
Abstract
Altered activity of glycogen synthase kinase-3 (GSK-3) is associated with psychiatric diseases and neurodegenerative diseases. GSK-3 is a key regulator in multiple aspects of neuronal differentiation in the brain. However, little is known about the role of GSK-3 in astrocyte development. To examine the role of GSK-3 in astrocytes, we generated a conditional knockout mouse using a glial fibrillary acidic protein (GFAP)-cre driver, in which the GSK-3 alpha and beta genes are deleted in astrocytes. We found that GFAP-cre-mediated GSK-3 deletion led to a larger brain. The number and size of astrocytes were increased in GSK-3 mutant brains. The levels of GFAP and phospho-STAT3, indicators of astrogenesis, were elevated in GSK-3 mutants. Furthermore, we found upregulation of astrocyte regulatory molecules such as phospho-AKT, phospho-S6, and cyclin D in GSK-3 mutant brains. Finally, GSK-3 mutant mice exhibited aberrant anxiety and social behavior. Our results suggest that GSK-3 plays a significant role in astrocyte development and behavioral control in mice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Astrocytes are multifunctional nonneuronal cells abundant in the central nervous system (CNS). Recent studies have shown that astrocytes are critically involved in a variety of essential activities in the normal CNS, including neuronal protection and support, neuronal migration, synaptic transmission, and information processing [1–4]. Furthermore, astrocytes respond to pathological lesions and environmental insults in the CNS via a process called reactive astrogliosis [5–7, 2]. Thus, intrinsic regulation of astrogenesis is implicated in pathogenic progression of CNS diseases. A better understanding of astrogenesis could improve therapeutic approaches to CNS diseases.
Glycogen synthase kinase-3 (GSK-3) is a serine/threonine kinase and has two isoforms, GSK-3α and GSK-3β. Both isoforms are highly expressed in the nervous system [8]. Studies have shown that GSK-3 plays a role in neuronal development [9, 10]. For example, GSK-3 controls neural progenitor homeostasis and neurogenesis in the developing brain [11]. Changes in local and global GSK-3 activity play a critical role in axon/dendritic outgrowth and specification [12, 8, 13] as well as neuronal migration [14–16]. However, whether GSK-3 plays a role in astrocyte regulation during development and in adults is unclear.
Members of the Janus kinase signal transducers and activators of transcription (Jak-STAT) signaling family are implicated as triggers of astrogenesis [17]. The STAT3 gene plays a major role in cellular phospho-signaling cascades to stimulate astrocyte differentiation [18]. In addition, the mammalian target of rapamycin (mTOR) functions as a molecular regulator in astrocyte cell growth and motility [19]. mTOR is involved in cross-talk with AKT for gene transcriptional expression. Activation of AKT/mTOR signaling directly influences the GSK-3 activity in the brain [20].
Here, we investigated the function of GSK-3 in astrogenesis in the brain using a conditional loss of function approach. By crossing with a glial fibrillary acidic protein (GFAP)-cre line, we deleted GSK-3 in astrocytes in the brain. GSK-3 deletion markedly enhanced astrocyte proliferation and induced astrocyte hypertrophy. Phosphorylation of STAT3, AKT, and S6 were associated with the morphological changes induced by GSK-3 deletion in astrocytes. Furthermore, the abnormal regulation of astrocytes in GSK-3 mutant mice was accompanied by behavioral abnormalities. Together, our findings reveal a novel function of GSK-3 in astrocyte development and behavioral control in mice.
Materials and Methods
Mice
Mice were handled according to our animal protocol approved by the University Nebraska Medical Center. GSK-3α null [21] and GSK-3β floxed mice [22] were described previously. GFAP-cre (B6.Cg-Tg(GFAP-cre)73.12Mvs/J) mice were purchased from Jackson Laboratory and were used to generate Gsk3α -/− ; Gsk3β loxP/loxP ;GFAP-cre mice.
BrdU Administration
Bromodeoxyuridine (BrdU, Sigma-Aldrich) was dissolved in 0.007 N NaOH and administered into postnatal day (P) 7, 35, and 84 mice by intraperitoneal injection. Control and GSK-3 mutant mice were injected with BrdU (100 mg/kg) for two consecutive days before they were sacrificed. T98G human glioblastoma cells were cultured in 24-well plates (5 × 104/well), followed by treatments of GSK3-inhibitor IX for 72 h. The cells were then incubated with 10 μM BrdU for 6 h, followed by fixation in 4 % paraformaldehyde.
Real-Time PCR
Total RNA was extracted from control and GSK-3 mutant brains using the TRIzol reagent (Invitrogen), and the concentration of total RNA was determined by measuring the absorbance at 260 nm. First-strand complementary DNA (cDNA) was prepared by subjecting total RNA (1 μg) to reverse transcription using Verso cDNA synthesis kit (Thermo Fisher Scientific). Real-time PCR was performed with 1 μl of the cDNA template added to 10 μl of 2× SYBR Premix Ex Taq (Applied Biosystems) and specific primers (10 pM each). The primers for cyclin D were 5′-TCCTCTCCAAAATGCCAGAG-3′ (sense) and 5′-GCAGGAGAGGAAGTTGTTGG-3′ (anti-sense) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were 5′-AAGGTCATCCCAGAGCTGAA-3′ (sense) and 5′-AGGAGACAACCTGGTCCTCA-3′ (anti-sense). Real-time PCR (Applied Biosystems) was carried out for 40 cycles of denaturation at 95 °C for 15 s, annealing at 58 °C for 15 s, and extension at 72 °C for 15 s. Fluorescence intensity was measured at the end of the extension phase of each cycle. The threshold value for fluorescence intensity of all the samples was set manually. The cycle at which fluorescence intensity of the PCR products exceeded this threshold during the exponential phase of PCR amplification was identified as the threshold cycle (CT).
