
Abstract
Epigenetic modifiers can work in concert with transcription factors to control the transition of cells from proliferating progenitors into quiescent terminally differentiated cells. This transition involves changes in histone methylation and one of the key regulators of this is the H3K4me2/1 histone demethylase LSD1. Here, we show that the highest expression of LSD1 occurs in postmitotic retinal cells during the peak period of rod photoreceptor differentiation. Pharmacological inhibition of LSD1 in retinal explants cultured from PN1 to PN8 had three major effects. It prevented the normal decrease in expression of genes associated with progenitor function, it blocked rod photoreceptor development, and it increased expression of genes associated with other retinal cell types. The maintained expression of progenitor genes was associated with a maintained level of H3K4me2 over the gene and its promoter. Among the genes whose expression was maintained was Hes1, a repressor known to block rod photoreceptor development. The inhibition of rod photoreceptor gene expression occurred in spite of the normal expression of transcription factors CRX and NRL, and the normal accumulation of H3K4me2 marks over the promoter and gene body. We suggest that LSD1 acts in concert with a series of nuclear receptors to modify chromatin structure and repress progenitor genes as well as to inhibit ectopic patterns of gene expression in the differentiating postmitotic retinal cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The reprogramming of dividing progenitors into terminally differentiated cells in the central nervous system requires large-scale repression of progenitor genes and activation of genes characteristic of each differentiated cell type. A key element in the reprogramming of cellular states is the change of histone modifications by enzymes in complexes whose binding to promoters and enhancers is regulated by the specificity of their associated DNA-binding proteins [1]. One of the best-characterized histone-modifying enzymes is lysine-specific demethylase 1 or LSD1 (also known as KDM1A or AOF2) [2]. LSD1 demethylates mono- and dimethylated H3K4 and H3K9, but does not alter trimethylated H3K4 and H3K9 [2, 3].
A variety of studies suggest that LSD1 plays a key role at multiple stages of development. In human embryonic stem cells, high levels of LSD1 are necessary to maintain their undifferentiated state [4]. Both knockdown and pharmacological inhibition of LSD1 prevented the epithelial to mesenchymal transition in a mouse cell culture model [5]. Conditional knockouts of LSD1 in different blood lineages led to disrupted terminal differentiation of granulocytes and erythrocytes [6, 7], and its inhibition blocked estrogen-induced expression of specific genes in mouse Sertoli cells [8]. While the actions of LSD1 may promote or inhibit differentiation in various cell types, it is clear that its action is critical at these phenotypic switchpoints during development.
Within the developing nervous system, there is evidence for the involvement of LSD1 in neural stem cell differentiation in culture and in the early stages of development in vivo. To understand the epigenetic regulation of terminal differentiation in the CNS, we have been studying the retina. The postnatal mouse retina provides a valuable model of development as over the first 2 postnatal weeks a large proportion of mouse retinal cells undergo a terminal mitosis and change from a dividing progenitor to a postmitotic differentiated cell. The large majority of cells formed during this period are rod photoreceptors, the cell type that constitutes 85 % of the adult retina [9]. Retina maturation is accompanied by a large-scale switch in gene expression as genes characteristic of progenitors are repressed and genes specific for retinal cell types are upregulated [10]. We have recently shown that the downregulation of progenitor genes and the upregulation of cell type-specific genes correlate with a genome-wide decrease and increase, respectively, of an active epigenetic mark H3K4me2 at the transcriptional start site and over the body of these genes [11]. This suggests that the terminal differentiation of retinal cells involves a dynamic regulation of histone methylation, particularly at the H3K4 position.
In the present study, we have examined the role of the LSD1 demethylase during retinal development by using pharmacological inhibitors that have been well documented and used in a variety of functional studies [12–15]. We found that LSD1 is most highly expressed as the outer retinal cells become postmitotic and begin to differentiate, and that inhibition of LSD1 blocked the differentiation of rod photoreceptors. Analysis of the changes in the retinal transcriptome indicated that blocking LSD1 prevented both the cessation of progenitor programs and the expression of terminal differentiation programs, including those linked to rod photoreceptor development. LSD1 would appear to be a key enzyme allowing cells to pass from progenitor to terminally differentiated states.
Materials and Methods
Mice
All animal experiments were conducted in accordance with NIH and ARVO guidelines and were approved by the Animal Care and Use Committee of Pennsylvania State University School of Medicine (Protocol # 2009–061). C57BL/6j mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and are free of known retinal degeneration mutations.
Antibodies and Reagents
Chemicals were purchased from Fisher Scientific (Pittsburgh, PA), unless otherwise noted. Mouse recombinant IGF1 (working concentration 50 ng/ml), LSD1 inhibitors: trans-2-Phenylcyclopropylamine (parnate or tranylcypromine, TCP, working concentration-50 μm), and N-methyl-N-propargylbenzylamine hydrochloride (Pargyline, working concentration 2.5 mM) were purchased from Sigma (St. Louis, MO). Because we used LSD1 inhibitors in tissue samples, working concentration for inhibitors were increased two-fold over the IC100 for recombinant LSD1 inhibition by TCP and pargyline or used previously in cultured cells [12, 15, 16]. Anti-rhodopsin monoclonal antibodies have been described previously [17] and react with an N-terminal sequence shared by many species [18]. HNK-1 (VC1.1) antibody recognizes a carbohydrate epitope on N-CAM and other neural proteins [19, 20]. Anti-H3K4me2 (07–030, Upstate, Charlottesville, VA), and anti-H3K9me2 (ab1220, Abcam Cambridge, MA) were used in our previous ChIP studies and have passed validation (htt://compbio.med.harvard.edu/antibodies/) [21]. Anti-PCNA and anti-HES1 antibodies were from Cell Signaling (Danvers, MA), and anti-PKCα antibody were from Abcam (Cambridge, MA). Anti-GNAT2 antibody was a gift from Arshavsky VY [22]. Anti-LSD1 antibody from Abcam (ab17721; rabbit polyclonal) was raised against of a synthetic peptide derived from residue 800 to the C-terminus of human LSD1 and conjugated to KLH. The specificity of this antibody has previously been well documented [6, 14, 23]. Alexa Fluor 488-labeled mouse anti-BrdU antibody (BD Biosciences) was used in cell replication assays.
SDS-PAGE Gel Electrophoresis and Western Blotting
Homogenized tissue or isolated nuclei were dissolved in SDS-containing loading buffer and the electrophoresis was carried out in 15 % polyacrylamide SDS-containing gels [24]. Proteins were transferred to Immun-Blot PVDF membrane (Bio-Rad, Hercules, CA) and detected with primary and secondary HRP-conjugated antibodies as described [25]. For semi-quantitative analysis of relative protein levels in nuclear samples, the Coomassie-stained gels or autoradiographs after ECL (Thermo Scientific, Rockford, IL) detection were scanned and digitized and the intensity of protein bands was quantitated using the OptiQuant version 03.00 (Packard Instrument Co, Meriden, CT) or Image J (NIH) software packages.
