
Abstract
To examine the association between the sphingomyelin phosphodiesterase 1, acid lysosomal (SMPD1) gene, and Parkinson’s disease (PD) in Han Chinese from Central South part of Mainland China, we performed systematic genetic analysis in 502 Chinese Han patients with PD and 637 gender-, age-, and ethnicity-matched normal controls from Central South part of the Mainland China. We identified 11 single nucleotide variants and Leu-Ala (Val) repeat variants in the SMPD1 gene in our large cohort. Two novel missense variants, c.638A > C (p.H213P) and c.1673T > C (p.L558P), and a rare known missense variant, c.1805G > A (p.R602H, rs370129081), were identified in three sporadic PD cases. None of these three variants were observed in controls. Additionally, case-control analysis showed association between Leu-Ala (Val) repeat variants in SMPD1 and Chinese Han patients with PD (P = 0.015, χ 2 = 8.451). Our data provide supportive evidence that some genetic variants in SMPD1 increase the risk of PD in the Chinese Han population.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD; MIM 168600) is the second most common neurodegenerative disorder affecting 1–2 % of the population above age 60 [1]. It is characterized by the loss of dopamine-producing neurons in the substantia nigra and other brainstem nuclei, ultimately resulting in troublesome symptoms, including rest tremor, bradykinesia, rigidity, postural instability, and other motor and nonmotor symptoms [2]. The etiology and pathogenic mechanism of PD are largely unknown. Previous studies have shown that mutations in lysosomal-related genes, such as the glucocerebrosidase gene (GBA) and the ATPase type 13A2 gene (ATP13A2), have been linked to PD [3–6], suggesting that lysosomal dysfunction may play an important role in the pathogenesis of PD. A founder mutation, p.L304P (previously named p.L302P), in another lysosomal enzyme, sphingomyelin phosphodiesterase 1, acid lysosomal (SMPD1), was reported to be associated with increased risk of PD in Ashkenazi Jewish population [7]. But, the association between the SMPD1-p.L304P mutation and PD has not been validated in additional population, possibly due to the ethnic specificity of the mutation and limited sample size [8]. A rare known variant, p.R591C, in SMPD1 was identified in two out of 806 PD cases, but not in 7481 control subjects from the southern part of China [9], suggesting that this variant may increase the risk of PD. To further evaluate the association between the SMPD1 variants and PD, we conducted a genetic analysis of 502 PD cases and 637 control subjects from the Han population in the Central South part of Mainland China.
Materials and Methods
Patients and Controls
Five hundred and two unrelated Chinese Han patients with PD (male/female = 308/194; age 66.0 ± 9.8 years; onset age 62.6 ± 11.2 years) and 637 unrelated gender-, age-, and ethnicity-matched healthy controls (male/female = 390/247; age 67.6 ± 9.6 years) from the Central South part of Mainland China were enrolled in this study. Among the 502 PD patients, 111 (22.1 %) had first- or second-degree relatives affected with PD (familial PD; mean age at onset 59.8 ± 12.9 years; male/female 69/42), and 391 cases (77.9 %) had no family history (sporadic PD; mean age at onset 63.1 ± 12.0 years; male/female 239/152). Thirty-eight percent (191/502) of the patients were screened and found to be negative for the vacuolar protein sorting 35 gene (VPS35) mutation [10]. The diagnosis of PD was made according to common diagnostic criteria [11]. The study was approved by the Ethics Committee of the Third Xiangya Hospital, Central South University, China. All participants have signed an informed consent.
Genetic and Statistical Analysis
Genomic DNA (gDNA) was extracted from lymphocytes using standard phenol-chloroform method. Polymerase chain reaction (PCR) products were generated with 100 ng of gDNA in 2.5 μL 10× PCR buffer, 2 μL of 2.5 mmol/L each dNTP, 1.5 μL of 25 mmol/L MgCl2, 1 μL of 10 μmol/L each primer, and 1 U Taq polymerase in a total volume of 25 μL. Primers covering all coding regions and intron/exon boundaries of the SMPD1 gene (NM_000543.4) were used for PCR amplification with a GeneAmp 9700 thermal cycler system (Applied Biosystems, Foster City, CA, USA). The sequences are available upon request. PCR conditions were 95 °C for 3 min, followed by 35 cycles of 95 °C for 40 s, 58 °C for 35 s, 72 °C for 40 s, and a final extension step at 72 °C for 5 min. The 8.5 μL PCR products were digested by 0.8-U shrimp alkaline phosphatase (SAP, Fermentas) and 8-U exonuclease I (Fermentas) in a 10-μL reaction volume and sequenced directionally using an 8-capillary 3500 genetic analyzer (Applied Biosystems, Foster City, CA, USA) [12].