Target gene expression was quantified relative to that of the internal control gene (GAPDH) based on the comparison of CTs at constant fluorescence intensity. The amount of transcript was inversely related to the observed CT, and the CT was expected to increase by 1 for every 2-fold dilution of the transcript. Relative expression (R) was calculated using the equation R = 2−[ΔCT sample − ΔCT control]. All data were normalized relative to GAPDH as well as to the respective controls.
Western Blot Analysis
Western blotting was performed as described previously [23, 24]. Brain lysates were extracted with RIPA buffer (Invitrogen). Proteins (40 μg/lane) were separated on 4–15 % SDS-PAGE gel and transferred to immobilon-P membrane (Millipore) by a Trans-Blot SD semidry transfer cell (Bio-Rad Laboratories). The resulting blot was blocked in Tris-buffed saline (TBS) containing 5 % BSA and 0.1 % Tween 20 for 60 min and then incubated with a primary antibody rabbit anti-GFAP (1:1000, DAKO), rabbit anti-p-STAT3 (1:600, Cell Signaling Technology), rabbit anti-activated caspase 3 (1:800, Cell Signaling Technology), PTEN (1:1000, Cell Signaling Technology), rabbit anti-p-AKT (1:1000, Cell Signaling Technology), rabbit anti-p-S6 (1:1000, Cell Signaling Technology), or GAPDH (1:2000, Millipore) for overnight at 4 °C. After washing in TBS containing 0.1 % Tween 20, the membrane was incubated with an appropriate horseradish-peroxidase-conjugated secondary antibody (1:2500, Santa Cruz) for 1 hour at room temperature. After washing, the membrane was developed by using ECL chemiluminescence reagent (Thermo Fisher Scientific) and imaged by the FluorChem HD2 imaging system (ProteinSimple). Signal specificity was ensured by omitting each primary antibody, and bands were normalized by GAPDH-immunoreactive bands in the same membrane after stripping. Density measurements for each band were performed with NIH ImageJ software. Background samples from an area near each lane were subtracted from each band to acquire mean band density.
Immunostaining
Immunostaining of brain sections or dissociated cells was performed as described previously [25, 26]. Primary antibodies used were mouse anti-GFAP (DAKO), rabbit anti-parvalbumin (Millipore), rabbit anti-olig2 (Millipore Co), rabbit anti-Ki67 (Abcam), and mouse anti-BrdU (BD Biosciences) antibodies. Appropriate secondary antibodies conjugated with Alexa dyes (Invitrogen) were used to detect primary antibodies. DAPI (Sigma-Aldrich) was used to stain nuclei.
Morphometry
Images of brain sections at periodic distances along the rostro-caudal axis were taken with Zeiss LSM510 and LSM710 confocal microscopes and a Nikon Eclipse epifluorescence microscope attached with a QImaging CCD camera. Ten mice for each experiment (control mice, n = 5; mutant mice, n = 5) were used. Stereological analysis of GFAP-positive cells was performed by analyzing with confocal microscopy one-in-six series of 40-μm coronal sections (240 μm apart). The total estimated number of cells within the cortical layer I–VI and dentate gyrus was obtained by multiplying the average number of positive cells per section by the total number of 40-μm sections comprising the entire cortex and dentate gyrus as previously described [27–30].
The area of GFAP-stained cell body was measured by using ImageJ software (NIH) [31–33]. GFAP-positive astrocytes were imaged using a confocal microscope with a ×40 objective. The images were converted to 8-bit grayscale and subjected to software-driven segmentation analysis with automatic machine-set thresholding in ImageJ, thus eliminating subjective investigator bias. Then, a particle parameter enumeration analysis was followed for size exclusion at minimum of 10 pixel2.
For analyzing cultured cells, more than 20 fields scanned horizontally and vertically were examined in each condition. Cell numbers were described in figure legends. The images were analyzed by using ZEN (Zeiss), LSM image browser (Zeiss), QCapture software (QImaging), and ImageJ. The calculated values were averaged, and some results were recalculated as relative changes versus control.
Elevated Plus-Maze Test
The apparatus (EB Instrument) includes two open arms (35 × 5 cm), two enclosed arms (35 × 5 × 15 cm), and a central platform (5 × 5 cm). The entire apparatus was elevated 45 cm above the floor. A mouse was placed on the central platform, facing the same open arm, and allowed to roam freely for 5 min. The number of entries into and the time spent on open and closed arms were recorded. Percent open-arm time was calculated as time spent in the open arms per total time. Additionally, rearing frequency was recorded.
Three-Chamber Test for Social Interaction and Novelty Behavior
Social behavior was evaluated as described previously [34]. A rectangular and transparent Plexiglas box divided by walls into three equally sized compartments (Ugobasile) was used. Circular holes in the Plexiglas walls provide access between the chambers. For sociability test, the test mouse was moved to center chamber 2 with the connecting holes blocked. A stimulus mouse (unfamiliar mouse) designated as “stranger 1” was placed in the wire enclosure in chamber 1. Then, chambers 1 and 3 were opened and the test mouse was allowed to explore the entire apparatus for 10 min. For social novelty test, stranger 1 was randomly placed in one of the enclosures in which the test mouse had the choice of what to investigate stranger 1 or a novel mouse. A novel mouse was designated as “stranger 2” and was taken from a different home cage and placed into the remaining empty enclosure. Time spent to sniff a partner by the test mouse was recorded for 10 min in both sociability and social novelty behavior test. The chambers of the apparatus were cleaned with water and dried with paper towels between each trial. At the end of each test day, the apparatus was sprayed with 70 % ethanol and wiped clean with paper towels.