Retina Isolation and Explant Culture
Whole retinas were isolated from pups at postnatal day 1 (PN1) by removing the sclera and most of the retinal pigmented epithelium (RPE) layer, and were cultured in UltraCulture™ (Cambrex Bio Science Rockland, ME, USA) serum-free medium supplemented with gentamycin antibiotic (10 μg/ml), as previously described [26–28]. Retinas were cultured individually in 1 ml of media in a 24-well culture dish at 37 °C in a 5 % CO2, balance air, atmosphere. Medium was changed every other day by replacing 0.5 ml with fresh medium.
Histology and Immunofluorescence Staining
Whole eyes or explanted retinas were fixed with 4 % paraformaldehyde in PBS for 24 h at 4 °C. For paraffin sections, after three washes with PBS, fixed explants were dehydrated through a series of graded ethanols and embedded in paraffin. All samples for one experiment were placed in the same blocks and sectioned for immunohistochemistry (5–7 μm). For cryosections, after three washes with PBS, tissue samples were incubated 15 min in 5 % sucrose solution/PBS, then overnight in 20 % sucrose solution/PBS and embedded in 2:1 mix of 20 % sucrose and OCT. Blocks with tissue samples were stored at −80 °C and section to 10–12 μm on Cryostat Microtome HM550 (Thermo Fisher Scientific). Antigen retrieval was performed using 6.5 mM sodium citrate pH 6 and boiling for 45 min. A standard immunohistochemistry protocol [29, 30] was employed for single or double labeling using fluorescent dye-conjugated secondary antibodies (Jackson Immuno-Research Laboratory, West Grove, PA). Sections were imaged using an Olympus Fluoview FV1000 confocal microscope (Olympus Center Valley, PA). For each set of experiments, acquisition parameters for each antibody were maintained constant.
cDNA Synthesis
Total RNA was isolated by TRIzol (Invitrogen, Carlsbad, California) or by RNeasy mini kits (Qiagen, Germantown, MD). Final concentrations were determined spectrophotometrically using a GeneSpect III (Hitachi Tokyo, Japan). cDNA was synthesized with SuperScriptII First-Strand Synthesis System kit according to manufacturer’s protocol (Invitrogen).
Chromatin Immunoprecipitation
The method has been described in detail previously [11].
Quantitative PCR
For quantitative real-time PCR, we used 2x iQ-SYBR Green PCR supermix from Bio-Rad. Samples in triplicate were run on iQ5 Multicolor Real Time PCR Detection System (Bio-Rad). Each run included suitable standard curve samples using wide range of concentration for genomic DNA prepared similar to inputs. Primers design for ChIP Q-PCR was based on our previous whole genome ChIP-Seq data [11]. Primer sequences are listed in Online Resource Table 1.
Analysis of ChIP-Seq Data
Reads from a published set of ChIP-Seq experiments [11] were retrieved from the Gene Express Omnibus data repository (accession number GSE38500) and mapped to the mouse genome NCBI37/mm9 using the program Bowtie (version 0.12.4) as previously described [11]. Reads over regions containing genes of interest were analyzed using NextGENE software (version 2.10).
Expression Array for Retina Explants Culture
RNA was isolated by RNeasy mini kits (Qiagen) from three biological replicas of retinas explant cultures for each condition: control explant cultured for 24 h, TCP-treated explant cultured for 24 h, control explant cultured for 8 days, TCP-treated explant cultured for 8 days. Each biological replica consists of two retinas. RNA quality and quantity were analyzed using an Agilent Bioanalyzer 2110, and RINs for RNA samples were 7.7 on average. Illumina MouseRef8 v2.0 Expression BeadChip arrays (BD-202-0202) were processed in the PSU Genome Science Facility starting with 500 ng of template and using the standard Illumina Total Prep protocol with 18 h hybridization. Following hybridization, beadchips were washed and fluorescently labeled. Beadchips were scanned with a BeadArray Reader (Illumina, San Diego, CA). A project was created with resultant scan data imported into GenomeStudio 1.0 (Illumina). Results were exported to GeneSpring Gx11 (Agilent Technologies). Measurements less than 0.01 were then set to 0.01, arrays normalized to the 50th percentile, and individual genes normalized to the median of controls.
Apoptosis Assay
TUNEL assays used the ApoBrdU DNA fragmentation Assay kit from BioVision (Milpitas, CA) according to the manufacturer’s protocol.
Cell Proliferation Assay
A final concentration of 10 mM of BrdU (Sigma) was used for labeling explant culture at PN4. After 20 h incorporation into the DNA of replicating cells, explants were processed for paraffin or cryo- sections. DNA was denatured by incubating slides for 30 min in 4 N HCl. The acid was neutralized by immersing for 3 × 10 min in 0.1 M sodium borate buffer (pH 8.5) and washing in PBS 2 × 5 min. Proliferating cells were detected by incubating slides overnight at 4 °C with Alexa Fluor 488-conjugated mouse anti-BrdU antibody.
Statistical Analysis
The CLC Genomics Workbench 4.8 package was used to compute statistics for the 24 h and for the 8 days case vs. control microarray samples. For each category, quantile normalization was performed followed by pairwise homogeneous t test resulting in normalized fold changes and p values. Gene expressions with an absolute fold change greater than 1.5 and a p value <0.05 were considered significant. Statistical analyses for other experiments were performed using the GraphPad Prism software. Student’s t test (two-tailed, unpaired) was used to compare two groups and one-way ANOVA (with Newman–Keuls post test) was used to compare more than two groups.
Results
LSD1 Expression Peaks During the Transition From Late Progenitors to Rod Photoreceptors in the Mouse Retina
LSD1 is an enzyme active at key stages of development in a number of tissues, including the CNS. To begin studying whether LSD1 might have a role in late retinal development, we followed its expression through the postnatal period using both Western blots and immunocytochemistry using an antibody whose specificity has been well characterized [6, 14, 23]. In Western blots, low levels of LSD1 were detected at E17.5 and the amount increased through the first postnatal week before decreasing to low levels that were maintained in the adult (Fig. 1a). Immunofluorescence staining of retinal sections (Fig. 1b) indicated that LSD1 was expressed in the nuclei of a subpopulation of cells in all layers of the retina. At PN3, the proportion of LSD1-labeled cells had increased in the outer retina without a change in labeling of cells in the inner retina. A higher power view of the labeling at this age clearly shows that almost all of the LSD1 positive cells were distinct from the dividing progenitor cells labeled with antibodies to PCNA, although a small number of cells expressed both proteins (arrows). We interpret these labeling patterns as the transition of a PCNA+ve/LSD1-ve progenitor population to a PCNA-ve/LSD1+ve postmitotic population, with a rapid switch between the cell phenotypes resulting in only a few double-labeled cells. Interestingly, LSD1 appeared to be absent at the outer margin of the retina where mitoses occur as well as in the portion of the outer retina that contained the densest accumulation of PCNA positive cells.