Pearson’s χ 2 tests were applied to test for significance in differences of gene frequencies. A value (P < 0.05, two-tailed) was considered to be significant. The Hardy-Weinberg equilibrium was performed as to ascertain the normal heterogeneity of the population.
Results
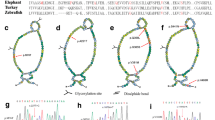
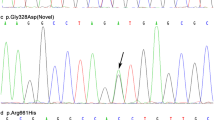
Eleven single nucleotide variants and Leu-Ala (Val) repeat variants in the SMPD1 gene were identified in the 502 patients with PD. Distributions of the genotypes in PD and control groups were both in Hardy-Weinberg equilibrium. One novel missense variant, c.638A > C (p.H213P), was found in a sporadic female PD patient with onset age of 47 years, and the other novel missense variant, c.1673 T > C (p.L558P), was observed in a sporadic male PD patient with onset age of 56 years. A known rare missense variant, c.1805G > A (p.R602H, rs370129081), was identified in a sporadic male patient of PD with onset age of 60 years (Fig. 1). None of the three variants were observed in the control cohort that consists of 637 subjects. In addition, none of these variants were identified in 2375 ethnicity-matched controls from exome sequencing data from BGI-Shenzhen. Besides the p.R602H variant recorded in the database of single nucleotide polymorphisms (rs370129081) and Human Gene Mutation Database, the other two mutations were absent in both databases. The p.H213, p.L558, and p.R602 are highly conserved amino acid residuals spanning the species from rat to human (Fig. 1), suggesting the structural and functional importance of these residuals. MutationTaster predicted the three variants to be disease-causing. Case-control analysis showed association between Leu-Ala (Val) repeat variants in SMPD1 and Chinese Han patients with PD (P = 0.015, χ 2 = 8.451, Table 1), and this association remained to be statistically significant after Bonferroni correction. The allele with more than seven Leu-Ala (Val) repeats conferred susceptibility for PD.

Sequencing and conservation analysis of c.638A > C (p.H213P), c.1673T > C (p.L558P), and c.1805G > A (p.R602H, rs370129081) in the SMPD1 gene. a The arrow shows the novel missense variant c.638A > C (p.H213P) identified in a sporadic female PD patient with onset age of 47 years. b The arrow shows the novel missense variant c.1673T > C (p.L558P) identified in a sporadic male PD patient with onset age of 56 years. c The arrow shows the known rare missense variant c.1805G > A (p.R602H, rs370129081) identified in a sporadic male PD patient with onset age of 60 years. d Conservation analysis of the SMPD1 p.H213, p.L558, and p.R602 amino acid residues
Other eight known variants (c.107C > T, p.A36V, rs1050228; c.636T > C, p.D212D, rs7951904; c.900C > T, p.T300T, rs368353322; c.995C > G, p.P332R, rs202081954; c.1133G > A, p.R378H, rs559088058; c.1522G > A, p.G508R, rs1050239; c.1598C > T, p.P533L, rs199915216; c.1632C > T, p.T544T, rs201659696) were identified in both PD and control cohorts. None of these variants were predicted to result in alteration of splicing site (http://www.fruitfly.org/seq_tools/splice.html). There were no significant differences in genotypic distribution between the PD cohort and control cohort for any of these eight known variants (all P > 0.05, Table 2), indicating that they were not pathogenic variants.
Discussion
Accumulating evidence indicates that genetic abnormalities play an important role in the etiopathogenesis of PD. The past 16 years have witnessed dramatic progress in the genetic basis of PD by discovery of at least 15 disease-related genes for parkinsonism [2, 13–16]. Recently, the variants identified in the SMPD1 gene were reported to be associated with PD [7, 17]. The observation of high OR value (9.4) to develop PD in SMPD1 p.L304P carriers indicated that this variant is a robust susceptibility factor in multigenic inheritance or monogenic disease-causing mutation with incomplete penetrance [7].
