
Abstract
Several plant-derived natural compounds are known to exhibit anti-amyloid aggregation activity which makes them attractive as potential therapies to treat Alzheimer’s disease. The mechanisms of their anti-amyloid activity are not well known. In this regard, many natural compounds are known to exhibit direct binding to various amyloid species including oligomers and fibrils, which in turn can lead to conformational change in the beta-sheet assembly to form nontoxic aggregates. This review discusses the mechanism of anti-amyloid activity of 16 natural compounds and gives structural details on their direct binding interactions with amyloid aggregates. Our computational investigations show that the physicochemical properties of natural products do fit Lipinski’s criteria and that catechol and catechol-type moieties present in natural compounds act as lysine site-specific inhibitors of amyloid aggregation. Based on these observations, we propose a structural template to design novel small molecules containing site-specific ring scaffolds, planar aromatic and nonaromatic linkers with suitably substituted hydrogen bond acceptors and donors. These studies will have significant implications in the design and development of novel amyloid aggregation inhibitors with superior metabolic stability and blood-brain barrier penetration as potential agents to treat Alzheimer’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview of Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that slowly destroys memory and cognition in the elderly population [1]. The major histopathological hallmarks of AD are extracellular senile plaques, consisting of abnormally aggregated amyloid-beta (Aβ) peptide [2]. To date, available drugs only treat the symptoms of AD and no therapies are able to prevent or fundamentally treat the disease.
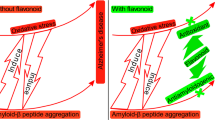
Amyloid aggregation is a critical step for Aβ to form neurotoxic species and their deposition in the brain, leading to neuroinflammation, neurite degeneration, and neuronal death causing cognitive decline [3]. The therapeutic strategies for AD mainly focus on inhibiting Aβ production (such as β- and γ-secretase inhibitors), facilitating Aβ clearance (such as Aβ immunotherapy [4]), and blocking Aβ neurotoxicity by inhibiting the formation of various forms of Aβ aggregates. However, most researchers focus on the first two strategies but have encountered various challenges. As the monomeric Aβ is less toxic, keeping Aβ from self-assembling into toxic dimers, oligomers, protofibrils, and fibrils can reduce its toxicity. Aggregation of monomeric Aβ could be reduced in the presence of many natural compounds such as epigallocatechin gallate [5], curcumin [6], resveratrol [7], and scyllo-inositol [8], which offer examples for us to screen and design inhibitors of Aβ aggregation. In this review, we discuss the effects of the natural compounds on Aβ aggregation and neurotoxicity, analyze the active motif of their structures, and propose perspectives for designing new AD therapeutic compounds to target Aβ aggregation and its toxicity.
Role of Aβ Aggregation in AD Pathogenesis
The 4-kD Aβ monomer is consistently produced by proteolytic cleavage of the amyloid precursor protein (APP) by β-secretase (BACE 1) and γ-secretase. The recent finding that a rare mutation in the APP decreases production of Aβ and protects against AD strongly supports the fact that Aβ plays a crucial role in the pathogenesis of AD [9]. The neurotoxicity of the amyloid peptide depends on its peptide length and conformation. Aβ40 accounts for more than 90 % of total Aβ, while Aβ42 is more prone to aggregate and is the predominantly more toxic peptide [10, 11]. A little increase in the Aβ42:Aβ40 ratio stabilizes toxic Aβ oligomeric species [12]. In vitro and in vivo studies suggest that Aβ causes synaptic dysfunction, abnormal iron homeostasis, oxidative stress, mitochondrial dysfunction, energy hypometabolism, and memory impairment [13–19]. Monomeric Aβ was shown to be nontoxic and prevents neuronal cell death caused by oxidative stress [20]. The toxic effects of aggregation-prone Aβ result from conformational transition of Aβ monomers from predominantly α-helical to β-sheet structures that result in monomeric Aβ aggregation and fibrillization. The assembly forms of Aβ include oligomers, protofibrils, fibrils, and plaques, the neurotoxicity of which is different from one another. Numerous studies indicate that soluble Aβ oligomers play important causal roles in AD (reviewed in [21]), and Aβ42 oligomers are thought to be the most toxic form [22]. Therefore, the aggregation of amyloid peptide is a pivotal step in AD pathogenesis, and inhibiting Aβ aggregation is an attractive neuroprotective strategy for the prevention and treatment of this disease.