LSD1 is expressed more strongly during the transition from late progenitor to rod photoreceptor. a Western blot of samples isolated from mouse retina probed with antibody against LSD1 with Coomassie staining of core histones as loading control. b Immunofluorescence microscopic images of sections of developmental mouse retina tissue array stained with PCNA (green), LSD1 (red), and nuclear counterstained with Hoechst33358 (blue). ONBL, outer neuroblast layer; INBL, inner neuroblast layer; GCL, ganglion cell layer; ONL, outer nuclear layer; INL, inner nuclear layer. The higher power view of a PN3 section indicates the low number of double-labeled cells (arrows)
At PN5, the proportion of positive cells and the intensity of LSD1 labeling in the outer retina were greater, with positive cells intermingled with PCNA positive cells at the inner edge of the outer neuroblast layer. The labeling in the outer retina continued to increase such that at PN7 essentially all cells in this layer strongly expressed LSD1, with only a few PCNA positive cells remaining. By PN13, the labeling had decreased such that the outer nuclear layer had barely detectable labeling of the photoreceptor nuclei and cells of the inner nuclear layer and ganglion cell layers showed a uniform light labeling.
The processes of horizontal cells in the developing outer plexiform layer demarcate the outer and inner nuclear layers and can be labeled by the antibody HNK-1 [20]. Double labeling of PN7 retinal sections with antibodies against HNK-1 and LSD1 detected LSD1-positive cells on both sides of the outer plexiform layer demarcated by HNK-1, indicating that they were part of both the late developing portion of the inner nuclear layer and the developing outer nuclear layer (arrows in Fig. 2a, b). The HNK-1 positive horizontal cells were themselves LSD1 positive (Fig. 2a, b, asterisks). At least some developing bipolar cells were LSD1 positive, as indicated by the co-expression of LSD1 and the bipolar marker PKC-alpha (Fig. 2c, marked by #). Examination of the developing outer nuclear layer at PN1 (Fig. 2d), 7 (Fig. 2e, f), and 28 (Fig. 2g) indicated that cells imaged in the correct plane through the nucleus showed both rhodopsin and LSD1 labeling (Fig. 2d, arrowhead), although the latter was very faint and became weaker as the intensity of rhodopsin labeling increased such that it was barely detectable in the mature retina. Through this period, a small number of cells in the inner nuclear and ganglion cell layers remained strongly labeled with faint labeling in the others (Fig. 2g, h). From these studies, we conclude that LSD1 is expressed predominantly in postmitotic cells, its highest level occurs in the outer retina as cells transition from progenitor to terminally differentiated cell, and it then remains expressed at detectable levels in only a minority of inner retinal cells.

LSD1 expression in different retina cell types. a, b Immunofluorescence microscopic images of sections of mouse retina at PN7 stained with HNK-1 (green), LSD1 (red), and nuclear counterstained with Hoechst33358 (blue). c Immunofluorescence microscopic images of sections of mouse retina at PN7 stained with PKCα (green), LSD1 (red), and nuclear counterstained with Hoechst33358 (blue). d–h Immunofluorescence microscopic images of sections of mouse retina stained with Rhodopsin (green), LSD1 (red), and nuclear counterstained with Hoechst33358 (blue) at PN1 (d), PN7 (e, f), and PN28 (g, h). RPE, retinal pigment epithelium; GCL, ganglion cell layer; OPL, outer plexiform layer; IPL, inner plexiform layer; ONL, outer nuclear layer; INL, inner nuclear layer. Arrows show LSD1 expression in different retinal layers
Inhibition of LSD1 Blocks Rod Photoreceptor Development
To begin investigating the function of LSD1 in late retinal development, we blocked its activity during the period of its maximum expression using the inhibitors trans-2-phenylcyclopropylamine hydrochloride (TCP, tranylcypromine or parnate) and pargyline hydrochloride [13]. Retinal explants were established from PN1 animals, cultured for 4–8 days in the presence of 50 μM TCP or 2.5 mM pargyline hydrochloride and then labeled with a rhodopsin antibody to quantitate the number of rod photoreceptors. Most of the explants retained a normal morphology throughout the experimental period but after 8 days, they began to lose their laminar structure.
As shown in Fig. 3a, c, after 8 days of culture, TCP had blocked rhodopsin expression by 91 % compared to control. Similar results were obtained using the second LSD1 inhibitor, pargyline hydrochloride (Fig. 3b). Even in the presence of IGF1, a peptide known to stimulate rod formation [26], TCP reduced rhodopsin expression by 82 % (Fig. 3c). To ensure that the changes in rhodopsin expression caused by TCP were not secondary to changes in cell proliferation or cell death, we measured BrdU incorporation and TUNEL-positive cells, respectively. A similar number of BrdU-labeled cells and TUNEL-positive cells were seen in retina explants untreated or treated with TCP and harvested after 5 and 8 days of culture, respectively (Fig. 3d, e). Analysis of multiple samples found no statistical difference between control and TCP-treated for either BrdU or TUNEL labeling.

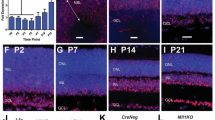
LSD1 inhibition blocked rod photoreceptor development. a, b Rhodopsin (green) expression in PN1 retinal explants cultured for 8 days in the presence of LSD1 inhibitors, TCP (a) or pargyline (b), with nuclear counterstained with Hoechst33358 (blue). c. Rhodopsin expression by RT-PCR in PN1 retinal explants cultured for 8 days in the presence of IGF1 or alone and in combination with LSD1 inhibitor TCP (*p < 0.01 vs. control; *** p < 0.0001 vs. control). d, e Changes in rhodopsin expression caused by TCP were not due to changes in cell death or cell proliferation. TUNEL labeling (green) for apoptotic cell in 8 days control or TCP-treated explants culture (d). BrdU incorporation (green) in 5 days control or TCP-treated explants culture (e). Nuclear counterstained with Hoechst33358 (blue)
To determine whether the effect of inhibiting LSD1 was on the generation of rod photoreceptors or selectively on rhodopsin expression, we performed global detection of transcript levels using expression microarrays that sample 25,600 transcripts. We collected RNA from retinal explants cultured for 24 h or 8 days with or without TCP. To analyze the array data, we used two standard methods of normalizing and analyzing array data and only genes that showed statistically significant differences between control and TCP-treated samples by both methods were incorporated into the final table (Online Resource Table 2). Using the criteria of a >1.5 fold change and a p value <0.05, 111 genes were expressed at higher levels in retinas treated with TCP for 24 h, and this number increased to 565 after 8 days of treatment. By the same criteria, 70 genes were expressed at lower levels after 24-h treatment and 342 genes were expressed at lower levels after 8 days. Previous work has defined a group of genes expressed by rod photoreceptors, many of which are known to be rod-specific [10, 11, 31]. We identified 33 of these genes that were expressed at significantly (>1.5, p < 0.05) lower levels following LSD1 treatment for 8 days (Table 1). Among these genes were a group of rod-specific genes associated with visual transduction, including Gnat1, Pde6, Prph2, Rho, Rom1, Rtbdn, and Sag. We verified these microarray data by RT-PCR analysis of a subset of these genes and each showed decreased expression following TCP treatment (Table 2). In addition, we examined the transcription factors Nrl and Crx that orchestrate the transcription of rod-specific genes. However, expression levels of these key regulators of retina development were not changed following TCP treatment (Online Resource Table 2, Table 2).
