after brain ischemia remained widely unknown. The herein used experimental approach revealed that the scaffold function of PI3Kγ is responsible to prevent microglial cAMP upregulation after ischemic conditions (Fig. 8, see below). The time course of infarct size enlargement in the current study suggests that the genotype-related differences in brain damage may likewise induce brain intraparenchymal processes in response to PI3Kγ-dependent neuroinflammatory activation of microglial cells via modulated cAMP signaling, leading to varying extracellular matrix decompaction by MMP-9 secretion and removal of dying cells and cell debris by phagocytosis.

Schematic representation of PI3Kγ-dependent microglial responses after transient brain ischemia. Microglial activation appeared early after brain ischemia induced by numerous factors including adrenergic transmitters and ATP released from damaged cells, being a universal danger signal as well as its degrading products formed by endonucletidases also acting as signaling molecules [33]. Among a multitude of diverse immune-relevant channels and receptors activated microglial cells express different G protein-coupled purinergic receptors with stimulating as well as inhibitory effects on membrane-bound adenylate cyclase activity affecting intracellular cAMP content [34, 35]. Our results indicate a relevant role for PI3Kγ in controlling microglial cAMP signaling as key functions of the neuroinflammatory response after transient brain ischemia. The catalytic subunit (p110γ) of PI3Kγ anchors protein kinase A through a site in its N-terminal region and thereby provokes a stimulation of phosphodiesterases and cAMP degradation [43]. In turn, the retained cAMP signaling warrant sufficient phagocytotic activity and unaltered MMP production of activated microglial cells after brain ischemia (left panel). In contrast, loss of PI3Kγ’s scaffold function by p110γ knockout resulted in sustained increase of microglial cAMP levels and signaling with accompanied suppression of phagocytotic activity, enhanced MMP-9 release, and enlarged brain infarction (right panel). The suppressive effect of PI3Kγ on cAMP levels appears critical for the restriction of ischemia-induced immune cell functions and in turn tissue damage
Previous reports showed that MMP-9 expression and activity after brain ischemia are induced first in reactive microglia [36] and contribute, among other factors, to infarct extent [37, 38]. Indeed, a pathogenetic role of microglial MMP-9 release is well-defined. MMP-9 acts as executing protease for degrading matrix substrates and interrupting cell–cell or cell–matrix homeostatic interactions, which may directly trigger anoikis-like neuronal cell death by interrupting cell–matrix survival signaling [39] and may further disturb scar formation as a key process of infarct demarcation for preventing inflammatory spreading into perilesional tissue [40]. Furthermore, it has been shown that upregulation of microglial phagocytic activity occurs rapidly after brain ischemia [41] and represents the predominant phagocytic activity [42] for removal of released danger signals resulting from dying cells and debris after necrotic cell death. These damaged cells are responsible for infarct enlargement by an overwhelming neuroinflammatory response [1].
Our data reveal for the first time that microglial MMP-9 upregulation and secretion as well as microglial phagocytic activity after focal brain ischemia and recirculation is controlled by PI3Kγ-dependent cAMP signaling. Our genetic approach suggests that the catalytic subunit p110γ of PI3Kγ signals through its earlier characterized scaffold function in this context. PI3Kγ has the capacity to anchor protein kinase A through a site in its N-terminal region thereby mediating stimulation of phosphodiesterases and cAMP degradation [43]. Loss of PI3Kγ in p110γ-deficient mice impedes formation of this multiprotein complex resulting in enhanced microglial cAMP signaling after OGD/reoxygenation challenge (Fig. 4). Uncoupling of this signal transduction network led to increased microglial MMP-9 release and suppression of microglial phagocytic activity during a critical period of infarct expansion after temporal focal brain ischemia, which is mainly driven by proinflammatory mechanisms [3].
In conclusion, our findings characterize the lipid kinase-independent scaffold function of PI3Kγ as a key mediator for controlling the neuroinflammatory response after temporary MCAO in mice. PI3Kγ serves a protective role in that it suppresses MMP-9 release and reinforces phagocytosis leading to improved brain tissue survival.