Aggregation and Neurotoxic Properties of Aβ and the Related Motifs
The amino acid sequence of Aβ42 is NH2-DAEFRHDSGYEVHHQKLVFFAEDVGSNKGA IIGLMVGGVVIA-COOH. Residues 1–28 and 29–42 respectively represent a hydrophilic domain and a hydrophobic domain. The natively unfolded monomeric Aβ primarily have a random-coil structure in their soluble state when they are released into extracellular matrix [23]. However, conformational transition of Aβ from native state to β-sheet happens rapidly, which initiates the aggregation process and forms toxic Aβ oligomers [24, 25]. The monomeric Aβ peptide self-assembles through multiple pathways, and aggregated Aβ conformers range from dimers, soluble oligomers to fibrillar oligomers, protofibrils, and fibrils [26–29]. As toxic Aβ oligomers are not an obligate intermediate on the pathway to fibril formation [30], selectively inhibiting oligomerization but not affecting fibrillization is also a possible target to attenuate Aβ toxicity. Although the exact mechanism of Aβ aggregation is not completely clear, the key structure related to Aβ aggregation will be discussed, which may assist in understanding the aggregation process and to design new compounds as Aβ aggregation inhibitors and thus attenuate its toxicity.
The Aβ region consisting of amino acids 12–24 and 30–40, known as the fibril-forming Aβ fragment, is responsible for their native states self-assembling into a β-sheet structure [4], and N-methylated residues at this fragment effectively inhibit the formation of intermolecular hydrogen bonds required for peptide oligomerization into β-sheet aggregates [31]. Several small molecules that bind to the C-terminus of Aβ42 and exhibit hydrophobic and hydrogen-bonding interactions have also been designed to inhibit β-sheet formation [32, 33]. Some studies suggest that β-sheet conformation in residues 24–28 promote Aβ oligomerization [34, 35]. Thus, a β-sheet structure is necessary for Aβ oligomerization and neurotoxicity. Therefore, preventing the conformational transition of the Aβ monomer from an initial random coil or α-helix into a β-sheet is the primary goal of blocking Aβ toxicity by small-molecule inhibitors.
The Aβ monomer consists of aromatic residues, phenylalanine (Phe4, Phe19, and Phe20) and tryptophan (Tyr10) [36]. Pi-stacking interactions between aromatic residues were suggested to play a significant role in the Aβ self-assembly process by enhanced fibril assembly kinetics [37, 38]. The rodent Aβ peptide sequence is identical to the human Aβ sequence except three amino acids located within the N-terminal domain of the Aβ peptide (Arg-to-Gly, Tyr-to-Phe, and His-to-Arg). However, most of these rodent species lack AD-like pathology [39]. Thus, the three specific sites are believed to be important in Aβ aggregation or Aβ-induced neurotoxicity. Substitution of the amino acid residue Tyr10 with alanine or phenylalanine in the Aβ peptide led to a dramatic decrease in neurotoxicity, suggesting that tyrosine is important in Aβ-induced neurotoxicity [40, 41]. Substitution of histidine (His13) in rodent Aβ can disrupt the zinc binding site and subsequently alleviates zinc-induced aggregation in vitro [42]. In addition, oxidation of methionine (Met35) can promote toxic conformations in Aβ oligomers [43]. Although their role in Aβ aggregation still needs to be further clarified, these findings help us better understand the structural perturbation of Aβ to its aggregation.
It has been shown that biometals (e.g., iron, zinc, and copper) are significantly enriched in the brains of AD patients compared with normal age-matched subjects [44, 45], which may promote Aβ oligomerization [46] and stabilize soluble Aβ oligomers [47]. The residues Tyr10, His13, and His14 of Aβ are investigated as potential metal ion coordination centers [48, 49]. So compounds that could act as biometal chelators may also reduce Aβ oligomerization.
Natural Compounds Targeting Aβ Aggregation and Toxicity
A wide range of natural and synthetic molecules (both large and small molecules) has been investigated for their ability to counteract Aβ aggregation and toxicity [50]. In this review, we will focus on some natural compounds that are known to exhibit anti-Aβ activity and will address the mechanisms of their inhibition. The discussion on synthetic molecules with anti-Aβ activity is beyond the scope of this review. Polyphenol-containing natural compounds are known to exhibit multi-target effects on AD including antioxidant, free radical-scavenging properties, anti-amyloidogenic activity, cell signaling modulation, telomere length, and modulation of the sirtuin proteins [51]. Many compounds were shown to inhibit the formation of Aβ fibrils or to destabilize preformed Aβ fibrils by binding directly to monomeric Aβ or mature aggregates [52]. But disassembly of large fibrils raises concerns of increasing load of Aβ oligomers and causing secondary damage to neurons, which is defined as the “raising dust” effect [4]. Thus, compounds that convert mature Aβ fibrils into smaller or nontoxic aggregates and avoid regeneration of toxic aggregation intermediates are attractive candidates to attenuate Aβ toxicity. In the following section, some natural compounds with amyloid aggregation inhibition properties are discussed.
