
Abstract
Cerebral autoregulation is defined as the mechanism by which constant cerebral blood flow is maintained despite changes of arterial blood pressure, and arterial blood pressure represents the principle aspect of cerebral autoregulation. The impairment of cerebral autoregulation is reported to be involved in several diseases. However, the concept, mechanisms, and pathological dysfunction of cerebral autoregulation are beyond full comprehension. Nitric oxide control and sympathetic control are main contributors to cerebral autoregulation. Although impaired cerebral autoregulation after nitric oxide inhibition or sympathetic ganglia blockade is reported, managing the inhibition or blockade can have negative consequences and needs further exploration. Additionally, impaired cerebral autoregulation following subarachnoid hemorrhage and traumatic brain injury has been proven by several descriptive studies, although without corresponding explanations. As the most important mechanisms of cerebral autoregulation, the changes of nitric oxide and sympathetic stimulation play significant roles in these insults. Therefore, the in-depth researches of nitric oxide and sympathetic nerve in cerebral autoregulation may help to develop new therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebral autoregulation refers to an important physiological control of cerebrovascular function. The impairment of cerebral autoregulation has been reported to be involved in several diseases, such as cerebral stroke [1], Alzheimer’s disease [2], congenital patent foramen ovale [3], etc. However, the concept, mechanism, or pathological alteration of cerebral autoregulation is far beyond comprehension. This review aims to discuss the precise concept of cerebral autoregulation, with elaboration of its regulation by nitric oxide and sympathetic nerves in both basic research and clinical conditions, such as subarachnoid hemorrhage and traumatic brain injury.
Cerebral Autoregulation: Precise Concept
Before a more in-depth elaboration, the concept of cerebral autoregulation needs to be clarified. Cerebral autoregulation is defined as the mechanism by which constant cerebral blood flow is maintained despite changes of arterial blood pressure [4–6], and arterial blood pressure represents the principle aspect of cerebral autoregulation. Cerebral autoregulation can be evaluated by measuring relative blood flow changes in response to a steady state in the blood pressure (static cerebral autoregulation) or to a rapid change in blood pressure (dynamic cerebral autoregulation) [7]. The related physiological parameters involved in cerebral autoregulation include sympathetic control, cerebral metabolism [8, 9], myogenic responses [8, 10, 11], and endothelium function, which are still largely unknown.
In previous studies, cerebral autoregulation may be confused with other physiological mechanisms that act simultaneously to regulate cerebral blood flow, as cerebrovascular reactivity and neurovascular coupling. Generally, they are independent definitions with relevant principles in regulation of cerebral blood flow. Cerebrovascular reactivity is recognized as the compensatory constriction or dilation of distal cerebral arteries as a result of vasoactive stimuli [12]. Briefly, cerebrovascular reactivity is associated with carbon dioxide, acetazolamide, and other vasoactive substances. In human subjects, it has been reported that results of cerebral autoregulation are not consistent with those of cerebrovascular reactivity, suggesting different mechanisms involved between the two phenomena [12, 13]. Another concept that may be confused with cerebral autoregulation is neurovascular coupling. Neurovascular coupling consists of three cell types in the brain: neurons, supporting cells (astrocytes), and vascular cells (vascular smooth muscle, pericyte, and endothelial cells). All of which are tightly associated to modulate regional blood flow in response to local metabolic consumption. This modulation ensures rapid spatial and temporal increase of local cerebral blood flow in response to neuronal activation, despite relatively constant global blood flow [14]. According to the findings of previous studies, these three mechanisms may interact with each other to produce their function via autonomic pathway, calcium channel activity, etc. [14–20]. Research regarding the interactions of the above mechanisms during past 10 years is listed in Table 1.
Clinically, the arterial blood pressure is volatile in patients with acute neurological diseases, such as cerebral infarction, subarachnoid hemorrhage, and traumatic brain injury. If the cerebral autoregulation is impaired, the arterial blood pressure variation may significantly affect cerebral blood flow, even to the point of causing secondary brain injury, and thereby contributing to the poor prognosis. This suggests that monitoring of the cerebral autoregulation is of great importance for the management of arterial blood pressure to guide normalization, improving the treatment and prognosis for patients with these acute neurological diseases.
Mechanisms
There are four main mechanisms involved in the regulation of cerebral autoregulation: sympathetic control, cerebral metabolism, myogenic response, and endothelium function (represented by nitric oxide). Sympathetic control and nitric oxide concentrations, rather than cerebral metabolism and myogenic responses, can be detected and regulated in the human body using existing technologies. Concerning these two mechanisms, contradictory conclusions have been reported. This review aims to investigate, summarize, and clarify the role of nitric oxide and sympathetic control in cerebral autoregulation.
The Role of Nitric Oxide
Theoretical Basis
Nitric oxide is considered one of the main endothelium-derived vasodilation factors, which exert a potential role in the regulation of cerebral autoregulation [21–23]. Theoretically, nitric oxide regulates the vascular tone of small arteries. Nitric oxide diffuses into the adjacent smooth muscle cells and relaxes them by increasing cyclic guanosine monophosphate [24]. Nitric oxide synthase is widely expressed in various cell types in the brain other than vascular endothelial cells, such as neurons, glia cells, and the perivascular fibers that innervate cerebral vessels. This suggests that nitric oxide can mediate local vasodilation in response to the activation of non-endothelial cells as well. Some studies indicate that the secretion of nitric oxide in the brain increases in response to a sudden drop in arterial blood pressure; this leads to the dilation of the small cerebral arteries and thereby provides adequate blood supply to the brain [24].