Cell Culture
Primary cortical culture was described previously [11, 8]. Briefly, cerebral cortices were isolated from E12.5-14.5 mice. Meninges were removed and cortical cells were dissociated with trituration after trypsin/EDTA treatment. The cells were plated onto poly-d-lysine/laminin-coated coverslips and cultured in the medium containing Neurobasal medium (Invitrogen), 2 mM glutamine, 2 % (v/v) B27 supplement (Invitrogen), 1 % (v/v) N2 supplement (Invitrogen), 5 % fetal bovine serum (Invitrogen), and 50 U/ml penicillin/streptomycin (Invitrogen). The cells were exposed to GSK3-inhibitor IX (Calbiochem). After days in vitro (DIV) 6 or DIV 9, whole cells were fixed for immunostaining. All experiments were performed in triplicate.
T98G cells were grown as monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM, Gibco), supplemented with 10 % FBS (Invitrogen), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Gibco) [35, 36]. All cells were grown at 37 °C in a humidified atmosphere of 95 % O2 and 5 % CO2.
Statistical Analysis
Normal distribution was tested using a Kolmogorov-Smirnov test and variance was compared. Unless otherwise stated, statistical significance was determined by two-tailed unpaired Student’s t test for two-population comparison or one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons. Data were analyzed using the GraphPad Prism and presented as means ± SEM.
Results
GSK-3 Deletion Using GFAP-cre Driver Increases Brain Size and Astrocyte Number
To examine the role of GSK-3 in astrocyte development, we deleted GSK-3 in astroglial lineage cells using a GFAP-cre mouse line. The GFAP-cre driver expresses Cre recombinase under the promoter of the regulatory sequence of GFAP gene [37, 38]. GSK-3α null and GSK-3β floxed mice were crossed with the GFAP-cre driver to generate Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre mice. The GSK-3 mutant brain exhibited gross morphological abnormalities. Compared to control littermate mice (Gsk3α +/+ ; Gsk3β +/+ ;GFAP-cre), Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre mice had larger brains (Fig. 1a). The brain weight was not different at P7. However, Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre brains weighed significantly more than controls at P35, P70, and P84 (Fig. 1b). The difference in brain/body weight ratio was also noticeable after the age of P35. Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre brains weighed 17, 46, and 58 % more than control brains at P35, P70, and P84, respectively (Fig. 1c). Western blotting showed that the GSK-3β level in GSK-3 mutant astrocytes was markedly reduced (Supplemental Fig. 1).

GSK-3 deletion results in a larger brain: a representative brains of Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre (wild type; left), Gsk3α +/− ; Gsk3β +/loxP ;GFAP-cre (heterozygous; middle), and Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre (homozygous; right) mice. Compared to wild-type control, Gsk3α −/− ;Gsk3β loxP/loxP ; GFAP-cre brains were noticeably larger at P84. Gsk3α +/− ;Gsk3β +/loxP ;GFAP-cre mice did not show different brain size to control mice. Scale bar = 2.5 mm. b Brain weight of wild-type control and Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice was measured at P7, P35, P70, and P84. c Quantification of the brain/body weight ratio. Weight of the brain per total body weight was measured and presented as relative changes versus control. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. Data shown are mean ± SEM. N = 5 mice for each condition. Statistical significance was determined by a two-tailed Student’s t test. *Indicates a significant difference when compared with WT controls at p < 0.05, **p < 0.01
To explore the basis of the enlarged brain, we examined if there is a change in the number of astrocytes in Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre brains. We assessed astrocyte numbers by using immunostaining of control and GSK-3 mutant brain tissues with an antibody to an astrocyte marker, GFAP. We observed no changes in astrocyte numbers in the cerebral cortex and the hippocampus of control and Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre mice at P7 (Fig. 2a, b). At P35, however, GSK-3 mutant brains showed 104 % increase in astrocyte number in the cerebral cortex compared with controls. The number of GFAP-positive cells was further increased by approximately 220 % in the cerebral cortex of GSK-3 mutants at P84. In the hippocampus, the increased number of astrocytes became obvious at P84 (Fig. 2a, b). Additionally, we assessed the numbers of oligodendrocytes and interneurons in control and Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre cerebral cortex. We found no changes in the total number of either olig2-positive oligodendrocytes or parvalbumin-positive neurons in the cerebral cortex and the hippocampus (Supplemental Fig. 2), suggesting that GFAP-cre-mediated GSK-3 deletion is specific in the astrocyte population. However, the densities of these cells were decreased by 60 and 44 %, respectively, indicating the increase in brain volume in GSK-3 mutant mice. Together, these results show that GSK-3 deletion in astrocytes increases brain size and astrocyte numbers in mice.