Epigallocatechin Gallate
Epigallocatechin-3-gallate (EGCG, Fig. 1), the principal polyphenol present in green tea, has been shown to promote the formation of unstructured, nontoxic Aβ aggregates by inhibiting its conformational change from a random-coil to a β-sheet structure [5, 53]. The small Aβ oligomers formed in the presence of EGCG were nontoxic and unable to seed further fibril growth [54]. Solid-state nuclear magnetic resonance (NMR) analysis revealed that the N-terminus residues 1–20 become unstructured, whereas residues 22–39 adopt a β-sheet conformation. Furthermore, EGCG is able to remodel mature Aβ fibrils and toxic oligomers into smaller nontoxic aggregates with loss of β-sheet content [55]. This remodeling effect may be via alternative hydrogen bonding facilitated by the hydroxyl-rich EGCG [5]. There are 12 important residues of Aβ that strongly interact with EGCG (Phe4, Arg5, Phe19, Phe20, Glu22, Lys28, Gly29, Leu34 to Gly37, and Ile41), while the side chains of some hydrophobic residues (Phe, Met, and Ile) and the main chains of Lys28 and Gly29 provide nonpolar interactions that contributes more than 71 % to the binding free energy of the EGCG-Aβ complex [56]. Another study shows that when free amines or thiols of Aβ are proximal to the EGCG hydrophobic binding sites, the EGCG-based quinones are capable of covalently modifying Aβ through the formation of Schiff base [57]. Therefore, the combination of hydrophobic interaction and Schiff base formation between oxidized EGCG and Aβ may be responsible for its remodeling effect. EGCG displays the anti-aggregation effect from mainly two pathways: first, EGCG binds to the native form of Aβ through interactions with the side chains of specific residues, and second, EGCG binds to the misfolded Aβ species with noncovalent interactions involving Aβ backbone and subsequently remodels toxic aggregates into small nontoxic, off-pathway oligomers.

Chemical structures of epigallocatechin gallate (EGCG), curcumin tautomers, and resveratrol
Curcumin
Curcumin ((1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione, Fig. 1), a diphenol isolated from the rhizomes of Curcuma longa (turmeric), was first shown to decrease the levels of insoluble and soluble Aβ and plaque burden in Tg2576 transgenic mice [58]. It binds to both Aβ oligomers and Aβ fibrils [59]. The anti-aggregating activity of curcumin against Aβ has been actively investigated. Curcumin is believed to inhibit Aβ oligomerization, promote the deposition of nontoxic Aβ fibrils, and decrease the neurotoxicity of Aβ aggregates [6, 60]; thus, it may be capable of changing or accelerating the pathway from toxic Aβ oligomers to nontoxic Aβ forms. In addition, curcumin was shown to accelerate Aβ fibril conversion by reducing the pre-fibrillary species of Aβ, and hence alleviate Aβ toxicity in the transgenic Drosophila model [61]. Modeling studies have shown that curcumin can act as an efficient β-sheet breaker [62]. Furthermore, curcumin was shown to bind Cu2+ and Fe2+ ions, which could also play a role in inhibiting Aβ aggregation [63].
Numerous studies have tried to identify structural features that contribute to the anti-oligomerization activity of curcumin and its analogs. An early study indicated that Aβ inhibitors interact with GxxxG motifs (i.e., Gly33 in Aβ40 and Gly37 in Aβ42) in the C-terminus of the Aβ fibrils [64]. The enolic center and the two phenolic polar groups separated by a hydrophobic bridge make it prone to binding to Aβ oligomers [65]. Solid-state NMR analysis reveals that the methoxy and/or hydroxy groups of curcumin interact with the V12 and 16KLVFFA21 residues of the Aβ [66, 67]. They emphasized that the enone group and unsaturated carbon spacer between aryl rings are essential for its anti-Aβ aggregation activity. Recent studies show that the pi-stacking interaction between curcumin and the aromatic residues (Tyr, Phe, and His) of Aβ contributes to the reduction of the β-sheet content in the Aβ dimer [62]. According to the previous findings, the coordination of copper with imidazoles on His13 and His14 of Aβ could initialize Aβ aggregation [68, 69]. This led to the design of imidazole-containing curcumin analogs as new tracers of Aβ oligomers and aggregation inhibitors of Aβ capable of specifically interrupting the coordination of copper and imidazoles from His13 and His14 [70].