Controversial Results and Possible Reconciliation
It is understood that nitric oxide plays a primary role in the regulation of vascular tone [24]. Consequently, cerebral autoregulation is likely to be impaired following inhibition of nitric oxide synthase. However, the results from previous studies about the effects of nitric oxide on cerebral autoregulation in animal models are conflicting. Some authors reported impaired cerebral autoregulation following nitric oxide inhibition [22, 23], while others reported no change [25]. With regard to the effect of nitric oxide on cerebral autoregulation in human, the information is limited. White et al. studied the role of nitric oxide on cerebral autoregulation in healthy subjects using the nitric oxide synthase inhibitor NG-monomethyl-l-arginine. When using a control group with an equally titrated, increased level of blood noradrenaline, they found that the autoregulatory index was significantly lower in nitric oxide synthase inhibitor group than that in the noradrenaline titrated control. This study indicated that reduced nitric oxide secretion was associated with impaired cerebral autoregulation, suggesting that nitric oxide has a role in the native regulation of cerebral autoregulation in humans [21]. In contrast, Zhang et al. revealed that both arterial blood pressure and cerebral blood flow velocity variability did not alter after using NG-monomethyl-l-arginine to block nitric oxide synthase, which suggested that inhibition of nitric oxide production had no discernable effect on cerebral autoregulation in humans [26].
The conclusions from the referenced studies differ, which can be attributed to the following reasons. First of all, cerebral autoregulation was detected under different circumstances. White et al. used a thigh tourniquet release maneuver to manipulate arterial blood pressure [21], whereas Zhang et al. conducted a head-up tilt [26]. The two methods likely cause different degrees of change in cerebral arterial blood pressure. Secondly, there were less than 20 subjects enrolled in each study; the sample sizes were relatively small to reach consistent statistical results. Last but not least, the effect of transmural pressure intervened with the autoregulatory response of nitric oxide on cerebral arterioles. Kajita et al. reported that the vasogenic secretion of nitric oxide was significantly increased when transmural pressure decreased below 60 mmHg, thereby promoting autoregulatory vasodilation. If transmural pressure was elevated higher than 60 mmHg, the effect of nitric oxide became limited [24].
Sympathetic Control of Cerebral Autoregulation
Theoretical Basis, Controversial Results, and Possible Reconciliation
One of the important roles of sympathetic nervous system is to control the vasomotor function. Theoretically, sympathetic nervous stimulation causes the vasoconstriction of blood vessels as a result of activation of alpha-1 adrenergic receptors by norepinephrine released by post-ganglionic sympathetic neurons. These changes may affect the function of blood vessels. The cerebrovascular bed is widely innervated by sympathetic nerve fibers [27, 28]. Indeed, current theory holds that sympathetic nerves regulate cerebral blood flow by managing cerebral vascular resistance. However, the role of the sympathetic nervous system in cerebral autoregulation remains poorly characterized, since research results were inconsistent in both human and animal models. In 1961, Sagawa and Guyton found that the resection of carotid sinus nerves completely abolished the effect of cerebral autoregulation in dog models, suggesting that sympathetic pathways were involved in cerebral autoregulation [29]. However Eklöf et al. reported that cerebral autoregulation was preserved in monkeys with chronic sympathetic denervation [30], which suggested the opposite. Existing research in human subjects also reaches a debatable conclusion. Using trimethaphan to block the autonomic ganglionic pathways in 11 healthy subjects, Zhang et al. found that sympathetic vasoconstriction was not the primary mechanism in regulation of cerebral blood flow [31]. Gierthmühlen et al. enrolled 17 patients with infarction of the dorsolateral medulla oblongata, which affects central sympathetic pathways. They demonstrated that the sympathetic nervous system did not impact cerebral autoregulation if perfusion pressure decreased under normotonous conditions [32]. In contrast, Zhang et al., in another study, reported a significant change of cerebral autoregulation after ganglion blockade with trimethaphan [33]. Similarly, the study of Hamner indicated that sympathetic regulation for cerebral and brachial blood flow was dependent on frequency in pressure oscillations [28].
The role of sympathetic nerves in cerebral autoregulation remains controversial for the following reasons: (1) cerebral autoregulation includes static cerebral autoregulation and dynamic cerebral autoregulation. Dynamic cerebral autoregulation is calculated from a mathematical model, which is more sensitive and able to distinguish the subtle differences among experimental groups [28, 31, 33]. However, these two indexes were arbitrarily adopted in previous studies, which contributed to the inconsistency. (2) Compensatory and regenerative mechanisms are confounding factors in sympathetic control. For example, chronic sympathetic denervation [30] and chronic central sympathetic deficit [32] barely impacted cerebral autoregulation, whereas acute carotid sinus nerve removal resulted in total loss of cerebral autoregulation [29]. (3) The distribution of sympathetic innervations is heterogeneous. It is suggested that only in certain areas of the brain, sympathetic control has the priority in regulating blood flow [34]. (4) The sample sizes of their work are relatively too small to reach consistent statistic results.