References
Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17(7):796–808
Kono H, Rock KL (2008) How dying cells alert the immune system to danger. Nat Rev Immunol 8(4):279–289
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22(9):391–397
Lo EH (2008) A new penumbra: transitioning from injury into repair after stroke. Nat Med 14(5):497–500
Nathan C, Ding A (2010) Nonresolving inflammation. Cell 140(6):871–882
Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, Witte OW, Frahm C (2013) Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS ONE 8(1), e52982
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87(5):779–789
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91(2):461–553
Gehrmann J, Mies G, Bonnekoh P, Banati R, Iijima T, Kreutzberg GW, Hossmann KA (1993) Microglial reaction in the rat cerebral cortex induced by cortical spreading depression. Brain Pathol 3(1):11–17
Mies G (1993) Inhibition of protein synthesis during repetitive cortical spreading depression. J Neurochem 60(1):360–363
Nedergaard M, Hansen AJ (1988) Spreading depression is not associated with neuronal injury in the normal brain. Brain Res 449(1–2):395–398
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19(8):312–318
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J (2012) Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43(11):3063–3070
Patel AR, Ritzel R, McCullough LD, Liu F (2013) Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol 5(2):73–90
Biber K, Owens T, Boddeke E (2014) What is microglia neurotoxicity (Not)? Glia 62(6):841–854
Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS (1998) Activation of Akt/protein kinase B by G protein-coupled receptors. A role for alpha and beta gamma subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinasegamma. J Biol Chem 273(30):19080–19085
Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, Hawkins PT (1997) The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89(1):105–114
Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nurnberg B, Gierschik P, Seedorf K, Hsuan JJ, Waterfield MD, Wetzker R (1995) Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269(5224):690–693
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP (2000) Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287(5455):1049–1053
Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E (2004) PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 118(3):375–387
Frister A, Schmidt C, Schneble N, Brodhun M, Gonnert FA, Bauer M, Hirsch E, Muller JP, Wetzker R, Bauer R (2014) Phosphoinositide 3-Kinase gamma Affects LPS-Induced Disturbance of Blood–brain Barrier Via Lipid Kinase-Independent Control of cAMP in Microglial Cells. Neuromol Med 16(4):704–713
Schmidt C, Schneble N, Muller JP, Bauer R, Perino A, Marone R, Rybalkin SD, Wymann MP, Hirsch E, Wetzker R (2013) Phosphoinositide 3-kinase gamma mediates microglial phagocytosis via lipid kinase-independent control of cAMP. Neuroscience 233:44–53
Jin R, Song Z, Yu S, Piazza A, Nanda A, Penninger JM, Granger DN, Li G (2011) Phosphatidylinositol-3-kinase gamma plays a central role in blood–brain barrier dysfunction in acute experimental stroke. Stroke 42(7):2033–2044
Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA (1994) Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 265(5180):1883–1885
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17(3):472–476
Popp A, Jaenisch N, Witte OW, Frahm C (2009) Identification of ischemic regions in a rat model of stroke. PLoS ONE 4(3), e4764
del Zoppo GJ, Frankowski H, Gu YH, Osada T, Kanazawa M, Milner R, Wang X, Hosomi N, Mabuchi T, Koziol JA (2012) Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J Cereb Blood Flow Metab 32(5):919–932
Hawkins PT, Stephens LR (2007) PI3Kgamma is a key regulator of inflammatory responses and cardiovascular homeostasis. Science 318(5847):64–66
Rotshenker S, Reichert F, Gitik M, Haklai R, Elad-Sfadia G, Kloog Y (2008) Galectin-3/MAC-2, Ras and PI3K activate complement receptor-3 and scavenger receptor-AI/II mediated myelin phagocytosis in microglia. Glia 56(15):1607–1613
Chen B, Friedman B, Cheng Q, Tsai P, Schim E, Kleinfeld D, Lyden PD (2009) Severe blood–brain barrier disruption and surrounding tissue injury. Stroke 40(12):e666–674. doi:10.1161/STROKEAHA.109.551341