Resveratrol
Resveratrol (3, 5, 4′-trihydroxystilbene, Fig. 1), a nonflavonoid polyphenol abundant in red wine and many plants, consists of two aromatic rings joined by a methylene bridge and is known to possess a wide range of biological effects [71]. Several studies indicate that the neuroprotective effects of resveratrol on AD is attributed to a number of factors, including attenuation of Aβ-induced toxicity [72], reduction in secreted and intracellular Aβ via BACE-1 inhibition [73], and intracellular degradation of Aβ via proteasome [74]. It can also lower extracellular Aβ accumulation by increasing AMPK activity to trigger autophagy and lysosomal degradation of Aβ [75]. It reduced Aβ-stimulated NF-kappa B signaling by overexpressing and activating SIRT-1 [76]. But how resveratrol interacts with the neural cell and enhances its intracellular degradation of Aβ is unclear. Whether there exists a receptor of resveratrol on the neural cell membrane deserves further consideration.
Most notably, resveratrol was able to directly interfere with the Aβ aggregation. Resveratrol was proved to directly interact with Aβ monomers and fibrils, inhibit Aβ fibrillization, and convert toxic oligomers into nontoxic species [77, 78]. Resveratrol derivatives were also shown to inhibit Aβ42 aggregation by targeting the C-terminus and inhibiting the formation of β-sheets [79]. Others found resveratrol selectively remodeled certain Aβ conformers that possess β-sheet structures into nontoxic, unstructured species [7]. The remodeling activity may result from the interaction between resveratrol and aromatic side chains (e.g., Phe4, Tyr10, Phe19, Phe20) of Aβ. Thus, the interaction between resveratrol and the N-terminal of Aβ is able to disaggregate mature aggregates into nontoxic species, although the N-terminal of Aβ is hidden in mature aggregates. A glycoside of resveratrol was shown to disaggregate Aβ soluble oligomers at sub-stoichiometric concentrations [7]. Researchers found that polyphenol aglycones and glycosides were able to remodel toxic Aβ oligomers into nontoxic, off-pathway conformers [80]. Thus, the phenolic rings are responsible for its remodeling activity and the minimal structural requirement for phenolic aglycones to remodel Aβ oligomers consists of two aromatic rings, of which at least one must possess a hydroxyl substituent.
Flavonoids
Flavonoids, especially myricetin and quercetin (Fig. 2), exhibit neuroprotection in AD by multiple mechanisms, such as antioxidative effect [81, 82], AMPK activation [83], and inhibition of BACE-1 activity [84]. Quercetin possesses a strong ability to inhibit Aβ fibril formation and protects neuronal cells against Aβ-induced toxicity [85]. Myricetin was shown to prevent conformational change of Aβ from a random-coil to a β-sheet-rich structure, and its inhibitory effects against Aβ aggregation were directly proportional to the number of hydroxyl groups on the B ring [86]. Another study reached the same conclusion that the 3′,4′-dihydroxyl group was essential for their inhibition of Aβ aggregation [87]. A multi-methodological study also proved that myricetin inhibits Aβ aggregation by targeting preferentially Aβ monomers and short transient oligomers [88]. Computational analysis showed that myricetin interacts with the surface of the β-sheet via H-bonding, weakening the interstrand hydrogen bonds and eventually disrupting the outer layer of the aggregate [89]. In addition, the integrated catechol moiety of flavonoids performs a bifunctional inhibitory activity (metal chelation and Aβ interaction) against Aβ aggregation by interacting with the metal binding site on Aβ via their α-keto enolate group [90, 91]. Furthermore, oxidizing the residue methionine (Met35) in Aβ monomers, which hinders the amyloidogenic conformation, is also the potential mechanism for anti-aggregation of myricetin as revealed by mass spectrometry experiments [92]. Other flavonoids with potential Aβ aggregation inhibition include baicalein [93], morin [94], apigenin [95], and kaempferol [89] shown in Fig. 2.

Chemical structures of flavonoids myricetin, quercetin, baicalein, morin, apigenin, and kaempferol
Others
Extra virgin olive oil (EVOO) was shown to improve cognitive decline associated with aging and disease [96–98]. Oleuropein, the functional component of EVOO, was investigated to inhibit Aβ aggregation and toxicity in vitro and in vivo. It was shown to interact with the monomeric form of Aβ or its oxidized form Aβ Met35(O) by noncovalent binding [99], and the hydrophobic region of Aβ (16KLVFFA21), a critical Aβ sequence implicated in its aggregation, was most likely to interact with the nonpolar moiety of oleuropein [100, 101]. Caenorhabditis elegans, a simplified model of AD, showed significant reduction of Aβ plaque deposition and toxic oligomer formation after the administration of oleuropein aglycone (OLE, Fig. 3) [102]. This compound also strongly improved cognitive performance of a young/middle-aged TgCRND8 mouse model of AD. They found that OLE was able to induce autophagy possibly by the regulation of the mTOR pathway in cell cultures [103].