The SMPD1 gene, mapped on chromosome 11p15.1–p15.4, spans about 4.6 kb and contains six exons encoding sphingomyelin phosphodiesterase 1 (acid sphingomyelinase, ASM). The protein is a lysosomal enzyme that cleaves the phosphocholine head group of sphingomyelin to generate ceramide [18]. The deficient activity of ASM may result in types A or B Niemann-Pick disease (NPD) [19, 20]. The phenotype of homozygous Smpd1 −/− mice mimics the neurovisceral form of human NPD: extensive accumulation of sphingomyelin in the reticuloendothelial system of the liver, spleen, bone marrow, lung, and brain, and complete degeneration of the ganglionic cell layer of Purkinje cells of the cerebellum, leading to severe impairment of neuromotor coordination [21, 22]. Alterations of calcium (Ca2+) homeostasis were detected in ASM knockout mice brain [23], which may change the vulnerability of substantia nigra pars compacta dopaminergic neurons to genetic and environmental factors and play a crucial role in 1-methyl-4-phenylpyridinium (MPP+)-induced dopaminergic neuronal cell death.
Usually, mutation in both alleles of a disease-causing gene may cause loss of function of the gene, resulting in autosomal recessive disease, while a mutant carrier may result in autosomal dominant disease or enhance susceptibility to a complex disease. Mutation in an important domain of a gene may cause a monogenic form of disease, whereas a nucleotide variant in a noncritical region may enhance susceptibility to polygenic disease. Recent studies have identified that mutations in lysosomal storage disorders disease-causing genes, including the SMPD1 gene, the GBA gene, and the Niemann-Pick 1 gene (NPC1), could act as risk variants for PD.
The first reported PD-associated SMPD1 p.L304P mutation leads to dramatically reduced SMPD1 enzymatic activity and causes a fatal infantile type A NPD in homozygous condition [24], suggesting that loss of function in SMPD1 is involved in the pathogenesis of both NPD and PD. In this study, we identified two novel mutations (p.H213P and p.L558P) and a known but rare variant (p.R602H) in our PD cohort consisting of 502 subjects. None of these variants were identified in 3012 control subjects. All the three amino acids at p.H213, p.L558, and p.R602 are highly conserved. Six variants including p.V36A, p.P332R, p.R378H, p.G508R, p.P533L, and p.R602H were reported in NPD patients. However, the pathogenicity or association between the variants and NPD remains unclear except that p.P533L was considered as one severe mutation of the two disease-causing alleles [25–27]. Additionally, case-control analysis showed association between PD and SMPD1 Leu-Ala (Val) repeat variants, which may affect the product structure and function, eventually resulting in PD. Our data, therefore, support the notion that some genetic variants in SMPD1 increase the risk of PD in the Chinese Han population. Further genetic studies in different PD populations and functional analysis of the variants are warranted to better understand SMPD1-associated PD and the role of SMPD1 variants in the pathogenesis of PD.