We next investigated whether the effect of inhibiting LSD1 on rod gene expression might be due to perturbed histone methylation patterns at these genes. First, we determined if there were global effects on the LSD1 substrates, H3K4me2 and H3K9me2, by Western blot. Inhibition of this enzyme had no significant effect on overall levels of either the active mark H3K4me2 (Fig. 4a, b) or the inhibitory mark H3K9me2 (Fig. 4c, d), suggesting that changes in gene expression resulting from LSD1 inhibition must be due to actions at specific genes.

Global level of LSD1 substrates, H3K4me2 and H3K9me2 during LSD1 inhibition. a Western blot of samples isolated from mouse PN1 retinal explants cultured for 8 days with or without LSD1 inhibitor probed with antibody against H3K4me2. Coomassie staining of core histones was used as loading control. b Quantification of H3K4me2 Westerns, n = 3. c Western blot of samples isolated from mouse PN1 retinal explants cultured for 8 days with ±LSD1 inhibitor probed with antibody against H3K9me2. Beta-actin was used as loading control. d Quantification of H3K9me2 Westerns, n = 3. e, f. Comparison of H3K4me2 (e) or H3K9me2 (f) accumulation on gene promoters in 8 days retina explants cultured with TCP vs. with control media only. ChIP experiments were done on 2–3 biological replicas and quantified by real-time PCRs, y-axis represent fold changes relative to control
During normal retina development, H3K4me2 marks increase on the promoters of rod-specific genes [11]. We evaluated what happened with this epigenetic mark under LSD1 inhibition with ChIP and Q-PCR for the promoter for rhodopsin (Rho) as well as for the promoters of the rhodopsin regulators Nrl and Crx. At these sites, H3K4me2 accumulation increased ∼2-fold (Fig. 4e). H3K9me2 is the other major substrate of LSD1, so we additionally assessed whether LSD1 inhibition could increase of H3K9me2 on rod-specific promoters in retina and thus lead to downregulation of rod-specific genes. We found no difference between control and TCP treatment in H3K9me2 occupancy on promoters of rod-specific genes (Fig. 4f), ruling out an involvement of the inhibitory mark H3K9me2 in blocking rod photoreceptor development under LSD1 inhibition.
From these results, we conclude that LSD1 activity is necessary for cells to undergo terminal differentiation into rod photoreceptors, but this is probably not achieved either by altering expression of known key rod-specific transcription factors or by preventing the normal developmental changes in histone methylation patterns at the rod genes.
Inhibition of LSD1 Changes Expression of Many Retinal Genes
The early postnatal period of retinal development is accompanied by large changes in the program of retinal gene expression centered around PN5 [10]. During this developmental window, large numbers of genes associated with cell proliferation are downregulated. The largest group of genes with higher expression under TCP treatment was a group of cell cycle genes, such as Ccnd1, Yap1, and genes involved in the progression from progenitors to differentiated cell types, such as Atoh7 and Eif4g2 at 24 h treatment (Online Resource Table 2) and Ascl1 (Mash1), Vsx1 and 2 (Chx10), Dkk3, Foxn4, Hes1 and 5, Irx5 and 6, Sox5 and 9 (Listed in Table 3) at 8 days treatment. We chose several genes to confirm the microarray data by RT-PCR (Table 4). In each case, the genes showed significantly higher expression in the presence of the LSD1 inhibitor. We then used Ingenuity System (Qiagen Silicon Valley, Redwood City CA) to perform pathways analysis of all 565 genes that showed higher expression in the presence of the LSD1 inhibitor. Selected top IPA canonical pathways enriched by these genes are presented in Fig. 5a and show that the majority of these genes are involved in CNS development and retinogenesis including such modules as Axonal Guidance Signaling, Cell Cycling Signaling, Wnt signaling, Nanog signaling, and Notch signaling. We then used the IPA’s gene network analysis functionality to generate networks of interacting genes and gene products of all genes that had significantly higher expression under TCP treatment. A list of the networks identified is given in Online Resource Table 3. Two examples of the networks strongly identified and related to development and retina differentiation, STAT3 signaling and HES1 signaling are illustrated in Figs. 5b, c. Interestingly, almost all genes identified in these networks are affected by LSD1 inhibition (Shadowed shapes).

LSD1 inhibition prevented the normal decrease in expression of progenitor genes. a Selected top IPA canonical pathways enriched with genes that were significantly upregulated at retina explants after LSD1 inhibitor treatment for 8 days compared to untreated control. Data from the expression microarray were analyzed by IPA’s pathway enrichment functionality. p value showed –log on y-axis and the line represents –log 2.0 = p < 0.01. List of pathways: 1. Axonal guidance signaling; 2. Cell cycle signaling; 3. Cxrc signaling; 4. Wnt signaling; 5. Phospholipase C signaling; 6. Nanog signaling; 7. Protein kinase A signaling; 8. Notch signaling. b IPA gene network analysis related to STAT3 signaling. Genes in close boxes represented upregulated expression in retina explant cultures following LSD1 inhibitor treatment compared to their untreated controls (Genes are listed in Online Resource table 3.2 under network “Cell-To-Cell Signaling and Interaction, Inflammatory Response, Cell Cycle”). c IPA gene causal network analysis related to HES1 signaling. Genes in close boxes represented upregulated expression in retina explant cultures following LSD1 inhibitor treatment compared to their untreated controls (Genes are listed in Online Resource table 3.2 under network “Nervous System Development and Function, Tissue Morphology, Visual System Development and Function”). Enlarged version of B and C is in Online Resource Figs. 1 and 2
Comparison of the group of genes showing higher expression under TCP treatment with the results of a previous developmental gene expression study [10, 32] suggests that a major effect of inhibiting LSD1 is the maintained expression of a group of key regulatory genes that are normally shut off as the retina develops. We hypothesized that this could be achieved by keeping active histone modification mark H3K4me2 on promoter and gene body of progenitor genes and promoted transcription of these genes. To test this, we investigated whether the effect of inhibiting LSD1 on progenitor gene expression might be due to perturbed H3K4me2 patterns at these genes. We first used the genome-wide database of retinal H3K4me2 marks during normal development (Gene Express Omnibus data repository (Accession number GSE38500)) to construct the pattern of H3K4me2 over the promoter and gene body of three example genes whose expression was maintained by LSD1 inhibition (Fig. 6a). In each case, the level and extent of the H3K4me2 marks over the promoter and gene showed a developmental decrease that correlated with decreased gene expression. We confirmed the ChIP-Seq data for these, and other progenitor genes that showed higher expression under TCP treatment in the expression array, by regular ChIP on retina extracts at PN1 and PN7, followed by Q-PCR with primers for sequences on either the promoter or the gene body. The ratios of H3K4me2 at PN1 to PN7 are indicated on Fig. 6b and confirm a reduction in H3K4me2 level over the first postnatal week. However, in retinal explants cultured for eight days with or without TCP treatment ChIP–qPCR showed a higher degree of H3K4 methylation under TCP treatment compare to control over both the promoters and bodies of progenitor genes (Fig. 6c). Similar studies for two of the genes showed that under the same conditions H3K9me2 was not changed (Fig. 6d). These results indicate that inhibition of LSD1 lessened the normal developmental decrease in H3K4me2 marks associated with decreased gene expression and suggest that LSD1 action is an important component of the developmental inhibition of these genes.