Chemical structures of oleuropein aglycone (OLE), isoliquiritigenin (ISL), and cyanidin-3-O-glucopyranoside
Isoliquiritigenin (ISL, compound 11 in Fig. 3), a trihydroxychalcone, is one of the components of licorice. It possesses an α,β-unsaturated system similar to curcumin. A series of new ISL derivatives were designed as inhibitors of Aβ aggregation and cytotoxicity by decreasing the β-sheet structure [104]. Yet another natural product, cyaniding 3-O-glucopyranoside (Cy-3G, Fig. 3), derived from fruit and vegetables, prevents the formation of soluble Aβ25–35 oligomers and their neurotoxicity in SH-SY5Y cells [105], which is probably due to the presence of a polyphenol-like structure with many hydroxyl groups, which could interact with amino acid residues in the Aβ25–35 peptide through hydrogen bonds. Other natural compounds that exhibit anti-Aβ aggregation properties include rosmarinic acid (RA), nordihydroguaiaretic acid (NDGA), and ferulic acid (FA) as shown in Fig. 4. The large molecular weight natural compound tannic acid [106] is metabolically unstable and is expected to degrade to smaller fragments (gallic acid, a trihydroxybenzoic acid) whereas the inositol isomer scyllo-inositol is known to bind to amyloid-forming peptide fragment Aβ (16KLVFFA21), coat the surface of Aβ protofibrils, and disrupt their lateral stacking into amyloid fibrils [107].

Chemical structures of rosmarinic acid, nordihydroguaiaretic acid, ferulic acid, and tannic acid
Overall, the anti-Aβ aggregation mechanisms of all these natural compounds discussed can be due to their ability to
-
a)
Increase the stability of amyloid peptides in the native state
-
b)
Inhibit the formation of toxic oligomers by preventing the required conformational transition and or by acting as a biometal chelator
-
c)
Disassemble toxic Aβ species into nontoxic forms
-
d)
Inhibit toxic Aβ oligomer interaction with the cell membrane by selective neutralization of the toxic Aβ structure epitope
Computational Studies of Natural Compounds with Aβ
In order to understand the anti-amyloid activity of natural compounds shown in Figs. 1, 2, 3, and 4, we first investigated the binding interactions of ligands with Aβ. This was carried out by molecular docking studies using the CDOCKER program (BIOVIA/Accelrys Inc) [108]. The steric zipper assembly of Aβ hexapeptide 16KLVFFA21 was used to obtain the most stable ligand-peptide complex [109]. We focused our attention on the flavonoid class of polyphenolic compounds represented by myricetin, quercetin, baicalein, morin, apigenin, and kaempferol (Fig. 2). These molecules contain a common pharmacophore 2-phenylchromen-4-one. The Aβ hexapeptide 16KLVFFA21 steric zipper assembly is a useful model to study the binding modes of ligands with anti-amyloid activity [110, 111]. The spine of the steric zipper assembly contains amino acid side chains of lysine, leucine, and phenylalanine. The flavonoids underwent polar and nonpolar interactions with these amino acids. Investigating the binding modes of flavonoids shows that in general the 2-phenylchromen-4-one pharmacophore was oriented in the core of the steric zipper assembly. Furthermore, in the case of both myricetin and quercetin, their C-2 3,4,5-trihydroxylphenyl and 3,4-dihydroxyphenyl substituents respectively were oriented in the vicinity of lysine amino acid side chains and underwent polar contact (Fig. 5). The binding mode of baicalein shows that the 5,6,7-trihydroxy-2-phenylchromen-4-one was oriented closer to lysine amino acid side chains at the steric zipper interface. Interestingly, the common feature seen with these flavonoids was the fact that the 3,4-dihydroxy substituents underwent polar contacts with lysine amino acid side chains as shown in Fig. 5. It should be noted that the KLVFFA region of Aβ is known to play a significant role as a seeding point in the nucleation-dependent aggregation [110, 112]. Docking studies suggest that flavonoids that contain catechol groups or adjacent dihydroxy (3,4-dihydroxy) substituents such as myricetin, quercetin, and baicalein are able to exhibit superior anti-Aβ aggregation properties due to their ability to undergo polar contacts with lysine chains compared to flavonoids that lack catechol, catechol-type, or 3,4-dihydroxy substituents such as morin, epigenin, and kaempferol (Fig. 2). These observations are further supported by a recent work from Sato and coworkers which show that catechol-containing flavonoids can undergo oxidation in vivo to form quinone intermediates. These quinone intermediates can potentially link covalently to lysine side chains and prevent Aβ aggregation [113].