The number of astrocytes is increased in Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre brains. a Changes in astrocyte numbers in GSK-3 mutant brains. Sections of the cerebral cortex and dentate gyrus from wild-type control and Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice at P7, P35, and P84 were immunostained with an antibody to GFAP. Top panels: cerebral cortex samples. Bottom panels: hippocampus samples. GFAP-positive cells were increased in Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice compared to controls. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. Scale bar = 100 μm. b Quantification of GFAP-positive astrocytes. The numbers of GFAP-positive astrocytes to the DAPI-stained cells per field were counted. Data were presented as percent versus Wild-type controls. The number of astrocytes in the cerebral cortex was elevated in P35 and P84 in GSK-3 mutants, while the number was not different at P7. Astrocyte number was also significantly increased by 57 % around the dentate gyrus of P84 GSK-3 mutants (left). Histogram shows the number of GFAP-positive astrocytes per 0.04-mm2 area, one section or total sections at P84 (right). WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. N = 5 mice for each condition; cell counts = 6382 cells for control and 8207 cells for KO. Statistical significance was determined by a two-tailed Student’s t test. *p < 0.05, ***p < 0.001
Increased Proliferation of Astrocyte in the GSK-3 Mutant Brain
The increased number of astrocytes suggests changes in cell proliferation in GSK-3 mutant brains. To test this possibility, we assessed the number of proliferating cells around the hippocampus after BrdU injection in control and Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre mice. The number of BrdU-positive cells was increased by approximately 50 % in GSK-3 mutant mice at P35 compared to control mice, while there was no change at P7 (Fig. 3a, b). The relative change in BrdU-positive cells was further elevated in P84 control and GSK-3 mutant samples. We confirmed the effect of GSK-3 in astrocyte proliferation by examining astrocyte numbers in primary cortical cultures. We cultured cortical progenitors from E13.5 mice for 6 and 9 days with the treatment of a GSK-3 inhibitor and then immunostained with a GFAP antibody. The treatment with a GSK-3 inhibitor markedly increased GFAP-positive cells (Fig. 3c, d). In addition to mouse cells, we examined the GSK-3 effect on human astrocytes. T98G astroglioma cells were cultured and treated with a GSK-3 inhibitor. The treatment with a GSK-3 inhibitor showed a significant increase in the number of GFAP and BrdU-double-positive cells (Supplemental Fig. 3).

Loss of GSK-3 activity increases astrocyte proliferation in the developing brain. a BrdU labeling of brain tissues shows that astrocyte proliferation is promoted in the absence of GSK-3 in vivo. Control and GSK-3 mutant mice at P7, P35, and P84 were injected with BrdU. After 2 days, BrdU immunostaining was performed in the hippocampus from wild type and Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. Scale bar = 100 μm. b Quantification of a. Histogram shows that the number of BrdU-positive cells in GSK-3 mutant brains is increased at P35 or P84. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. N = 5 mice for each condition; cell counts = 1476 cells for control and 2604 cells for KO. Statistical significance was determined by a two-tailed Student’s t test. *p < 0.05. c Suppression of GSK-3 activity using a GSK-3 inhibitor increases astrocyte proliferation in vitro. Cortical cultures were made from E14.5 mouse brain in the absence (control) or presence of a GSK-3 inhibitor (10 or 50 nM). At DIV 6 or DIV 9, cultures were fixed and immunostained with a GFAP antibody. Scale bar = 50 μm. d Quantification of GFAP-positive astrocytes. The treatment of GSK-3 inhibitor increased astrocyte numbers by approximately 60 % at either concentration. Statistical significance was determined by one-way ANOVA with a Bonferroni correction test. *p < 0.05. e Double immunostaining using GFAP and BrdU antibodies shows the increase in newly generated astrocytes in the dentate gyrus from wild type and Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice at P84. Higher magnification images of white-boxed regions (middle panels) were shown in right panels. Scale bar = 40 μm. f Quantification of e. Histogram shows that the number of both GFAP and BrdU-positive cells in GSK-3 mutants was increased at P84. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. Statistical significance was determined by a two-tailed Student’s t test. *p < 0.05
Next, we investigated whether the increased number of GFAP-positive cells in mutant brains results from newly generated astrocytes or from an elevation of GFAP expression in other cells. We double-labeled brain tissues with antibodies to BrdU and GFAP antibodies to trace and identify newly generated cells. We found that the number of overall BrdU and GFAP-double-positive cells in mutant brains was increased by 78 % compared with controls, while GFAP-negative and BrdU-positive cells were not significantly different (Fig. 3e, f). Interestingly, most BrdU-positive cells in the mutant brain were GFAP positive, suggesting that the increase in GFAP-positive cells in GSK-3 mutant brain resulted from astrocyte proliferation.
The increase in astrocyte number is not associated with cell death because there was no change in the level of activated caspase-3 in mutant brain tissues (Fig. 4). Together, these data show that inhibition of GSK-3 activity leads to the increase in astrocyte proliferation in the brain.

The level of activated caspase-3 in GSK-3 mutant brains. a There is no change in the activation of caspase-3 in GSK-3 mutant brains. Western blotting with an antibody to activated caspase-3 was performed using cortical lysates from P7, P35, and P84 wild type and GSK-3 mutant mice. b Histograms show quantification of blots. The band intensity of each protein was normalized to the GAPDH level. Data were shown as relative changes versus Wild-type control. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. N = 3 independent experiments using three mice for each condition
GSK-3 Deletion Induces Astrocyte Hypertrophy
Changes in cell size can contribute to the larger brain in GSK-3 mutants. Thus, we examined the size of GFAP-labeled astrocytes in control and Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre cerebral cortex. GFAP-positive cells were enlarged in the cerebral cortex and the dentate gyrus of GSK-3 mutant mice (Fig. 5a). There was a 3.6-fold increase in the size of GSK-3 mutant astrocytes compared to control (Fig. 5b). These results suggest that elimination of GSK-3 using the GFAP-cre line leads to the enlargement of the brain via astrocyte hypertrophy as well as the increase in astrocyte numbers.

Altered size of astrocytes in GSK-3 mutant brains. a The area of GFAP-positive cell body is increased in GSK-3 mutant brains. GFAP immunostaining was performed on wild-type control (Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre) and GSK-3 mutant (Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre) brain sections at P84. Mutant brain samples showed larger GFAP-positive astrocytes than control cells. Scale bar = 5 μm. b Quantification of a. The area of cell body was increased by 264 % in GSK-3 mutant brains compared to wild-type samples. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. N = 5 mice for each condition; cell counts = 200 cells for control and 200 cells for KO. Statistical significance was determined by a two-tailed Student’s t test. ***p < 0.001
Signaling Molecules Associated with GSK-3-Mediated Regulation of Astrocyte
Studies have shown that STAT3 and AKT/mTOR signaling are associated with astrocyte proliferation and size determination [39]. Thus, we first examined the expression level of phospho-STAT3 in control and GSK-3 mutant brains. Western blots showed that the phosphorylation of STAT3 was not altered in P7 brain tissues of GSK-3 mutants compared to control (Fig. 6a). The level of astrocyte marker GFAP was also not changed at this age. No changes in the levels of phospho-STAT3 and GFAP appear to be consistent with no morphological alterations shown in Figs. 1, 2, and 3 at P7. However, the levels of phospho-STAT3 and GFAP were increased by 57 and 45 %, respectively, at P35 (Fig. 6a). At P84, we found more increases in their levels.