Levels of H3K4me2 maintained over progenitor genes under LSD1 inhibition. a Combined genome-wide tracks of H3K4me2 accumulation at different stages of normal retina development for factors important for general retinogenesis and cell type specification; gene TSS is marked by arrow. b Comparison of H3K4me2 accumulation on promoter and body at PN1 and PN7 during normal retina development for selected genes upregulated after LSD1 inhibition. ChIP experiments were done in 2–3 biological replicates; quantitative real-time PCRs were performed with primers specific for promoter and gene body (Online Resource Table 1) of progenitor genes. c–d Comparison between H3K4me2 (c) and H3K9me2 (d) accumulation on gene promoters and gene body areas in retina explants cultured for 8 days with TCP or in media only. ChIP experiments were done in 2–3 biological replicates; quantitative real-time PCRs were done with same primers as in (b)
Among the genes whose expression was higher after TCP treatment (Table 3) was the transcriptional repressor Hes1. Previous studies have shown that the absence of HES1 leads to maintenance of progenitor cells and reduction in the generation of rod photoreceptors [33]. Analysis of Hes1 expression during retinal development showed decreasing levels of protein such, that by the end of the second postnatal week it was barely detectable (Fig. 7a). Treatment of PN1 retina explants with TCP for 8 days led to maintain the high levels of HES1 protein (Fig. 7b). The increased level of HES1 was due to an increased number of expressing cells in the outer retina, where HES1 is present as punctate foci in the euchromatin area of nuclei (Fig. 7c, e). Double labeling with HES1 and rhodopsin antibodies showed no overlap. Levels of H3K4me2 at the Hes1 promoter normally decrease during postnatal development in parallel with the decreased protein expression (Fig. 7d). Treatment of PN1 retinal explants with TCP for 8 days resulted in a significant increase in H3K4me2 at the Hes1 promoter (Fig. 7e). Together these studies suggest that inhibition of LSD1 prevents the normal decrease in H3K4me2 at the promoter that coincides with the decreased levels of HES1 protein. This maintains expression of HES1 in a population of cells in the outer retina and prevents them from becoming rod photoreceptors.

Upregulation of Hes1 under LSD1 inhibition. a Down regulation of HES1 protein during normal retina development. Western blot of samples isolated from mouse retina probed with antibody against HES1 with Coomassie staining of core histones as loading control. Quantification of HES1 band intensities normalized to histone loading. b Amount of HES1 protein is increased in PN1 retinal explants cultured for 8 days in the presence of the LSD1 inhibitor TCP. Western blot of explant culture samples probed with antibody against HES1 with Coomassie staining of core histones as loading control. Quantification of HES1 bands intensity normalized on histone loading, **p < 0.001; n = 3. c Amount of HES1 Immunofluorescence staining (red) is increased in PN1 retinal explants cultured for 6 days in the presence of LSD1 inhibitors, TCP, while Rhodopsin (green) staining is decreased (nuclear counterstained with Hoechst33358 (blue)). Enlarge inset of upper left panel is present at F. d. Combined genome-wide tracks of H3K4me2 accumulation at different stages of normal retina development for Hes1 gene, TSS is marked by arrow. e. Increased H3K4me2 accumulation on Hes1 gene promoters in 8 day retina explants cultured with TCP compare with control. ChIP experiments were done in 2 biological replicas; quantitative real-time PCRs were done with primers for Hes1 promoter area (Online Resource Table 1), location of the primers mark by arrowhead in d, *p < 0.05, **p < 0.01, ***p < 0.0005. F Enlarge inset of upper left panel from C
LSD1 has no DNA-binding domain and is thought to work in complexes with specific binding proteins, particularly those of the orphan nuclear receptor (NR) family. To examine a possible relationship between LSD1 and NRs we utilized the known NR2E1/ NR2E3 DNA-binding motif AAGTCA [34] for an in silico search of potential NR-binding sites. Such sites were found for all the genes whose developmental decrease in expression was blocked by TCP treatment, with the interesting exception of NR2E1 itself (Table 3). For several genes we designed appropriate primer pairs to probe ChIP DNA fragments from PN1 and PN28 retina samples that were precipitated by anti-LSD1 (Fig. 8). We used primers inside Rho gene as an example of a gene whose expression is not maintained by LSD1 inhibition. All primer pairs for Gnat2 and Smad3 had NR-binding sites inside PCR amplified regions (Fig. 8a, b). Primer pairs for the Hes1 gene were originally chosen on basis of DNAse Hypersensitive Sites (DHS) and correspond to different regions of Hes1 genomic locus: 1, 3, and 4 have no NR-binding motif; 2, 5, 6 have NR-binding sites inside or in closed proximity to PCR amplified regions (DNA for chromatin immunoprecipitation was sonicated to fragments 300–700 bp length) (Fig. 8c, e). Not all potential NR-binding sites were in fragments precipitated by anti-LSD1, but primer pairs 5 and 6 for Hes1; primer pair 2 for Gnat2 and primer pairs 1 and 3 for Smad3 have LSD1 bindings at PN1 and statistical down regulation of bindings at PN28. From these data we conclude that LSD1 may interact with nuclear receptors NR2E1/ NR2E3 to bind to specific loci around progenitor genes and its promoters and that this binding is developmentally regulated.

LSD1 binding to genomic loci of selected genes upregulated by inhibition of LSD1. a–d LSD1 accumulation on loci of genes maintained after LSD1 inhibition in PN1 and PN28 retinal explants. Quantitative real-time PCRs after LSD1 ChIP were performed with primers specific for Gnat2 (a), Smad3 (b), Hes1 (c), and Rho (d) genes (Online Resource Table 1). All primer pairs for Gnat2 and Smad3 have NR-binding sites inside PCR amplified regions. e Hes1 genomic loci with primer pairs for Hes1 gene correspond to following regions: 1, 3, and 4 have no NR-binding motif; 2, 5, 6 have NR-binding sites inside or in closed proximity to PCR amplified regions. Dotted lines around primers 5 and 6 represent length of sonicated DNA fragments from chromatin immunoprecipitation that could be tested by these primers. DNAse Hypersensitive Sites (DHS) at 3 developmental stages at the Hes1 locus are taken from ENCODE for reference. Experiments done in three technical replicates, *p = 0.016; **p < 0.09; ***p = 0.002
LSD1 Inhibition Alters Expression of Genes Associated with Other Retinal Cell Types
There are fewer well-documented cell type-specific genes for late-generated retinal cell types other than rods but further analysis of the microarray data indicated that LSD1 inhibition did result in increased RNA levels of genes whose expression has been documented in bipolar and Muller glial cells, an effect opposite to that on rod photoreceptors (Table 5 and Fig. 9a). Most notably we found increased expression of a cluster of 13 genes associated with cone photoreceptors including the cone-specific genes Gnat2, Otop3, Pde6c, and Gnb3 (Table 5). Unlike many of the other changes in gene expression described above, these changes were rapid with 6 of the cone genes showing increased expression at 24 h. The change in RNA levels of Gnat2 and Pde6c, both cone-specific genes, was confirmed by PCR (Fig. 9a). Western blot analysis showed that LSD1 inhibition led to an increase in GNAT2 protein levels as well (Fig. 9b).