Binding modes of myricetin (green), quercetin (violet), and baicalein (blue) in the Aβ-hexapeptide KLVFFA tetramer steric zipper model (pdb id: 3OVJ)
Docking studies of other natural compounds containing catechol groups (EGCG 1, OLE 10, and Cy-3G 12, Figs. 1 and 3) exhibit similar binding modes with their catechol groups interacting with lysine side chains in the Aβ-hexapeptide model. The resorcinol containing natural compounds resveratrol 3 (Fig. 1) and ISL 11 (Fig. 3) binding indicates that the resorcinol groups were interacting with lysine side chains whereas the aromatic rings underwent nonpolar contacts with leucine, phenylalanine, and valine side chains. Our work has shown that the enol form of curcumin is primarily responsible for its anti-Aβ activity and the 4-hydroxy-3-methoxy substituents on the phenyl ring undergo polar contacts with lysine side chains whereas the aromatic rings were involved in nonpolar contacts with leucine, phenylalanine, and valine side chains in the Aβ-hexapeptide steric zipper assembly [114].
In order to understand the structural features that are required to exhibit anti-Aβ aggregation activity, we constructed a ligand-based pharmacophore model for flavonoids using the HipHopRefine algorithm in BIOVIA/Accelrys Inc. Structure-Based-Design software (Fig. 6a, b). The pharmacophoric map was generated by using a training set of flavonoids made up of their bioactive conformations (Fig. 6a) bound to the Aβ hexapeptide 16KLVFFA21 steric zipper model. The flavonoid myricetin, which is known to be the most potent inhibitor of amyloid aggregation, was used as the reference compound to generate the pharmacophore model by including hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), and aromatic ring (AR) parameters (Fig. 6b). The top-ranking pharmacophore model for flavonoids suggests that structural requirements for Aβ aggregation inhibition includes the presence of (i) three HBDs; (ii) two HBAs; and (iii) two ARs and that these features should be separated from a distance range of 2.73–7.40 Å (Fig. 6a). It should be noted that these features correlate with the molecular docking results of flavonoids myricetin, quercetin, and baicalein where the presence of adjacent HBDs (catechol and catechol-type groups respectively) leads to favorable interactions with lysine side chains.

a Pharmacophore model for flavonoids with HBDs, HBAs, ARs, and distance constraints; b myricetin mapped into the pharmacophore model. Color codes: pink = HBDs; green = HBAs, orange = ARs
Perspectives for the Design of New Compounds with Anti-Aβ Aggregation Properties Based on Natural Compounds
Natural compounds possessing polyphenolic groups are known to exhibit antioxidant and metal-chelating properties that are associated with their beneficial effects. Our studies on natural compounds 1–16 (Figs. 1, 2, 3, and 4) and their binding modes using the Aβ-hexapeptide model highlight the importance of certain ring scaffolds as structural requirement to exhibit anti-Aβ aggregation properties in the design of novel molecules. These requirements include substituents such as (i) phenol; (ii) catechol; (iii) resorcinol; (iv) 3,4,5-trihydroxyphenyl (pyrogallol); and (v) hydroxy-methoxyphenyl ring scaffolds linked together by planar core templates such as a 2-phenylchromen-4-one ring (flavonoids) or an α,β-unsaturated system (curcumin 2 and ISL 11, Figs. 1 and 2) and carbon-carbon double bonds (resveratrol) as shown in Fig. 7. This suggests that small molecules with planar geometry and capable of exhibiting linear extended conformation are able to bind at the β-sheet interface with the ability to convert neurotoxic Aβ aggregates to nontoxic forms. Furthermore, in the design of novel anti-Aβ agents, the presence of catechol or catechol-type substituents is expected to exhibit superior Aβ inhibition due to their potential to covalently link to lysine side chains at the β-sheet interface [110, 113]. Table 1 gives a summary of physicochemical properties of natural compounds 1–16. The general trend seen with the exception of compound 16 indicates that the minimum requirement for anti-Aβ activity is the presence of (i) one aromatic ring; (ii) two hydrogen bond donors; (iii) three hydrogen bond acceptors; (iv) one rotatable bond; (v) molecular volume range of 150–329 Å3; (vi) lipophilicity range with AlogP values 1.18–4.70; and (vii) molecular weight ranging from 194 to 458. These physicochemical parameters fit Lipinski’s rule of five [115]. It should be noted that polar polyphenolic natural compounds such as 4, 5, 11, and 12 (Figs. 2 and 3) exhibit weak blood-brain barrier (BBB) penetration and can undergo metabolic changes in the gut, while some lipophilic compounds are capable of entering the brain and reaching levels higher than the physiological concentration of soluble Aβ in the brain [60, 116–118]. Thus, the development of new compounds with superior in vivo metabolic stability and BBB penetration is critical.