Unresolved Issues in Sympathetic Control
Though there is limited proof that sympathetic activation causes direct change in cerebral autoregulation, it certainly causes indirect changes. Activated sympathetic neural pathways can enhance the production of renin, which results in downstream elevation of angiotensin II concentration [35]. Angiotensin II is a peptide hormone that causes vasoconstriction; it can increase the lower threshold of cerebral autoregulation, potentially shifting the lower limit of cerebral autoregulation toward higher blood pressure levels. Additionally, the concentrations of adrenaline and noradrenaline are increased after excessive sympathetic activation, which also induces vasoconstriction. One study of anxiety, a disease involving sympathetic activation, reported that sympathetic activation limited hypercapnic cerebral vasodilation [36]. Interestingly, another recent study revealed that the variation of cerebral blood flow velocity when changing position from supine to upright differed between the anxious and the healthy groups [37]. Anxious subjects exhibited pronounced reduction in cerebral blood flow velocity after abrupt standing, suggesting a deficit in alleviating low cerebral blood flow velocity by vasodilation [37]. Though these studies cannot come to any specific conclusion, they indicate that sympathetic activation may damage normal cerebral autoregulation. Further research is needed to investigate the relationship between sympathetic activation and cerebral autoregulation.
Cerebral Autoregulation in Subarachnoid Hemorrhage and Traumatic Brain Injury
The impairment of cerebral autoregulation has been reported in certain cerebral diseases such as subarachnoid hemorrhage and traumatic brain injury. Even though the mechanisms are unclear as mentioned above, sympathetic control and nitric oxide control are two of the main pathways in the regulation of cerebral autoregulation. Specific relationships between these pathways in subarachnoid hemorrhage and traumatic brain injury need to be investigated for designing new therapeutic methods. The following section aims to clarify the sympathetic and nitric oxide control of cerebral autoregulation in these specific diseases using both direct and indirect evidence.
Cerebral Autoregulation and Subarachnoid Hemorrhage
Overview
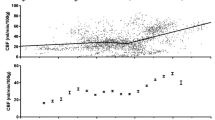
Subarachnoid hemorrhage is a subtype of stroke with a high mortality of nearly 50 % with one in eight patients dying outside of the hospital [38]. Delayed cerebral vasospasm and delayed cerebral ischemia are coupled to one another and are among the crucial causes of morbidity and mortality following subarachnoid hemorrhage [39]. It has been reported that cerebral autoregulation is associated with cerebral vasospasm/delayed cerebral ischemia (Fig. 1) [39, 40]. Budohoski et al. studied cerebral autoregulation in 98 consecutive patients with aneurysmal subarachnoid hemorrhage. They found that impaired cerebral autoregulation within the first 5 days after subarachnoid hemorrhage was associated with increased risk of delayed cerebral ischemia [40]. Otite et al. reported that patients with delayed cerebral ischemia had developed compromised cerebral autoregulation compared with those without complications [39]. Moreover, both the studies of Lang and Jaeger reached similar conclusions [41, 42]. In general, these studies indicate that comprehensive understanding of cerebral autoregulation can aid in identifying patients with high risk of secondary complications following subarachnoid hemorrhage.

Simulation diagram of dynamic cerebral autoregulation parameters in subarachnoid hemorrhage and traumatic brain injury. a The phase difference between arterial blood pressure and cerebral blood flow velocity in patient with subarachnoid hemorrhage is significantly lower than in the healthy subject, which indicates the compromised dynamic cerebral autoregulation. b Flatter slope of step response is observed in patient with traumatic brain injury, which demonstrates an impaired dynamic cerebral autoregulation. Phase difference and step response are two autoregulatory parameters derived from transfer function analysis. Phase difference is a frequency domain parameter. A large phase difference may suggest that cerebral blood flow velocity does not follow the changes of arterial blood pressure whereas cerebral blood flow velocity follows the change of arterial blood pressure passively when it changes in phase. Step response shows the response of cerebral blood flow velocity to a step change of arterial blood pressure. The quicker the step response drops back to the baseline level, the better autoregulation it indicates. SAH subarachnoid hemorrhage, TBI traumatic brain injury
Mechanisms
Nitric Oxide Control
Since relevant experiment research is extremely limited, the mechanism between cerebral autoregulation and cerebral vasospasm/delayed cerebral ischemia in subarachnoid hemorrhage is not fully understood. However, related research can offer some information. Sabri et al. found that the uncoupled endothelial nitric oxide synthase is upregulated in large cerebral arteries after subarachnoid hemorrhage, which led to decreased nitric oxide availability and increased superoxide and peroxynitrite concentrations [43]. Reduced availability of nitric oxide in blood and cerebrospinal fluid is one of the important mechanisms underlying cerebral vasospasm [44]. In addition, hemoglobin released from subarachnoid blood following aneurysmal rupture inhibits the activity of endothelial nitric oxide synthase, thereby reducing nitric oxide production which decreases nitric oxide availability for smooth muscle cells leading to vasoconstriction [44, 45]. In a later study, Sabri et al. discovered that recoupling endothelial nitric oxide synthase with simvastatin could reduce secondary complications such as vasospasm, microthromboemboli, and neuronal injury [46]. These studies indicated that the reduction in nitric oxide is crucial to induce cerebral vasospasm/delayed cerebral ischemia after subarachnoid hemorrhage. As previously described, nitric oxide plays a role in cerebral autoregulation, and the likely role is that nitric oxide deficiency disables proper cerebral autoregulation, leading to vasospasm/delayed cerebral ischemia and other related complications (Fig. 2).