Globus MY, Busto R, Dietrich WD, Martinez E, Valdes I, Ginsberg MD (1989) Direct evidence for acute and massive norepinephrine release in the hippocampus during transient ischemia. J Cereb Blood Flow Metab 9(6):892–896
Goyagi T, Nishikawa T, Tobe Y (2011) Neuroprotective effects and suppression of ischemia-induced glutamate elevation by beta1-adrenoreceptor antagonists administered before transient focal ischemia in rats. J Neurosurg Anesthesiol 23(2):131–137
Verkhratsky A, Krishtal OA, Burnstock G (2009) Purinoceptors on neuroglia. Mol Neurobiol 39(3):190–208
Bianco F, Fumagalli M, Pravettoni E, D'Ambrosi N, Volonte C, Matteoli M, Abbracchio MP, Verderio C (2005) Pathophysiological roles of extracellular nucleotides in glial cells: differential expression of purinergic receptors in resting and activated microglia. Brain Res Brain Res Rev 48(2):144–156
Dai SS, Zhou YG (2011) Adenosine 2A receptor: a crucial neuromodulator with bidirectional effect in neuroinflammation and brain injury. Rev Neurosci 22(2):231–239
Rivera S, Ogier C, Jourquin J, Timsit S, Szklarczyk AW, Miller K, Gearing AJ, Kaczmarek L, Khrestchatisky M (2002) Gelatinase B and TIMP-1 are regulated in a cell- and time-dependent manner in association with neuronal death and glial reactivity after global forebrain ischemia. Eur J Neurosci 15(1):19–32
Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH (2000) Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab 20(12):1681–1689
Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC (1998) Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29(5):1020–1030
Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA (2002) S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297(5584):1186–1190
Burda JE, Sofroniew MV (2014) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81(2):229–248
Stoll G, Schroeter M, Jander S, Siebert H, Wollrath A, Kleinschnitz C, Bruck W (2004) Lesion-associated expression of transforming growth factor-beta-2 in the rat nervous system: evidence for down-regulating the phagocytic activity of microglia and macrophages. Brain Pathol 14(1):51–58
Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R (2005) Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 196(2):290–297
Perino A, Ghigo A, Ferrero E, Morello F, Santulli G, Baillie GS, Damilano F, Dunlop AJ, Pawson C, Walser R, Levi R, Altruda F, Silengo L, Langeberg LK, Neubauer G, Heymans S, Lembo G, Wymann MP, Wetzker R, Houslay MD, Iaccarino G, Scott JD, Hirsch E (2011) Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol Cell 42(1):84–95
Acknowledgments
The authors acknowledge Mrs. M. Guenther, Mrs. S. Tausch and Mrs. R.-M., Zimmer for skillful technical assistance, and F. D. Boehmer, for his collegial editorial review of the manuscript. The study was supported by the Deutsche Forschungsgemeinschaft (Grant RTG 1715) and by the German Federal Ministry of Education and Research (BMBF; Grant FKZ 01EO1002; Center for Sepsis Control and Care). C.S and N.S. are PhD students of the Research Training Group 1715 “Adaptive Stress Responses” Grant RTG 1715 of the DFG, and C.S. was supported in part by the Integrated Research and Treatment Center, Center for Sepsis Control and Care, Jena University Hospital, Jena, Germany.
Authorship
C.S. designed and carried out the in vitro studies and analyzed the data. C.F. designed and supervised the animal experiments. N.S. carried out in vitro studies and analyzed the data. J.P.M. supervised the in vitro studies and revised the manuscript. M.B. carried out the immunohistochemical studies and analyzed the data. I.F. carried out the chimeric experiments. O.W.W. contributed to the study design, supervised the animal experiments, and revised the manuscript. E.H. designed and supervised the chimeric experiments. R.W. designed and coordinated the study and revised the manuscript. R.B. designed the study, supervised the experiments, analyzed the data, and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
Nothing to report.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 83.5 kb)
Rights and permissions
About this article

Cite this article
Schmidt, C., Frahm, C., Schneble, N. et al. Phosphoinositide 3-Kinase γ Restrains Neurotoxic Effects of Microglia After Focal Brain Ischemia. Mol Neurobiol 53, 5468–5479 (2016). https://doi.org/10.1007/s12035-015-9472-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9472-z