Schematic on designing novel small molecules with anti-Aβ activity inspired by natural compounds
In this regard, Fig. 7 gives a summary of critical structural features that can be incorporated in the design of novel small-molecule inhibitors of Aβ aggregation. It is anticipated that catechol and catechol-type scaffolds can act as site-specific Aβ aggregation inhibitors with the ability to link covalently to lysine side chains whereas other HBAs including C=O, C=S, and P=O can exhibit reversible binding to lysine side chains and the potential to reduce the toxicity of various Aβ aggregates [119]. These site-specific scaffolds can be fused to planar aromatic or nonaromatic rings or aliphatic linkers substituted with suitable HBDs and HBAs as shown in Fig. 7.
In summary, this review highlights the critical structural features present in natural compounds that are responsible for their ability to prevent Aβ aggregation and provides a structural template to design new small molecules as therapeutic agents to study and treat AD.
References
Wang YJ, Zhou HD, Zhou XF (2006) Clearance of amyloid-beta in Alzheimer's disease: progress, problems and perspectives. Drug Discov Today 11(19-20):931–8
Serrano-Pozo A et al (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1(1):a006189
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–6
Liu YH et al (2012) Immunotherapy for Alzheimer disease: the challenge of adverse effects. Nat Rev Neurol 8(8):465–9
Ehrnhoefer DE et al (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol 15(6):558–66
Liu KN et al (2012) Curcumin’s pre-incubation temperature affects its inhibitory potency toward amyloid fibrillation and fibril-induced cytotoxicity of lysozyme. Biochim Biophys Acta 1820(11):1774–86
Ladiwala AR et al (2010) Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Abeta into off-pathway conformers. J Biol Chem 285(31):24228–37
Sinha S et al (2012) Comparison of three amyloid assembly inhibitors: the sugar scyllo-inositol, the polyphenol epigallocatechin gallate, and the molecular tweezer CLR01. ACS Chem Neurosci 3(6):451–8
Jonsson T et al (2012) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488(7409):96–9
Wong PC et al (2002) Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci 5(7):633–9
Suzuki N et al (1994) An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264(5163):1336–40
Kuperstein I et al (2010) Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J 29(19):3408–20
Walsh DM, Selkoe DJ (2004) Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett 11(3):213–28
Crouch PJ et al (2005) Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci 25(3):672–9
Shankar GM et al (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14(8):837–42
Pierrot N et al (2004) Intraneuronal amyloid-beta1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem 88(5):1140–50
Golde TE, Janus C (2005) Homing in on intracellular Abeta? Neuron 45(5):639–42
Levi O et al (2007) Intraneuronal amyloid-beta plays a role in mediating the synergistic pathological effects of apoE4 and environmental stimulation. J Neurochem 103(3):1031–40
Lesne S et al (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440(7082):352–7
Zou K et al (2002) A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci 22(12):4833–41
Gilbert BJ (2013) The role of amyloid beta in the pathogenesis of Alzheimer’s disease. J Clin Pathol 66(5):362–6
Walsh DM, Selkoe DJ (2007) A beta oligomers—a decade of discovery. J Neurochem 101(5):1172–84
Danielsson J et al (2005) The Alzheimer beta-peptide shows temperature-dependent transitions between left-handed 3-helix, beta-strand and random coil secondary structures. FEBS J 272(15):3938–49
Yang DS et al (1999) Manipulating the amyloid-beta aggregation pathway with chemical chaperones. J Biol Chem 274(46):32970–4
Serpell LC et al (2000) Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A 97(9):4897–902
Rochet JC, Lansbury PT Jr (2000) Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol 10(1):60–8
Wetzel R (2006) Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res 39(9):671–9
Glabe CG (2008) Structural classification of toxic amyloid oligomers. J Biol Chem 283(44):29639–43
Ono K, Condron MM, Teplow DB (2009) Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci U S A 106(35):14745–50
Necula M et al (2007) Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 282(14):10311–24
Butterfield S et al (2012) Chemical strategies for controlling protein folding and elucidating the molecular mechanisms of amyloid formation and toxicity. J Mol Biol 421(2-3):204–36
Wang SS, Chen YT, Chou SW (2005) Inhibition of amyloid fibril formation of beta-amyloid peptides via the amphiphilic surfactants. Biochim Biophys Acta 1741(3):307–13
Yang C et al (2010) Exploration of the mechanism for LPFFD inhibiting the formation of beta-sheet conformation of A beta(1-42) in water. J Mol Model 16(4):813–21
Lazo ND et al (2005) On the nucleation of amyloid beta-protein monomer folding. Protein Sci 14(6):1581–96