LSD1 inhibition increased gene expression in other retinal cell types. a Confirmation of microarray data for examples of bipolar and cone-specific genes by RT-PCR. RNA in 3 biological replicas was purified from retinal explants cultured for 8 days with TCP or control. After cDNA isolation RT-PCRs were done with gene’s specific primers (Online Resource Table 1). b Amount of GNAT2 protein is increased in PN1 retinal explants cultured for 8 days in the presence of LSD1 inhibitors, TCP. Western blot of explant culture samples probed with antibody against GNAT2 with Coomassie staining of core histones as loading control. Quantification of GNAT2 band intensities normalized to histone loading, **p < 0.001; n = 3
Discussion
A major switch in gene expression from those involved in progenitor functions to those characteristic of postmitotic and differentiated cells accompanies early postnatal development in the mouse retina [10, 32]. Inactivation of many of the progenitor genes correlates with the decrease of an active epigenetic mark, H3K4me2, over the transcriptional start site and body of these genes [11].
Inhibition of LSD1 by either of two inhibitors caused a dramatic change in gene expression with a resulting halt in rod photoreceptor development. Both TCP and Pargyline have been well-characterized and TCP has been widely used as a LSD1 inhibitor [12, 14, 15]. We did not measure the loss of LSD1 activity directly and found no overall change in the levels of either H3K4me2 or H3K9me2. In spite of this, the observation of increased levels of H3K4me2 at some promoters does indicate that the enzyme was inhibited in our experimental conditions. Both TCP and Pargyline are known to inhibit monoamine oxidase (MAO) as well as LSD1. In the adult retina, MAO is present in only a minor subpopulation of amacrine cells and even here, it appears relatively late in development. Thus, we feel that this additional action of the LSD1 inhibitor has little influence on the transition of progenitors into rod photoreceptors.
The most obvious alterations of gene expression observed in retinal explants when treated with LSD1 inhibitors were maintained expression of genes associated with proliferating retinal progenitors, a block in normal differentiation of rod photoreceptors and abnormal enhancement of expression of genes characteristic of other retinal cell types.
In ES cells and other early progenitors, LSD1 maintains cells in a proliferative undifferentiated state [4, 15, 35]. In the retina, however, LSD1 is only weakly expressed in the embryonic proliferative neuroepithelium and shows maximum levels of expression in the outer retina at the postnatal period corresponding to the time of maximum rod photoreceptor differentiation. Since LSD1 was primarily expressed in postmitotic cells that had lost PCNA expression, we conclude that, unlike ES cells, LSD1 is not involved in the self-renewal of retinal progenitors. Rather, we suggest that LSD1 normally functions to switch off genes involved in cell cycle progression, progenitor and cell fate specification. This suggests that as cells exit their proliferative phase and become postmitotic, LSD1 is recruited to genes whose activity is no longer needed and presumably demethylation of H3K4 is part of this inactivation. A similar function for LSD1 on the transition from progenitor to differentiated cell has been described in the hematopoietic system, where removal of LSD1 activity was associated with increased H3K4me2 methylation on progenitor genes and maintained expression of these genes, leading to dramatic effects on stem cell differentiation and terminal blood cell maturation [6]. Because the changes in gene expression we observed correlate well with changes in histone methylation, we believe that the effects we observed are due to the action of LSD1 on histones. It has been reported that LSD1 also has non-histone targets such as p53 (TRP53) and DNMT1, but there is currently no evidence that actions on these targets can regulate retinal development [36–38]. While LSD1 appears to operate differently in different cell types, its action has the common feature that it is regulating specific developmental switchpoints.
Inhibition of LSD1 also blocked the normal expression of a group of rod photoreceptor specific genes. Residual expression of rod-specific genes even in the presence of LSD1 inhibitors is probably due to a population of early-born rods that became postmitotic before PN1 and had already initiated cell type-specific gene expression before application of the inhibitor. This suggests that inhibition of LSD1 is preventing the onset of expression rather than switching off expression. In addition to its action on H3K4 methylation, LSD1 can demethylate H3K9, and in some cases this has been suggested as a mechanism for derepression of genes [39, 40]. For Rho, however, inhibition of LSD1 did not change the H3K9me2 occupancy on its promoter indicating that this was not its mechanism of action in retina. Although Rho transcription is blocked by inhibition of LSD1, the expression of its major transcriptional activator, Crx, is relatively unchanged and the Rho gene shows increased H3K4me2, a mark that normally correlates with increased expression in the developing retina [11]. These results are compatible with the idea that LSD1 is normally either promoting the assembly of an additional activating complex or inhibiting the expression of a repressor molecule.
Our results are compatible with a model where the broad patterns of activity of LSD1 are determined by expression of the enzyme itself, but the specific genes on which it acts are a function of complexing with specific DNA-binding proteins [41, 42]. LSD1 has no DNA-binding domain and it can achieve gene specificity as a component of various transcriptional complexes with specific DNA-binding proteins [41, 42]. LSD1 can bind to orphan nuclear receptors (ONR) in CNS (NR2E1) [15, 43], blood (NR2C1 and NR2C2) [14, 44], and cell culture (NR2E1 and NR2E3) [45]. LSD1 also has a repressive action when it demethylates H3K4me2/1 as a part of complexes with CoREST (corepressor for element-1-silencing transcription factor) [42, 46–48], or NuRD (nucleosome remodeling and histone deacetylation) [23]. When complexed with estrogen or androgen nuclear receptors, LSD1 can also promote gene activation by demethylating H3K9me2/1 [49, 50]. LSD1 is recruited to the promoters of genes regulating proliferation of neural stem cells by the orphan nuclear receptors TLX or NR2E1 [15]. Similarly, in Y79 retinoblastoma cells, LSD1 interacted with NR2E1 through its SWIRM and amine oxidase domains and both proteins were essential for continued cell proliferation [45].
NR2E1 and NR2E3 are expressed in postnatal retina [34], where NR2E1 is expressed early [51, 52] and NR2E3 expression peaks around PN7 [10, 53, 54]. To study if LSD1/NR complexes bind to loci of the genes upregulated under LSD1 inhibition, we tried to perform ChIP with anti-NR antibody. Unfortunately, currently available antibodies for both NR2E1 and NR2E3 are not suitable for IP, ChIP, or even Western blots, so as proxy for NR ChIP, we carried out LSD1 ChIP and asked whether the precipitated DNA fragments had NR-binding sites. The results show that most of the genes that alter expression after LSD1 inhibition do contain NR-binding sites and that LSD1 antibodies can precipitate DNA fragments containing NR-binding sites. However, LSD1 antibodies can also precipitate DNA fragments with no known NR-binding site, and many genes with NR-binding sites are not affected by LSD1 inhibition. Thus, the relationship between LSD1 and nuclear receptors is complex and needs further investigation.