Altered mRNA and protein levels of astrocyte regulatory molecules in GSK-3 mutant brains. a GSK-3 deletion increases phospho-STAT and GFAP levels. Western blotting was performed using cortical lysates from P7, P35, and P84 wild type and GSK-3 mutant mice. Histograms show quantification of blots. The band intensity of each protein was normalized to GAPDH level. Data were shown as relative changes versus wild-type control. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. N = 3 independent experiments using three mice for each condition. b GSK-3 elimination activates the AKT/mTOR signaling pathway. Phosphorylation of AKT and S6 was examined by Western blotting using P84 wild type and GSK-3 mutant cortical lysates. c The level of cyclin D is increased in GSK-3 mutant brains. The mRNA level of cyclin D was assessed by using real-time PCR in several CNS tissues of P84 wild-type control and GSK-3 mutant mice. WT: Gsk3α +/+ Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. N = 3 independent experiments using three mice for each condition. Statistical significance was determined by a two-tailed Student’s t test.*p < 0.05, **p < 0.01, ***p < 0.001
Next, we examined if there are changes in expression and phosphorylation levels of AKT/mTOR signaling components associated with astrocyte proliferation in GSK-3 mutant brain. We measured the levels of phospho-AKT and phospho-S6 in control and GSK-3 mutant brains. Phosphorylation levels of AKT and S6 were significantly elevated by 34 and 120 %, respectively, in GSK-3 mutant brains at P84 (Fig. 6b). However, the level of PTEN was not changed. These results suggest that mTOR-mediated proliferation signaling is activated in GSK-3 mutant brains. Accordingly, when measured by real-time RT-PCR, the level of cyclin D mRNA in GSK-3 mutants was increased in several regions of the central nervous system including the cerebral cortex, the cerebellum, the olfactory bulb, and the ventral telencephalon at P84 age compared to control (Fig. 6c). Together, our data suggest that STAT3 and AKT/mTOR signaling pathways are associated with the GSK-3 regulation of astrocytes in the brain.
Abnormal Behavior in GSK-3 Mutant Mice
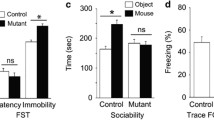
Global elimination of GSK-3α gene causes behavioral abnormalities [40–42]. We investigated if selective elimination of GSK-3α and GSK-3β in the astrocyte population leads to behavioral alteration. We first assessed anxiety in P70 control and Gsk3α −/− ; Gsk3β loxP/loxP ; GFAP-cre mice using the elevated plus-maze test. Control mice stayed most of their time in the closed arms. However, GSK-3 mutant mice spent markedly longer time in the open arms and were more likely to enter into open arms than control mice (Fig. 7a). Next, we assessed the social behavior of GSK-3 mutant mice including general sociability and social novelty using the three-chamber test. In a sociability test, control mice preferred a chamber containing an unfamiliar stranger 1 mouse to an empty chamber (Fig. 7b). Similarly, GSK-3 mutant mice stayed longer in the chamber with stranger 1 than in the empty chamber. In a social novelty test, stranger 2 mouse was newly introduced in the other chamber. Control mice preferred to enter into a chamber containing stranger 2 than a chamber containing a familiar stranger 1 (Fig. 7b). Importantly, GSK-3 mutant mice did not show statistical preference for stranger 2 mouse in favor of a familiarized stranger 1 in the social novelty test. Additionally, time spent sniffing a partner was markedly reduced in GSK-3 mutant mice (Fig. 7c). These results suggest that GSK-3 in astrocytes plays an important role in normal anxiety and social behavior.

GSK-3 mutant mice show altered anxiety and social behavior. a Anxiety behavior in GSK-3 mutant mice. Elevated plus-maze test was performed to assess anxiety-related behavior in GSK-3 mutant mice. The time spent in open arms and the number of entries into open arms were measured. GSK-3 mutant mice showed a reduction in anxiety-related behavior compared to wild-type control. b Social behavior in GSK-3 mutant mice. Social interaction behavior of control and GSK-3 mutant mice was assessed using the three-chamber social test. The time spent in each side chamber was measured. Compared to control mice, GSK-3 mutants stayed less time with a novel partner. c The time spent sniffing the wire cage in the chamber during the social test was recorded. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice. N = 3 independent experiments using three mice for each condition. Statistical significance was determined by a two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
GSK-3 is a critical regulator of neuronal development such as neurogenesis and neuronal differentiation in the brain [9, 10]. However, the function of GSK-3 in astrocyte development has been poorly understood. Furthermore, there is no in vivo evidence about selective loss of GSK-3 function in astrocytes. To study the function of GSK-3 in astrocyte development, we generated GSK-3α and GSK-3β double-knockout mice using a GFAP-cre driver, in which both GSK-3 genes are eliminated in the astrocyte population. Using in vivo BrdU proliferation assay and in vitro culture experiments, we found that elimination of GSK-3 increases proliferation of astrocytes in the brain. Our data suggest that overproliferation of astrocytes causes an enlarged brain in GSK-3 mutant mice. Our results are consistent with previous studies that have shown the role of GSK-3 in proliferation of multiple cell types including neural progenitors [11], hematopoietic stem cells [43], pancreatic mesenchymal stem cells [44], fibroblast [45], skin cancer cells [46], and mammary cancer cells [47]. Pharmacological inhibition or genetic elimination of GSK-3 promotes proliferation of these cells. Particularly, genetic inhibition of GSK-3 using small interfering RNA promotes proliferation of astrocyte lineage glioblastoma cells in culture [48]. Furthermore, overexpression of wild type or constitutively active GSK-3 inhibits the progression of skin cancer cells in mice [46]. Thus, downregulation of GSK-3 activity appears to positively contribute to the proliferation of cells. However, some studies have shown a contrary role of GSK-3. Inhibition of GSK-3 activity suppresses ovary cancer cells [49], colon cancer cells [50], and prostate cancer cells [51]. While GSK-3 plays opposing roles as a suppressor or a promoter of cell proliferation depending on cell types and cellular contexts, our current and previous studies [11] show an inhibitory function of GSK-3 in proliferation of neural cells such as astrocytes and neural progenitors.