Nitric oxide and sympathetic controls of cerebral autoregulation in subarachnoid hemorrhage. eNOS endothelial nitric oxide synthase, NO nitric oxide, CBF cerebral blood flow, CA cerebral autoregulation
Based on the conclusions above, some studies investigated the change in cerebral vasospasm/delayed cerebral ischemia by increasing the production of nitric oxide or activating nitric oxide synthase. Osuka et al. found that adiponectin, which plays an important role against cerebral vasospasm via the AMPK/endothelial nitric oxide synthase signaling pathway, was significantly elevated in the cerebrospinal fluid after subarachnoid hemorrhage [47]. The study of Vellimana indicated that hypoxic preconditioning protected against vasospasm and neurological deficits by increasing both nitric oxide availability and endothelial nitric oxide synthase activity [48]. Based on its effect against vasoconstriction, Kim et al. delivered ultrasound-facilitated nitric oxide to treat vasospasm following subarachnoid hemorrhage; improved neurologic function was observed in their treated animals [44]. Moreover, estrogen, specifically 17β estradiol (E2), has potential therapeutic implications for ameliorating the delayed neurological deterioration following aneurysmal subarachnoid hemorrhage via activating endothelial nitric oxide synthase [49]. These studies all together indicate the connection between nitric oxide and vasospasm after subarachnoid hemorrhage.
To sum up, nitric oxide, potentially by improving cerebral autoregulation, can attenuate cerebral vasospasm/delayed cerebral ischemia after subarachnoid hemorrhage. It can be implied that normalized or increased concentrations of nitric oxide contribute to a better prognosis in subarachnoid hemorrhage via alleviating cerebral autoregulation impairment.
Sympathetic Control
The acute stage following subarachnoid hemorrhage is accompanied by pronounced sympathetic activation, which contributes to the development of cerebral vasospasm [50, 51]. However, there is no study about the role of sympathetic nerves in cerebral autoregulation after subarachnoid hemorrhage. As previously described, excessive sympathetic activation is associated with cerebral autoregulation dysfunction. Thus, sympathetic activation after subarachnoid hemorrhage may disturb cerebral autoregulation and thereby induce cerebral vasospasm and delayed cerebral ischemia (Fig. 2).
Cerebral Autoregulation and Traumatic Brain Injury
Overview
Clinically, 49–87 % of patients with severe traumatic brain injury have an absence or impairment of cerebral autoregulation [52]. It is reported that even a minor head injury may impair cerebral autoregulation and increase the risk of secondary ischemic neuronal damage [53]. More recently, there has been investigation of the correlation between traumatic brain injury and cerebral autoregulation impairment [54, 55]. In a series of 36 severe traumatic brain injury patients, 30 of them were reported to have impaired cerebral autoregulation 3–5 days after injury by Sviri et al. (Fig. 1). Though recovered some, there were still 19 (53 %) and 9 (25 %) individuals with cerebral autoregulation deficit 9–11 and 12–14 days after injury, respectively. In their study, it was indicated that cerebral autoregulation recovery after severe traumatic brain injury was delayed, in particular cases more than 2 weeks [55]. Interestingly, similar results were demonstrated in professional boxers, a type of chronic traumatic brain injury. These results help explain why chronic traumatic brain injury is a progressive disease with the worsening cerebral autoregulation impairment [56]. The loss of cerebral autoregulation in the cerebral vasculature can lead to both the hypoperfusion that contributes to cerebral ischemia and the hyperemia that results in increased intracranial pressure [52, 57]. Both potentially induce secondary injury after traumatic brain injury.
Mechanisms
Nitric Oxide Control
As previously indicated, the role of nitric oxide in these pathological conditions remains unclear. Theoretically, both overexpression and deficiency have harmful effects. Attenuated production of nitric oxide by endothelial nitric oxide synthase results in inadequate cerebral perfusion, whereas excessive production by neuronal nitric oxide synthase and inducible nitric oxide synthase leads to neurotoxicity and cellular injury [52]. The production of nitric oxide in traumatic brain injury changes along with the pathological process. In the first 5–30 min after traumatic brain injury, nitric oxide production was increased in injured brain tissue, probably via the activation of endothelial nitric oxide synthase and neuronal nitric oxide synthase [58–61]. From 30 min to 6 h after traumatic brain injury, the concentration of nitric oxide was reduced, probably as a consequence of the diminished activity of endothelial nitric oxide synthase [61]. Nitric oxide expression started to recover after 6 h following traumatic brain injury, further elevating to its peak between 20 and 42 h after traumatic brain injury [62, 63], which was attributed to the activity variation of inducible nitric oxide synthase [61] (Fig. 3). Pre-injury administration of the neuronal nitric oxide synthase inhibitor 3-bromo-7-nitroindazole has been shown to have protective effects since it decreases nitric oxide concentration during the first 5–30 min after traumatic brain injury [64]. During the following 6 h, the lower concentration of nitric oxide is associated with low cerebral blood flow. Accordingly, administration of l-arginine at this time has been shown to improve cerebral blood flow and neurological outcome in traumatic brain injury models [61, 65]. Afterward, inhibition of inducible nitric oxide synthase reduces the expression of nitric oxide which has been shown to exert neuroprotective effects in most experimental traumatic brain injury models [66, 67].

Nitric oxide and sympathetic controls of cerebral autoregulation in traumatic brain injury. eNOS endothelial nitric oxide synthase, nNOS neuronal nitric oxide synthase, iNOS inducible nitric oxide synthase, CA cerebral autoregulation
As previously described, studies have shown that nitric oxide concentration is crucial for regulating cerebral autoregulation [21, 22]. However, previous studies only investigated the impact of lower nitric oxide concentration on cerebral autoregulation by using nitric oxide inhibitors. As reported before, the concentration of nitric oxide declined at 30 min to 6 h after traumatic brain injury, which may disturb cerebral autoregulation [21, 22]. Some studies revealed that inhaled nitric oxide reduces secondary brain damage after traumatic brain injury [68–70], which may be associated with the improvement of cerebral autoregulation. However, there is limited information about cerebral autoregulation under high nitric oxide concentration. Thus, no direct evidence is available to indicate the effect of nitric oxide on cerebral autoregulation in the first 5–30 min or 6 h later after traumatic brain injury.