A hypothetical scheme for LSD1 functions during retina maturation is presented in Fig. 10. We suggest that as cells move from a proliferating progenitor to a postmitotic cell, LSD1 is upregulated and can become complexed with a nuclear receptor, such as NR2E1 (Fig. 10a). Because of the DNA sequence specificity of these nuclear receptors, this leads to demethylation of histones at developmentally important genes and downregulation of their expression. In the presence of LSD1 inhibitor, this demethylation does not occur and expression of these genes is maintained. Some of the progenitor genes that LSD1 normally represses during cell type specification are themselves repressors of rod genes. Candidates for this action would be genes such as Hes1 [33], a downstream target of NOTCH1 [46] whose deletion in mice leads to early onset of photoreceptor production [55, 56]. Maintained expression of a repressor such as Hes1 would explain why rod genes were not expressed even though their key transcription factors were present and the genes underwent their normal pattern of enhanced H3K4me2 levels around the gene promoters.

Schematic presentation for the model of LSD1 functions during retina development. a Transition from proliferating to postmitotic retina progenitors on promoter of genes that are responsible for cell cycle progression, progenitor and cell fate specification, such as Notch1 or Hes1, as example. b Transition from committed rod precursors to mature rod photoreceptor on promoter of cone genes inside rod
In contrast to the action on rod photoreceptor genes, inhibition of LSD1 led to the enhanced expression of genes associated with other retinal cell types, particularly cone photoreceptors. The enhanced expression of cone genes is similar to that seen in the rd7 mouse in which there is a mutation in the orphan nuclear receptor Nr2e3 gene, and this suggests a mechanism of action of LSD1 in the developing retina. LSD1 may interact with NR2E3 and become part of a repressor complex that blocks ectopic expression of cone genes (and perhaps others) by demethylating histones (Fig. 10b) [57, 58]. In the presence of LSD1 inhibitors, however, this repression is ineffective and the cone genes are ectopically expressed. Some genes characteristic of other retinal cell types also showed increased expression. While it is possible that this also represents derepression of genes in rod photoreceptors, other mechanisms can also account for the results. For example, LSD1 inhibition led to maintained expression of Hes5, a key regulator of Muller cell maturation that promotes transcription of Muller cell specific genes [59].
The broader implication of this model of LSD1 action is that terminal differentiation of CNS neurons involves the concerted activity of two parallel pathways. The first pathway is the classic combination of transcription factors at the promoters and enhancers of genes that promote the assembly of transcription complexes and hence serve as activators of gene expression. The second is an epigenetic control of gene expression that utilizes sequence-specific DNA-binding proteins and histone-modifying enzymes for a second network of regulatory complexes that shows unique temporal and spatial specificity. Clearly these two pathways interact and have to work in concert. Abnormalities in either can lead to abnormal gene expression and serious disruptions in development.
References
Kooistra SM, Helin K (2012) Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 13:297–311
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119:941–953
Hou H, Yu H (2010) Structural insights into histone lysine demethylation. Curr Opin Struct Biol 20:739–748
Adamo A, Sese B, Boue S, Castano J, Paramonov I, Barrero MJ, Izpisua Belmonte JC (2011) LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol 13:652–659
McDonald OG, Wu H, Timp W, Doi A, Feinberg AP (2011) Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol 18:867–874
Kerenyi MA, Shao Z, Hsu YJ, Guo G, Luc S, O’Brien K, Fujiwara Y, Peng C et al (2013) Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. eLife 2, e00633
Sprussel A, Schulte JH, Weber S, Necke M, Handschke K, Thor T, Pajtler KW, Schramm A et al (2012) Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 26:2039–2051
Grimaldi P, Pucci M, Di Siena S, Di Giacomo D, Pirazzi V, Geremia R, Maccarrone M (2012) The faah gene is the first direct target of estrogen in the testis: role of histone demethylase LSD1. Cell Mol Life Sci: CMLS 69:4177–4190
Jeon CJ, Strettoi E, Masland RH (1998) The major cell populations of the mouse retina. J Neurosci: Off J Soc Neurosci 18:8936–8946
Zhang SS, Xu X, Liu MG, Zhao H, Soares MB, Barnstable CJ, Fu XY (2006) A biphasic pattern of gene expression during mouse retina development. BMC Dev Biol 6:48
Popova EY, Xu X, DeWan AT, Salzberg AC, Berg A, Hoh J, Zhang SS, Barnstable CJ (2012) Stage and gene specific signatures defined by histones H3K4me2 and H3K27me3 accompany mammalian retina maturation in vivo. PLoS One 7, e46867
Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R (2006) Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol 13(6):563–567
Schmidt DM, McCafferty DG (2007) trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 46:4408–4416
Shi L, Cui S, Engel JD, Tanabe O (2013) Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med 19:291–294
Sun G, Alzayady K, Stewart R, Ye P, Yang S, Li W, Shi Y (2010) Histone demethylase LSD1 regulates neural stem cell proliferation. Mol Cell Biol 30:1997–2005
Zhang F, Xu D, Yuan L, Sun Y, Xu Z (2014) Epigenetic regulation of Atrophin1 by lysine-specific demethylase 1 is required for cortical progenitor maintenance. Nat Commun 5:5815
Barnstable CJ (1980) Monoclonal antibodies which recognize different cell types in the rat retina. Nature 286:231–235
Hargrave PA, Adamus G, Arendt A, McDowell JH, Wang J, Szaby A, Curtis D, Jackson RW (1986) Rhodopsin’s amino terminus is a principal antigenic site. Exp Eye Res 42:363–373
Abo T, Balch CM (1981) A differentiation antigen of human NK and K cells identified by a monoclonal antibody (HNK-1). J Immunol 127(3):1024–1029
Naegele JR, Barnstable CJ (1991) A carbohydrate epitope defined by monoclonal antibody VC1.1 is found on N-CAM and other cell adhesion molecules. Brain Res 559(1):118–129
Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS et al (2011) An assessment of histone-modification antibody quality. Nat Struct Mol Biol 18:91–93
Lobanova ES, Herrmann R, Finkelstein S, Reidel B, Skiba NP, Deng WT, Jo R, Weiss ER et al (2010) Mechanistic basis for the failure of cone transducin to translocate: why cones are never blinded by light. J Neurosci 30(20):6815–6824
Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA (2012) Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482:221–225
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Popova EY, Grigoryev SA, Fan Y, Skoultchi AI, Zhang SS, Barnstable CJ (2013) Developmentally regulated linker histone H1c promotes heterochromatin condensation and mediates structural integrity of rod photoreceptors in mouse retina. J Biol Chem 288(24):17895–17907