Cellular hypertrophy increases in organ volume. The PI3K/AKT/mTOR signaling pathway plays a critical role in the determination of cell size and cell number in the mammalian brain [52, 26]. For example, there are macrocephaly and neural hypertrophy in PTEN knockout mice [53, 54]. TSC1, a negative regulator of mTOR, contributes to mTOR signaling downstream of PI3K/AKT [55, 52]. TSC1 knockout mice show astrocyte hypertrophy [56]. In contrast, conditional mTOR deletion using a nestin-cre line results in a smaller brain with hypotrophic neural cells [26]. Changes in AKT activity modify phosphorylation of mTOR, TSC2, S6, and STAT3 but not caspase-3 in human glioblastomas [57]. Our results show that GSK-3 deletion leads to astrocyte hypertrophy. GSK-3 could mediate PI3K/AKT/mTOR signaling in the determination of astrocyte size and number in the brain. Importantly, GSK-3 is a downstream mediator of PI3K/AKT signaling and regulates mTOR activity in neural cells [58, 59, 26, 60]. GSK-3 is negatively regulated by PI3K-mediated activation of AKT. GSK-3 directly phosphorylates TSC proteins and modulates mTOR activity in neural progenitors, fibroblast cells, and lung and renal cancer cells [61, 59, 26]. Furthermore, GSK-3 controls homeostasis of neural progenitors in the brain by PI3K/AKT signaling [11]. Consistently, our results have shown that GSK-3 regulates activity of AKT/mTOR signaling components. Phosphorylation of AKT, S6, and STAT3 is increased in GSK-3 mutant brain tissues. These results support the idea that GSK-3 interacts with PI3K/AKT/mTOR signaling in proliferation and size control of astrocytes.
GSK-3 mull mice display abnormal behavior [42]. For example, GSK-3α null mice show increased anxiety and decreased aggression and locomotion [42]. Similarly, GSK-3β heterozygous mice exhibit reduced exploratory behavior [62], increased anxiety [63], and reduced aggressiveness [64]. There is no known behavioral phenotype of GSK-3β homozygous null mice because these mice die during embryogenesis due to liver degeneration [65, 66]. We examined anxiety and social behavior of GSK-3 double mutants (Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre). Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mutant mice showed a reduction in anxiety-related behavior in the elevated plus-maze test. This result is similar to the finding of anxiety-like behavior of amyloid precursor protein mutant mice, in which astrogliosis is active [67, 68]. However, reduced anxiety in GSK-3 double mutants appears to be opposite to the behavior in GSK-3 single mutants. It remains to be elucidated whether the difference is caused by elimination of single versus double GSK-3 isoforms. Astrocyte-specific GSK-3 deletion could also lead to the different anxiety phenotypes. Additionally, we have found that Gsk3α −/− ;Gsk3β loxP/loxP ;GFAP-cre mice revealed a decreased social interaction with novel partners. GSK-3α null mice also exhibit weak interaction with a new mouse in both sociability and social novelty tests [42]. Our data suggest that by regulating astrocyte number and size, GSK-3 contributes to the transmission of the astrocytic input into cortical circuits that control social behavior in mice.
In summary, GSK-3 deficiency in astrocytes results in an enlarged brain and astrocyte overproliferation and hypertrophy. Altered AKT/mTOR signaling is associated with the abnormal phenotype. Finally, there are changes in anxiety and social interaction behaviors in GSK-3 mutant mice. Our study provides important insights into the function of GSK-3 in astrocyte development and behavior.