Sympathetic Control
Increased intracranial pressure and activated sympathetic pathways following severe traumatic brain injury lead to elevated plasma catecholamine levels [71] which last for several weeks to months [72, 73]. Blocking of the hyperadrenergic response using beta-blockers has been demonstrated to be associated with significant improvements in mortality, especially in older and seriously injured patients [74]. As previously described, there is some indirect evidence showing disabled cerebral autoregulation from excessive sympathetic activation. Chronic sympathetic activation further interferes with cerebral autoregulation and thereby may induce secondary brain damage. However, further research about sympathetic control on cerebral autoregulation after traumatic brain injury is needed to verify this hypothesis.
Notably, during the 5–30 min and >6 h after traumatic brain injury, the high concentration of nitric oxide induces vasodilation, while excessive sympathetic activation has the opposite effect. Participation of these and other confounding mechanisms make the change in cerebral autoregulation during these two phases uncertain (Fig. 3).
Questions and Prospects
With increasing relevant research being accomplished, cerebral autoregulation is becoming the intriguing topic in regulation of cerebral blood flow. Nevertheless, some issues remain for further study. First of all, there is no standard method for the detection or measurement of cerebral autoregulation. Results from different methods are not comparable, which makes it difficult to draw a precise conclusion. Secondly, present research is not comprehensive. For example, there are few studies of cerebral autoregulation combining high concentration of nitric oxide and excessive sympathetic activation, which is required for complete understanding of underlying mechanisms. Thirdly, in particular diseases such as subarachnoid hemorrhage and traumatic brain injury, studies of cerebral autoregulation are descriptive with phenomenon reported only. In future, it is necessary to use relevant animal models to reproduce the results observed in humans and to study the mechanisms. Fourthly, cerebral autoregulation, cerebrovascular reactivity, and neurovascular coupling are recognized as components of a whole system in cerebral blood flow regulation. As mentioned above, there are inevitable interactions among them, which remain unclear. These particulars would be advantageous, especially for understanding the pathology of certain cerebral vascular diseases. Last but not least, according to the results from existing studies, the improvement of cerebral autoregulation is associated with alleviated brain injury and reduced incidence of complications; however, there are few drugs that can improve cerebral autoregulation so far. Targeting cerebral autoregulation is a prospective treatment for cerebral vascular diseases.
Conclusion
Intact cerebral autoregulation is crucial for appropriate cerebral blood flow. Cerebral autoregulation impairment is associated with many diseases and unsatisfactory outcomes. However, the mechanisms of cerebral autoregulation regulation are currently beyond full comprehension. Nitric oxide control and sympathetic control are two major underlying factors involved in regulating cerebral autoregulation in physiological and pathological conditions. Altering nitric oxide concentration or sympathetic activity may serve as new therapeutic targets in regulation of cerebral blood flow.
References
Immink RV, van Montfrans GA, Stam J, Karemaker JM, Diamant M, van Lieshout JJ (2005) Dynamic cerebral autoregulation in acute lacunar and middle cerebral artery territory ischemic stroke. Stroke 36(12):2595–2600
Claassen JA, Zhang R (2011) Cerebral autoregulation in Alzheimer’s disease. J Cereb Blood Flow Metab 31(7):1572–1577. doi:10.1038/jcbfm.2011.69
Guo ZN, Xing Y, Liu J, Wang S, Yan S, Jin H, Yang Y (2014) Compromised dynamic cerebral autoregulation in patients with a right-to-left shunt: a potential mechanism of migraine and cryptogenic stroke. Plos One 9(8):e104849. doi:10.1371/journal.pone.0104849
Bellapart J, Fraser JF (2009) Transcranial Doppler assessment of cerebral autoregulation. Ultrasound Med Biol 35(6):883–893. doi:10.1016/j.ultrasmedbio.2009.01.005
Murkin JM (2007) Cerebral autoregulation: the role of CO2 in metabolic homeostasis. Semin Cardiothorac Vasc Anesth 11(4):269–273. doi:10.1177/1089253207311159
Hamner JW, Tan CO, Tzeng YC, Taylor JA (2012) Cholinergic control of the cerebral vasculature in humans. J Physiol Lond 590(24):6343–6352. doi:10.1113/jphysiol.2012.245100
Tiecks FP, Lam AM, Aaslid R, Newell DW (1995) Comparison of static and dynamic cerebral autoregulation measurements. Stroke 26(6):1014–1019
Terashvili M, Pratt PF, Gebremedhin D, Narayanan J, Harder DR (2006) Reactive oxygen species cerebral autoregulation in health and disease. Pediatr Clin N Am 53(5):1029–1037, xi