References
Lautier C, Goldwurm S, Durr A , Giovannone B, Tsiaras WG, Pezzoli G, Brice A, Smith RJ (2008) Mutations in the GIGYF2 (TNRC15) gene at the PARK11 locus in familial Parkinson disease. Am J Hum Genet 82:822–833
Deng H, Liang H, Jankovic J (2013) F-box only protein 7 gene in parkinsonian-pyramidal disease. JAMA Neurol 70:20–24
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351:1972–1977
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E (2004) Parkinsonism among Gaucher disease carriers. J Med Genet 41:937–940
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661
Dehay B, Martinez-Vicente M, Ramirez A , Perier C, Klein C, Vila M, Bezard E (2012) Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy 8:1389–1391
Gan-Or Z, Ozelius LJ, Bar-Shira A , Saunders-Pullman R, Mirelman A, Kornreich R, Gana-Weisz M, Raymond D, Rozenkrantz L, Deik A, Gurevich T, Gross SJ, Schreiber-Agus N, Giladi N, Bressman SB, Orr-Urtreger A (2013) The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 80:1606–1610
Wu RM, Lin CH, Lin HI (2014) The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 82:283
Foo JN, Liany H, Bei JX , Yu XQ, Liu J, Au WL, Prakash KM, Tan LC, Tan EK (2013) A rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol Aging 34:2890.e13–2890.e15
Deng H, Xu H, Deng X , Song Z, Zheng W, Gao K, Fan X, Tang J (2012) VPS35 mutation in Chinese Han patients with late-onset Parkinson’s disease. Eur J Neurol 19:e96–e97
Jankovic J (2008) Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79:368–376
Zheng W, Deng X, Liang H , Song Z, Gao K, Yang Y, Deng H (2013) Genetic analysis of the fused in sarcoma gene in Chinese Han patients with essential tremor. Neurobiol Aging 34:2078.e3–2078.e4
Edvardson S, Cinnamon Y, Ta-Shma A , Shaag A, Yim YI, Zenvirt S, Jalas C, Lesage S, Brice A, Taraboulos A, Kaestner KH, Greene LE, Elpeleg O (2012) A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7:e36458
Kitada T, Asakawa S, Hattori N , Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Krebs CE, Karkheiran S, Powell JC , Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, De Camilli P, Yue Z, Paisan-Ruiz C (2013) The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34:1200–1207
Polymeropoulos MH, Lavedan C, Leroy E , Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Santana P, Pena LA, Haimovitz-Friedman A , Martin S, Green D, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z, Kolesnick R (1996) Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 86:189–199
Schuchman EH, Levran O, Pereira LV, Desnick RJ (1992) Structural organization and complete nucleotide sequence of the gene encoding human acid sphingomyelinase (SMPD1). Genomics 12:197–205
Levran O, Desnick RJ, Schuchman EH (1991) Niemann-Pick type B disease. Identification of a single codon deletion in the acid sphingomyelinase gene and genotype/phenotype correlations in type A and B patients. J Clin Invest 88:806–810
Levran O, Desnick RJ, Schuchman EH (1991) Niemann-Pick disease: a frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc Natl Acad Sci U S A 88:3748–3752
Horinouchi K, Erlich S, Perl DP , Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH (1995) Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet 10:288–293
Otterbach B, Stoffel W (1995) Acid sphingomyelinase-deficient mice mimic the neurovisceral form of human lysosomal storage disease (Niemann-Pick disease). Cell 81:1053–1061
Ledesma MD, Prinetti A, Sonnino S, Schuchman EH (2011) Brain pathology in Niemann Pick disease type A: insights from the acid sphingomyelinase knockout mice. J Neurochem 116:779–788
Levran O, Desnick RJ, Schuchman EH (1992) Identification and expression of a common missense mutation (L302P) in the acid sphingomyelinase gene of Ashkenazi Jewish type A Niemann-Pick disease patients. Blood 80:2081–2087
Rhein C, Muhle C, Kornhuber J, Reichel M (2015) Alleged detrimental mutations in the SMPD1 gene in patients with Niemann-Pick disease. Int J Mol Sci 16:13649–13652
Simonaro CM, Desnick RJ, McGovern MM, Wasserstein MP, Schuchman EH (2002) The demographics and distribution of type B Niemann-Pick disease: novel mutations lead to new genotype/phenotype correlations. Am J Hum Genet 71:1413–1419
Zhang H, Wang Y, Gong Z , Li X, Qiu W, Han L, Ye J, Gu X (2013) Identification of a distinct mutation spectrum in the SMPD1 gene of Chinese patients with acid sphingomyelinase-deficient Niemann-Pick disease. Orphanet J Rare Dis 8:15
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81271921, 81441033), “531” Program of Central South University, China (H.D.), Grant for the Foster Key Subject of the Third Xiangya Hospital Clinical Laboratory Diagnostics (H.D.), and Zhishan Lead Project of the Third Xiangya Hospital (H.D.). The authors thank the participating patients and the investigators at the Third Xiangya Hospital, Central South University, for their cooperation and their efforts in collecting the genetic information and DNA specimens.
Conflict of Interest
The authors declare no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Deng, S., Deng, X., Song, Z. et al. Systematic Genetic Analysis of the SMPD1 Gene in Chinese Patients with Parkinson’s Disease. Mol Neurobiol 53, 5025–5029 (2016). https://doi.org/10.1007/s12035-015-9426-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9426-5