References
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7(1):41–53
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119(1):7–35
Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH (2012) Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 26(9):891–907
Kaneko N, Marin O, Koike M, Hirota Y, Uchiyama Y, Wu JY, Lu Q, Tessier-Lavigne M et al (2010) New neurons clear the path of astrocytic processes for their rapid migration in the adult brain. Neuron 67(2):213–223
Seifert G, Schilling K, Steinhauser C (2006) Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci 7(3):194–206
Barres BA (2008) The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60(3):430–440
Takano T, Oberheim N, Cotrina ML, Nedergaard M (2009) Astrocytes and ischemic injury. Stroke 40(3 Suppl):S8–S12
Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, Kaibuchi K, Woodgett JR et al (2006) Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron 52(6):981–996
Hur EM, Zhou FQ (2010) GSK3 signalling in neural development. Nat Rev Neurosci 11(8):539–551
Kim WY, Snider WD (2011) Functions of GSK-3 signaling in development of the nervous system. Front Mol Neurosci 4:44
Kim WY, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR, Snider WD (2009) GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci 12(11):1390–1397
Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K (2005) GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120(1):137–149
Jiang H, Guo W, Liang X, Rao Y (2005) Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120(1):123–135
Asada N, Sanada K (2010) LKB1-mediated spatial control of GSK3beta and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing neocortex. J Neurosci 30(26):8852–8865
Morgan-Smith M, Wu Y, Zhu X, Pringle J, Snider WD (2014) GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. eLife 3:e02663
Ka M, Jung EM, Mueller U, Kim WY (2014) MACF1 regulates the migration of pyramidal neurons via microtubule dynamics and GSK-3 signaling. Dev Biol 395(1):4–18
Rajan P, McKay RD (1998) Multiple routes to astrocytic differentiation in the CNS. J Neurosci 18(10):3620–3629
Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K et al (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci 28(28):7231–7243
Pastor MD, Garcia-Yebenes I, Fradejas N, Perez-Ortiz JM, Mora-Lee S, Tranque P, Moro MA, Pende M et al (2009) mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J Biol Chem 284(33):22067–22078
Emamian ES (2012) AKT/GSK3 signaling pathway and schizophrenia. Front Mol Neurosci 5:33
MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR (2007) Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab 6(4):329–337
Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR (2008) Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol 28(20):6314–6328
Kim WY, Horbinski C, Sigurdson W, Higgins D (2004) Proteasome inhibitors suppress formation of polyglutamine-induced nuclear inclusions in cultured postmitotic neurons. J Neurochem 91(5):1044–1056
Kim WY, Gonsiorek EA, Barnhart C, Davare MA, Engebose AJ, Lauridsen H, Bruun D, Lesiak A et al (2009) Statins decrease dendritic arborization in rat sympathetic neurons by blocking RhoA activation. J Neurochem 108(4):1057–1071
Kim WY, Fayazi Z, Bao X, Higgins D, Kazemi-Esfarjani P (2005) Evidence for sequestration of polyglutamine inclusions by Drosophila myeloid leukemia factor. Mol Cell Neurosci 29(4):536–544
Ka M, Condorelli G, Woodgett JR, Kim WY (2014) mTOR regulates brain morphogenesis by mediating GSK3 signaling. Development 141(21):4076–4086
Jessberger S, Romer B, Babu H, Kempermann G (2005) Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp Neurol 196(2):342–351
Farioli-Vecchioli S, Micheli L, Saraulli D, Ceccarelli M, Cannas S, Scardigli R, Leonardi L, Cina I et al (2012) Btg1 is required to maintain the pool of stem and progenitor cells of the dentate gyrus and subventricular zone. Front Neurosci 6:124
Kee N, Teixeira CM, Wang AH, Frankland PW (2007) Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat Neurosci 10(3):355–362
Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ (1999) Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2(3):260–265
Collins TJ (2007) ImageJ for microscopy. Biotechniques 43(1 Suppl):25–30
Lee JE, Patel K, Almodovar S, Tuder RM, Flores SC, Sehgal PB (2011) Dependence of Golgi apparatus integrity on nitric oxide in vascular cells: implications in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 300(4):H1141–H1158
Lee JE, Yang YM, Liang FX, Gough DJ, Levy DE, Sehgal PB (2012) Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol 302(5):C804–C820
Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, Barbaro JR, Wilson LM et al (2007) Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res 176(1):4–20
Sayah S, Ischenko AM, Zhakhov A, Bonnard AS, Fontaine M (1999) Expression of cytokines by human astrocytomas following stimulation by C3a and C5a anaphylatoxins: specific increase in interleukin-6 mRNA expression. J Neurochem 72(6):2426–2436
Yue G, Shi G, Azaro MA, Yang Q, Hu G, Luo M, Yin K, Nagele RG et al (2008) Lipopolysaccharide (LPS) potentiates hydrogen peroxide toxicity in T98G astrocytoma cells by suppression of anti-oxidative and growth factor gene expression. BMC Genomics 9:608
Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, Verveer M, de Groot LR et al (2012) GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One 7(8):e42823
Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV (2004) GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci 7(11):1233–1241
Fumarola C, La Monica S, Alfieri RR, Borra E, Guidotti GG (2005) Cell size reduction induced by inhibition of the mTOR/S6K-signaling pathway protects Jurkat cells from apoptosis. Cell Death Differ 12(10):1344–1357
Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS (2010) GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One 5(3):e9706
Latapy C, Rioux V, Guitton MJ, Beaulieu JM (2012) Selective deletion of forebrain glycogen synthase kinase 3beta reveals a central role in serotonin-sensitive anxiety and social behaviour. Philos Trans R Soc Lond B Biol Sci 367(1601):2460–2474
Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberte C, Khan M, Okamoto K et al (2009) Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain 2:35
Huang J, Zhang Y, Bersenev A, O’Brien WT, Tong W, Emerson SG, Klein PS (2009) Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J Clin Invest 119(12):3519–3529
Cao Q, Mahalanabis M, Chang J, Carey B, Hsieh C, Stanley A, Odell CA, Mitchell P et al (2012) Microfluidic chip for molecular amplification of influenza A RNA in human respiratory specimens. PLoS One 7(3):e33176