Pinzon-Guzman C, Zhang SS, Barnstable CJ (2011) Specific protein kinase C isoforms are required for rod photoreceptor differentiation. J neurosci 31:18606–18617
Sparrow JR, Hicks D, Barnstable CJ (1990) Cell commitment and differentiation in explants of embryonic rat neural retina. Comparison with the developmental potential of dissociated retina. Brain research. Dev Brain Res 51:69–84
Zhang SS, Fu XY, Barnstable CJ (2002) Tissue culture studies of retinal development. Methods 28:439–447
Zhang SS, Wei J, Qin H, Zhang L, Xie B, Hui P, Deisseroth A, Barnstable CJ et al (2004) STAT3-mediated signaling in the determination of rod photoreceptor cell fate in mouse retina. Invest Ophthalmol Vis Sci 45:2407–2412
Zhang SS, Wei JY, Li C, Barnstable CJ, Fu XY (2003) Expression and activation of STAT proteins during mouse retina development. Exp Eye Res 76:421–431
Siegert S, Cabuy E, Scherf BG, Kohler H, Panda S, Le YZ, Fehling HJ, Gaidatzis D et al (2012) Transcriptional code and disease map for adult retinal cell types. Nat Neurosci 15(3):487–495
Liu MG, Li H, Xu X, Barnstable CJ, Zhang SS (2008) Comparison of gene expression during in vivo and in vitro postnatal retina development. J Ocul Biol Dis Informatics 1:59–72
Tomita K, Ishibashi M, Nakahara K, Ang SL, Nakanishi S, Guillemot F, Kageyama R (1996) Mammalian hairy and enhancer of split homolog 1 regulates differentiation of retinal neurons and is essential for eye morphogenesis. Neuron 16:723–734
Forrest D, Swaroop A (2012) Minireview: the role of nuclear receptors in photoreceptor differentiation and disease. Mol Endocrinol 26:905–915
Yin F, Lan R, Zhang X, Zhu L, Chen F, Xu Z, Liu Y, Ye T et al (2014) LSD1 regulates pluripotency of embryonic stem/carcinoma cells through histone deacetylase 1-mediated deacetylation of histone H4 at lysine 16. Mol Cell Biol 34:158–179
Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R et al (2007) p53 is regulated by the lysine demethylase LSD1. Nature 449(7158):105–108
Nicholson TB, Chen T (2009) LSD1 demethylates histone and non-histone proteins. Epigenetics 4(3):129–132
Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W et al (2009) The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 41(1):125–129
Choi J, Jang H, Kim H, Kim ST, Cho EJ, Youn HD (2010) Histone demethylase LSD1 is required to induce skeletal muscle differentiation by regulating myogenic factors. Biochem Biophys Res Commun 401:327–332
Musri MM, Carmona MC, Hanzu FA, Kaliman P, Gomis R, Parrizas M (2010) Histone demethylase LSD1 regulates adipogenesis. J Biol Chem 285:30034–30041
Benner C, Konovalov S, Mackintosh C, Hutt KR, Stunnenberg R, Garcia-Bassets I (2013) Decoding a signature-based model of transcription cofactor recruitment dictated by cardinal cis-regulatory elements in proximal promoter regions. PLoS Genet 9, e1003906
Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H (2006) Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell 23:377–387
Gui H, Li ML, Tsai CC (2011) A tale of tailless. Dev Neurosci 33:1–13
Cui S, Kolodziej KE, Obara N, Amaral-Psarris A, Demmers J, Shi L, Engel JD, Grosveld F et al (2011) Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic beta-type globin promoters in differentiated adult erythroid cells. Mol Cell Biol 31:3298–3311
Yokoyama A, Takezawa S, Schule R, Kitagawa H, Kato S (2008) Transrepressive function of TLX requires the histone demethylase LSD1. Mol Cell Biol 28:3995–4003
Mulligan P, Yang F, Di Stefano L, Ji JY, Ouyang J, Nishikawa JL, Toiber D, Kulkarni M et al (2011) A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Mol Cell 42:689–699
Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y (2005) Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19:857–864
Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA et al (2007) Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446:882–887
Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, Ju BG, Ohgi KA et al (2007) Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 128:505–518
Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R et al (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437:436–439
Miyawaki T, Uemura A, Dezawa M, Yu RT, Ide C, Nishikawa S, Honda Y, Tanabe Y et al (2004) Tlx, an orphan nuclear receptor, regulates cell numbers and astrocyte development in the developing retina. J Neurosci 24:8124–8134
Yu RT, Chiang MY, Tanabe T, Kobayashi M, Yasuda K, Evans RM, Umesono K, (2000) The orphan nuclear receptor Tlx regulates Pax2 and is essential for vision. Proceedings of the National Academy of Sciences of the UniteEpigenetic regulation of Atrophin1 by lysine-specific demethylase 1 is required for cortical progenitor maintenance.d States of America 97, 2621–2625
Corbo JC, Cepko CL (2005) A hybrid photoreceptor expressing both rod and cone genes in a mouse model of enhanced S-cone syndrome. PLoS Genet 1, e11
Haider NB, Mollema N, Gaule M, Yuan Y, Sachs AJ, Nystuen AM, Naggert JK, Nishina PM (2009) Nr2e3-directed transcriptional regulation of genes involved in photoreceptor development and cell-type specific phototransduction. Exp Eye Res 89:365–372
Jadhav AP, Mason HA, Cepko CL (2006) Notch 1 inhibits photoreceptor production in the developing mammalian retina. Development 133(5):913–923
Mizeracka K, DeMaso CR, Cepko CL (2013) Notch1 is required in newly postmitotic cells to inhibit the rod photoreceptor fate. Development 140(15):3188–3197
Cheng H, Khan NW, Roger JE, Swaroop A (2011) Excess cones in the retinal degeneration rd7 mouse, caused by the loss of function of orphan nuclear receptor Nr2e3, originate from early-born photoreceptor precursors. Hum Mol Genet 20:4102–4115
Peng GH, Ahmad O, Ahmad F, Liu J, Chen S (2005) The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum Mol Genet 14:747–764
Hojo M, Ohtsuka T, Hashimoto N, Gradwohl G, Guillemot F, Kageyama R (2000) Glial cell fate specification modulated by the bHLH gene Hes5 in mouse retina. Development 127:2515–2522
Acknowledgments
This research was supported by NEI/NIH grant EY013865 and Macula Vision Research Foundation. We thank Rob Brucklacher from Penn State Hershey Genome Sciences Facility for help with gene expression microarray.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Highlight
1. LSD1 is expressed at transition from late progenitors to rod photoreceptors
2. Under LSD1 inhibition level of H3K4me2 maintained over the progenitor genes
3. LSD1 inhibition prevented the normal decrease in expression of progenitor genes
4. LSD1 inhibition blocked rod photoreceptor development
5. LSD1 inhibition increased other retinal cell type’s genes expression
Evgenya Y. Popova and Carolina Pinzon-Guzman contributed equally to this work.
Rights and permissions
About this article

Cite this article
Popova, E.Y., Pinzon-Guzman, C., Salzberg, A.C. et al. LSD1-Mediated Demethylation of H3K4me2 Is Required for the Transition from Late Progenitor to Differentiated Mouse Rod Photoreceptor. Mol Neurobiol 53, 4563–4581 (2016). https://doi.org/10.1007/s12035-015-9395-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9395-8