Armstead WM, Kiessling JW, Cines DB, Higazi AA (2011) Glucagon protects against impaired NMDA-mediated cerebrovasodilation and cerebral autoregulation during hypotension after brain injury by activating cAMP protein kinase A and inhibiting upregulation of tPA. J Neurotrauma 28(3):451–457. doi:10.1089/neu.2010.1659
Bohlen HG, Harper SL (1984) Evidence of myogenic vascular control in the rat cerebral cortex. Circ Res 55(4):554–559
McCarron JG, Osol G, Halpern W (1989) Myogenic responses are independent of the endothelium in rat pressurized posterior cerebral arteries. Blood Vessels 26(5):315–319
Carrera E, Lee LK, Giannopoulos S, Marshall RS (2009) Cerebrovascular reactivity and cerebral autoregulation in normal subjects. J Neurol Sci 285(1-2):191–194. doi:10.1016/j.jns.2009.06.041
Gommer ED, Staals J, van Oostenbrugge RJ, Lodder J, Mess WH, Reulen JP (2008) Dynamic cerebral autoregulation and cerebrovascular reactivity: a comparative study in lacunar infarct patients. Physiol Meas 29(11):1293–1303. doi:10.1088/0967-3334/29/11/005
Tan CO, Taylor JA (2014) Integrative physiological and computational approaches to understand autonomic control of cerebral autoregulation. Exp Physiol 99(1):3–15. doi:10.1113/expphysiol.2013.072355
Ainslie PN, Lucas SJ, Fan JL, Thomas KN, Cotter JD, Tzeng YC, Burgess KR (2012) Influence of sympathoexcitation at high altitude on cerebrovascular function and ventilatory control in humans. J Appl Physiol (1985) 113(7):1058–1067. doi:10.1152/japplphysiol.00463.2012
Jordan J, Shannon JR, Diedrich A, Black B, Costa F, Robertson D, Biaggioni I (2000) Interaction of carbon dioxide and sympathetic nervous system activity in the regulation of cerebral perfusion in humans. Hypertension 36(3):383–388
Azevedo E, Castro P, Santos R, Freitas J, Coelho T, Rosengarten B, Panerai R (2011) Autonomic dysfunction affects cerebral neurovascular coupling. Clin Auton Res 21(6):395–403. doi:10.1007/s10286-011-0129-3
Phillips AA, Krassioukov AV, Zheng MM, Warburton DE (2013) Neurovascular coupling of the posterior cerebral artery in spinal cord injury: a pilot study. Brain Sci 3(2):781–789. doi:10.3390/brainsci3020781
Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E (2002) Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J Neurosci 22(21):9430–9444
Hamner JW, Tan CO (2014) Relative contributions of sympathetic, cholinergic, and myogenic mechanisms to cerebral autoregulation. Stroke 45(6):1771–1777. doi:10.1161/STROKEAHA.114.005293
White RP, Vallance P, Markus HS (2000) Effect of inhibition of nitric oxide synthase on dynamic cerebral autoregulation in humans. Clin Sci (Lond) 99(6):555–560
Preckel MP, Leftheriotis G, Ferber C, Degoute CS, Banssillon V, Saumet JL (1996) Effect of nitric oxide blockade on the lower limit of the cortical cerebral autoregulation in pentobarbital-anaesthetized rats. Int J Microcirc Clin Exp 16(6):277–283
Tanaka K, Fukuuchi Y, Gomi S, Mihara B, Shirai T, Nogawa S, Nozaki H, Nagata E (1993) Inhibition of nitric-oxide synthesis impairs autoregulation of local cerebral blood-flow in the rat. Neuroreport 4(3):267–270. doi:10.1097/00001756-199303000-00010
Kajita Y, Takayasu M, Dietrich HH, Dacey RG Jr (1998) Possible role of nitric oxide in autoregulatory response in rat intracerebral arterioles. Neurosurgery 42(4):834–841, discussion 841-832
Thompson BG, Pluta RM, Girton ME, Oldfield EH (1996) Nitric oxide mediation of chemoregulation but not autoregulation of cerebral blood flow in primates. J Neurosurg 84(1):71–78. doi:10.3171/jns.1996.84.1.0071
Zhang R, Wilson TE, Witkowski S, Cui J, Crandall GG, Levine BD (2004) Inhibition of nitric oxide synthase does not alter dynamic cerebral autoregulation in humans. Am J Physiol Heart Circ Physiol 286(3):H863–H869
Edvinsson L, Aubineau P, Owman C, Sercombe R, Seylaz J (1975) Sympathetic innervation of cerebral arteries: prejunctional supersensitivity to norepinephrine after sympathectomy or cocaine treatment. Stroke 6(5):525–530
Hamner JW, Tan CO, Lee K, Cohen MA, Taylor JA (2010) Sympathetic control of the cerebral vasculature in humans. Stroke 41(1):102–109. doi:10.1161/STROKEAHA.109.557132
Sagawa K, Guyton AC (1961) Pressure-flow relationships in isolated canine cerebral circulation. Am J Physiol 200:711–714
Eklof B, Ingvar DH, Kagstrom E, Olin T (1971) Persistence of cerebral blood flow autoregulation following chronic bilateral cervical sympathectomy in the monkey. Acta Physiol Scand 82(2):172–176. doi:10.1111/j.1748-1716.1971.tb04956.x
Zhang R, Levine BD (2007) Autonomic ganglionic blockade does not prevent reduction in cerebral blood flow velocity during orthostasis in humans. Stroke 38(4):1238–1244
Gierthmuhlen J, Allardt A, Sawade M, Baron R, Wasner G (2011) Dynamic cerebral autoregulation in stroke patients with a central sympathetic deficit. Acta Neurol Scand 123(5):332–338. doi:10.1111/j.1600-0404.2010.01424.x
Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD (2002) Autonomic neural control of dynamic cerebral autoregulation in humans. Circulation 106(14):1814–1820