Kapoor M, Liu S, Shi-wen X, Huh K, McCann M, Denton CP, Woodgett JR, Abraham DJ et al (2008) GSK-3beta in mouse fibroblasts controls wound healing and fibrosis through an endothelin-1-dependent mechanism. J Clin Invest 118(10):3279–3290
Ma C, Wang J, Gao Y, Gao TW, Chen G, Bower KA, Odetallah M, Ding M et al (2007) The role of glycogen synthase kinase 3beta in the transformation of epidermal cells. Cancer Res 67(16):7756–7764
Farago M, Dominguez I, Landesman-Bollag E, Xu X, Rosner A, Cardiff RD, Seldin DC (2005) Kinase-inactive glycogen synthase kinase 3beta promotes Wnt signaling and mammary tumorigenesis. Cancer Res 65(13):5792–5801
Li J, Zhu Z (2010) Research and development of next generation of antibody-based therapeutics. Acta Pharmacol Sin 31(9):1198–1207
Cao M, Nicola E, Portela VM, Price CA (2006) Regulation of serine protease inhibitor-E2 and plasminogen activator expression and secretion by follicle stimulating hormone and growth factors in non-luteinizing bovine granulosa cells in vitro. Matrix Biol 25(6):342–354
Shakoori A, Mai W, Miyashita K, Yasumoto K, Takahashi Y, Ooi A, Kawakami K, Minamoto T (2007) Inhibition of GSK-3 beta activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci 98(9):1388–1393
Zhu Q, Yang J, Han S, Liu J, Holzbeierlein J, Thrasher JB, Li B (2011) Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate 71(8):835–845
Chen J, Alberts I, Li X (2014) Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. Int J Dev Neurosci 35:35–41
Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ et al (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50(3):377–388
Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ (2004) Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res 64(21):7773–7779
Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4(9):648–657
Uhlmann EJ, Li W, Scheidenhelm DK, Gau CL, Tamanoi F, Gutmann DH (2004) Loss of tuberous sclerosis complex 1 (Tsc1) expression results in increased Rheb/S6K pathway signaling important for astrocyte cell size regulation. Glia 47(2):180–188
Riemenschneider MJ, Betensky RA, Pasedag SM, Louis DN (2006) AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res 66(11):5618–5623
Buller CL, Loberg RD, Fan MH, Zhu Q, Park JL, Vesely E, Inoki K, Guan KL et al (2008) A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol 295(3):C836–C843
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C et al (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126(5):955–968
Kitagishi Y, Kobayashi M, Kikuta K, Matsuda S (2012) Roles of PI3K/AKT/GSK3/mTOR Pathway in Cell Signaling of Mental Illnesses. Depress Res Treat 2012:752563
Mak BC, Kenerson HL, Aicher LD, Barnes EA, Yeung RS (2005) Aberrant beta-catenin signaling in tuberous sclerosis. Am J Pathol 167(1):107–116
O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, Klein PS (2004) Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci 24(30):6791–6798
Bersudsky Y, Shaldubina A, Kozlovsky N, Woodgett JR, Agam G, Belmaker RH (2008) Glycogen synthase kinase-3beta heterozygote knockout mice as a model of findings in postmortem schizophrenia brain or as a model of behaviors mimicking lithium action: negative results. Behav Pharmacol 19(3):217–224
Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC, Gainetdinov RR, Caron MG (2008) Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A 105(4):1333–1338
Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K et al (2008) Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 118(11):3609–3618
Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR (2000) Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406(6791):86–90
Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, Clark IA, Abdipranoto A et al (2013) Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One 8(4):e59586
Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D et al (2000) High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20(11):4050–4058
Acknowledgments
We are thankful to Drs. Robert Norgren, Anna Dunaevsky, and Shelley Smith for valuable advice and comments on the manuscript. Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103471, a grant from NE DHHS (Stem Cell 2012-05), and a grant from Alzheimer’s Association (NIRP-12-258440) to WYK.
Author Contributions
E.J. and W.K. designed, performed, and analyzed the experiments. M.K. performed the experiments. E.J. and W.K. wrote the paper. W.K. supervised the study.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplemental Figure 1
The level of GSK-3β in GSK-3 mutant brains (A) GSK-3β level in GSK-3 mutant mice. Western blotting was performed using astrocyte lysates from P84 wild type and GSK-3 mutant mice. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α -/- ;Gsk3β loxP/loxP ;GFAP-cre mice (PDF 39 kb).
Supplemental Figure 2
The numbers of oligodendrocytes and neurons in Gsk3α -/- ; Gsk3β loxP/loxP ;GFAP-cre brains (A) Cerebral cortex samples from wild type control and Gsk3α -/- ;Gsk3β loxP/loxP ;GFAP-cre mice at P84s were immunostained with an antibody to Olig2. WT: Gsk3α +/+ ;Gsk3β +/+ ;GFAP-cre. KO: Gsk3α -/- ;Gsk3β loxP/loxP ;GFAP-cre mice. Scale bar = 100 μm. (B) Quantification of (A). The number of Olig2-positive cells was not significantly different between control and Gsk3α -/- ;Gsk3β loxP/loxP ;GFAP-cre mice (left). However, the density was decreased in mutant brains (right). Cells in the white boxes (layer I-VI) in (A) were counted. (C) The number of parvalbumin-positive neurons was examined as described in (A). (D) Quantification of (C). N = 5 mice for each condition; cell counts = 1037 cells for control and 1035 cells for KO (PDF 232 kb).
Supplemental Figure 3
Suppression of GSK-3 activity increases proliferation of human astroglial cells. (A) T98G astroglial cells were cultured in the absence (control) or presence of a GSK-3 inhibitor (10 nM or 50 nM). The cells were then incubated with 10 μM BrdU for 6 hours, fixed and immunostained with GFAP and BrdU antibodies. Scale bar = 50 μm. (B) Quantification of BrdU-positive cells. The treatment with a GSK-3 inhibitor increased BrdU-positive cells by 111 % and 154 % at 10 nM or 50 nM concentration, respectively. Statistical significance was determined by one-way ANOVA with Bonferonni correction test. * p < 0.05, ** p < 0.01 (PDF 179 kb).
Rights and permissions
About this article

Cite this article
Jung, EM., Ka, M. & Kim, WY. Loss of GSK-3 Causes Abnormal Astrogenesis and Behavior in Mice. Mol Neurobiol 53, 3954–3966 (2016). https://doi.org/10.1007/s12035-015-9326-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9326-8