Ainslie PN, Brassard P (2014) Why is the neural control of cerebral autoregulation so controversial? F1000Prime Rep 6:14. doi:10.12703/P6-14
Gordon RD, Kuchel O, Liddle GW, Island DP (1967) Role of the sympathetic nervous system in regulating renin and aldosterone production in man. J Clin Invest 46(4):599–605. doi:10.1172/JCI105561
Mathew RJ, Wilson WH (1997) Intracranial and extracranial blood flow during acute anxiety. Psychiatry Res 74(2):93–107
Zhang HL, Guo ZN, Yang G, Yang L, Han K, Wu J, Xing Y, Yang Y (2012) Compromised cerebrovascular modulation in chronic anxiety: evidence from cerebral blood flow velocity measured by transcranial Doppler sonography. Neurosci Bull 28(6):723–728. doi:10.1007/s12264-012-1282-y
van Gijn J, Kerr RS, Rinkel GJ (2007) Subarachnoid haemorrhage. Lancet 369(9558):306–318
Otite F, Mink S, Tan CO, Puri A, Zamani AA, Mehregan A, Chou S, Orzell S et al (2014) Impaired cerebral autoregulation is associated with vasospasm and delayed cerebral ischemia in subarachnoid hemorrhage. Stroke 45(3):677–682. doi:10.1161/STROKEAHA.113.002630
Budohoski KP, Czosnyka M, Smielewski P, Kasprowicz M, Helmy A, Bulters D, Pickard JD, Kirkpatrick PJ (2012) Impairment of cerebral autoregulation predicts delayed cerebral ischemia after subarachnoid hemorrhage: a prospective observational study. Stroke 43(12):3230–3237. doi:10.1161/STROKEAHA.112.669788
Lang EW, Diehl RR, Mehdorn HM (2001) Cerebral autoregulation testing after aneurysmal subarachnoid hemorrhage: the phase relationship between arterial blood pressure and cerebral blood flow velocity. Crit Care Med 29(1):158–163. doi:10.1097/00003246-200101000-00031
Jaeger M, Soehle M, Schuhmann MU, Meixensberger J (2012) Clinical significance of impaired cerebrovascular autoregulation after severe aneurysmal subarachnoid hemorrhage. Stroke 43(8):2097–2101. doi:10.1161/STROKEAHA.112.659888
Sabri M, Ai J, Knight B, Tariq A, Jeon H, Shang X, Marsden PA, Loch Macdonald R (2011) Uncoupling of endothelial nitric oxide synthase after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab 31(1):190–199. doi:10.1038/jcbfm.2010.76
Kim H, Britton GL, Peng T, Holland CK, McPherson DD, Huang SL (2014) Nitric oxide-loaded echogenic liposomes for treatment of vasospasm following subarachnoid hemorrhage. Int J Nanomedicine 9:155–165. doi:10.2147/IJN.S48856
Hanggi D, Steiger HJ (2006) Nitric oxide in subarachnoid haemorrhage and its therapeutics implications. Acta Neurochir (Wien) 148(6):605–613. doi:10.1007/s00701-005-0721-1, discussion 613
Sabri M, Ai J, Marsden PA, Macdonald RL (2011) Simvastatin re-couples dysfunctional endothelial nitric oxide synthase in experimental subarachnoid hemorrhage. Plos One 6(2):e17062. doi:10.1371/journal.pone.0017062
Osuka K, Watanabe Y, Yasuda M, Takayasu M (2012) Adiponectin activates endothelial nitric oxide synthase through AMPK signaling after subarachnoid hemorrhage. Neurosci Lett 514(1):2–5. doi:10.1016/j.neulet.2011.12.041
Vellimana AK, Milner E, Azad TD, Harries MD, Zhou ML, Gidday JM, Han BH, Zipfel GJ (2011) Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage-induced cerebral vasospasm. Stroke 42(3):776–782. doi:10.1161/STROKEAHA.110.607200
Ding D, Starke RM, Dumont AS, Owens GK, Hasan DM, Chalouhi N, Medel R, Lin CL (2014) Therapeutic implications of estrogen for cerebral vasospasm and delayed cerebral ischemia induced by aneurysmal subarachnoid hemorrhage. Biomed Res Int 2014:727428. doi:10.1155/2014/727428
Moussouttas M, Lai EW, Huynh TT, James J, Stocks-Dietz C, Dombrowski K, Khoury J, Pacak K (2014) Association between acute sympathetic response, early onset vasospasm, and delayed vasospasm following spontaneous subarachnoid hemorrhage. J Clin Neurosci 21(2):256–262. doi:10.1016/j.jocn.2013.03.036
Banki NM, Kopelnik A, Dae MW, Miss J, Tung P, Lawton MT, Drew BJ, Foster E et al (2005) Acute neurocardiogenic injury after subarachnoid hemorrhage. Circulation 112(21):3314–3319
Rangel-Castilla L, Gasco J, Nauta HJ, Okonkwo DO, Robertson CS (2008) Cerebral pressure autoregulation in traumatic brain injury. Neurosurg Focus 25(4):E7. doi:10.3171/FOC.2008.25.10.E7
Junger EC, Newell DW, Grant GA, Avellino AM, Ghatan S, Douville CM, Lam AM, Aaslid R et al (1997) Cerebral autoregulation following minor head injury. J Neurosurg 86(3):425–432. doi:10.3171/jns.1997.86.3.0425
Liu X, Czosnyka M, Donnelly J, Budohoski KP, Varsos GV, Nasr N, Brady KM, Reinhard M et al (2014) Comparison of frequency and time domain methods of assessment of cerebral autoregulation in traumatic brain injury. J Cereb Blood Flow Metab. doi:10.1038/jcbfm.2014.192
Sviri GE, Aaslid R, Douville CM, Moore A, Newell DW (2009) Time course for autoregulation recovery following severe traumatic brain injury. J Neurosurg 111(4):695–700. doi:10.3171/2008.10.17686
Bailey DM, Jones DW, Sinnott A, Brugniaux JV, New KJ, Hodson D, Marley CJ, Smirl JD et al (2013) Impaired cerebral haemodynamic function associated with chronic traumatic brain injury in professional boxers. Clin Sci (Lond) 124(3):177–189. doi:10.1042/CS20120259
Rosenthal G, Sanchez-Mejia RO, Phan N, Hemphill JC 3rd, Martin C, Manley GT (2011) Incorporating a parenchymal thermal diffusion cerebral blood flow probe in bedside assessment of cerebral autoregulation and vasoreactivity in patients with severe traumatic brain injury. J Neurosurg 114(1):62–70. doi:10.3171/2010.6.JNS091360
Cherian L, Goodman JC, Robertson CS (2000) Brain nitric oxide changes after controlled cortical impact injury in rats. J Neurophysiol 83(4):2171–2178
Sakamoto KI, Fujisawa H, Koizumi H, Tsuchida E, Ito H, Sadamitsu D, Maekawa T (1997) Effects of mild hypothermia on nitric oxide synthesis following contusion trauma in the rat. J Neurotrauma 14(5):349–353
Wada K, Chatzipanteli K, Kraydieh S, Busto R, Dietrich WD (1998) Inducible nitric oxide synthase expression after traumatic brain injury and neuroprotection with aminoguanidine treatment in rats. Neurosurgery 43(6):1427–1436
Cherian L, Hlatky R, Robertson CS (2004) Nitric oxide in traumatic brain injury. Brain Pathol 14(2):195–201
Clark RS, Kochanek PM, Obrist WD, Wong HR, Billiar TR, Wisniewski SR, Marion DW (1996) Cerebrospinal fluid and plasma nitrite and nitrate concentrations after head injury in humans. Crit Care Med 24(7):1243–1251
Uzan M, Tanriover N, Bozkus H, Gumustas K, Guzel O, Kuday C (2001) Nitric oxide (NO) metabolism in the cerebrospinal fluid of patients with severe head injury. Inflammation as a possible cause of elevated no metabolites. Surg Neurol 56(6):350–356
Wada K, Chatzipanteli K, Busto R, Dietrich WD (1999) Effects of L-NAME and 7-NI on NOS catalytic activity and behavioral outcome after traumatic brain injury in the rat. J Neurotrauma 16(3):203–212
Liu H, Goodman JC, Robertson CS (2002) The effects of L-arginine on cerebral hemodynamics after controlled cortical impact injury in the mouse. J Neurotrauma 19(3):327–334. doi:10.1089/089771502753594891
Louin G, Marchand-Verrecchia C, Palmier B, Plotkine M, Jafarian-Tehrani M (2006) Selective inhibition of inducible nitric oxide synthase reduces neurological deficit but not cerebral edema following traumatic brain injury. Neuropharmacology 50(2):182–190. doi:10.1016/j.neuropharm.2005.08.020
Jafarian-Tehrani M, Louin G, Royo NC, Besson VC, Bohme GA, Plotkine M, Marchand-Verrecchia C (2005) 1400W, a potent selective inducible NOS inhibitor, improves histopathological outcome following traumatic brain injury in rats. Nitric Oxide 12(2):61–69
Terpolilli NA, Kim SW, Thal SC, Kuebler WM, Plesnila N (2013) Inhaled nitric oxide reduces secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab 33(2):311–318. doi:10.1038/jcbfm.2012.176
Liu P, Li YS, Quartermain D, Boutajangout A, Ji Y (2013) Inhaled nitric oxide improves short term memory and reduces the inflammatory reaction in a mouse model of mild traumatic brain injury. Brain Res 1522:67–75. doi:10.1016/j.brainres.2013.05.032
Papadimos TJ (2008) The beneficial effects of inhaled nitric oxide in patients with severe traumatic brain injury complicated by acute respiratory distress syndrome: a hypothesis. J Trauma Manag Outcomes 2(1):1. doi:10.1186/1752-2897-2-1
Patel MB, McKenna JW, Alvarez JM, Sugiura A, Jenkins JM, Guillamondegui OD, Pandharipande PP (2012) Decreasing adrenergic or sympathetic hyperactivity after severe traumatic brain injury using propranolol and clonidine (DASH After TBI Study): study protocol for a randomized controlled trial. Trials 13:177. doi:10.1186/1745-6215-13-177
Baguley IJ, Heriseanu RE, Felmingham KL, Cameron ID (2006) Dysautonomia and heart rate variability following severe traumatic brain injury. Brain Inj 20(4):437–444
Baguley IJ, Nicholls JL, Felmingham KL, Crooks J, Gurka JA, Wade LD (1999) Dysautonomia after traumatic brain injury: a forgotten syndrome? J Neurol Neurosurg Psychiatry 67(1):39–43
Heffernan DS, Inaba K, Arbabi S, Cotton BA (2010) Sympathetic hyperactivity after traumatic brain injury and the role of beta-blocker therapy. J Trauma 69(6):1602–1609. doi:10.1097/TA.0b013e3181f2d3e8
Conflict of Interest
The authors declare that they have no conflict of interest.
Funding
This article was supported by Jilin Provincial government (Changbai mountain scholars, 440020031172) to Yi Yang and Youth development foundation of First Hospital of Jilin University to Zheni-Ni Guo.
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhen-Ni Guo, Anwen Shao, Lu-Sha Tong and Weiyi Sun contributed equally to this work.
Rights and permissions
About this article

Cite this article
Guo, ZN., Shao, A., Tong, LS. et al. The Role of Nitric Oxide and Sympathetic Control in Cerebral Autoregulation in the Setting of Subarachnoid Hemorrhage and Traumatic Brain Injury. Mol Neurobiol 53, 3606–3615 (2016). https://doi.org/10.1007/s12035-015-9308-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9308-x