Xu Y et al (2005) Conformational transition of amyloid beta-peptide. Proc Natl Acad Sci U S A 102(15):5403–7
Gazit E (2002) A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J 16(1):77–83
Gazit E (2002) Mechanistic studies of the process of amyloid fibrils formation by the use of peptide fragments and analogues: implications for the design of fibrillization inhibitors. Curr Med Chem 9(19):1725–35
Gazit E (2002) Global analysis of tandem aromatic octapeptide repeats: the significance of the aromatic-glycine motif. Bioinformatics 18(6):880–3
Sarasa M, Pesini P (2009) Natural non-transgenic animal models for research in Alzheimer’s disease. Curr Alzheimer Res 6(2):171–8
Dai X et al (2012) Abeta-40 Y10F increases betafibrils formation but attenuates the neurotoxicity of amyloid-beta peptide. Int J Mol Sci 13(5):5324–37
Barnham KJ et al (2004) Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease beta-amyloid. FASEB J 18(12):1427–9
Yang DS et al (2000) Examining the zinc binding site of the amyloid-beta peptide. Eur J Biochem 267(22):6692–8
Schoneich C (2005) Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer’s disease. Biochim Biophys Acta 1703(2):111–9
Pepeu G, Giovannini MG (2009) Cholinesterase inhibitors and beyond. Curr Alzheimer Res 6(2):86–96
Bush AI (2008) Drug development based on the metals hypothesis of Alzheimer’s disease. J Alzheimers Dis 15(2):223–40
Tougu V et al (2009) Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-beta (1-42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J Neurochem 110(6):1784–95
Sharma AK et al (2013) The effect of Cu and Zn on the Abeta peptide aggregation and cellular toxicity. Metallomics 5(11):1529–36
Smith DG, Cappai R, Barnham KJ (2007) The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim Biophys Acta 1768(8):1976–90
Tickler AK et al (2005) Methylation of the imidazole side chains of the Alzheimer disease amyloid-beta peptide results in abolition of superoxide dismutase-like structures and inhibition of neurotoxicity. J Biol Chem 280(14):13355–63
Cheng B et al (2013) Inhibiting toxic aggregation of amyloidogenic proteins: a therapeutic strategy for protein misfolding diseases. Biochim Biophys Acta 1830(10):4860–71
Jayasena T et al (2013) The role of polyphenols in the modulation of sirtuins and other pathways involved in Alzheimer’s disease. Ageing Res Rev 12(4):867–883
Ono K et al (2003) Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: implications for the prevention and therapeutics of Alzheimer’s disease. J Neurochem 87(1):172–81
Rezai-Zadeh K et al (2005) Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci 25(38):8807–14
Lopez del Amo JM et al (2012) Structural properties of EGCG-induced, nontoxic Alzheimer’s disease Abeta oligomers. J Mol Biol 421(4-5):517–24
Bieschke J et al (2010) EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A 107(17):7710–5
Liu FF et al (2011) Molecular insight into conformational transition of amyloid beta-peptide 42 inhibited by (-)-epigallocatechin-3-gallate probed by molecular simulations. J Phys Chem B 115(41):11879–87
Palhano FL et al (2013) Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J Am Chem Soc 135(20):7503–10
Lim GP et al (2001) The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci 21(21):8370–7
Yanagisawa D et al (2011) Curcuminoid binds to amyloid-beta1-42 oligomer and fibril. J Alzheimers Dis 24(Suppl 2):33–42
Hamaguchi T et al (2009) Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-beta aggregation pathway. Am J Pathol 175(6):2557–65
Caesar I et al (2012) Curcumin promotes A-beta fibrillation and reduces neurotoxicity in transgenic Drosophila. PLoS One 7(2):e31424
Zhao LN et al (2012) The effect of curcumin on the stability of Abeta dimers. J Phys Chem B 116(25):7428–35
Baum L, Ng A (2004) Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J Alzheimers Dis 6(4):367–77, 443-9
Sato T et al (2006) Inhibitors of amyloid toxicity based on beta-sheet packing of Abeta40 and Abeta42. Biochemistry 45(17):5503–16
Balasubramanian K (2006) Molecular orbital basis for yellow curry spice curcumin’s prevention of Alzheimer’s disease. J Agric Food Chem 54(10):3512–20
Masuda Y et al (2011) Solid-state NMR analysis of interaction sites of curcumin and 42-residue amyloid beta-protein fibrils. Bioorg Med Chem 19(20):5967–74
Orlando RA et al (2012) A chemical analog of curcumin as an improved inhibitor of amyloid Abeta oligomerization. PLoS One 7(3):e31869
Streltsov VA et al (2008) The structure of the amyloid-beta peptide high-affinity copper II binding site in Alzheimer disease. Biophys J 95(7):3447–56
Lu Y et al (2010) Copper(I) and copper(II) binding to beta-amyloid 16 (Abeta16) studied by electrospray ionization mass spectrometry. Metallomics 2(7):474–9
Zhang X et al (2013) Design and synthesis of curcumin analogues for in vivo fluorescence imaging and inhibiting copper-induced cross-linking of amyloid beta species in Alzheimer’s disease. J Am Chem Soc 135(44):16397–409
Baur JA, Sinclair DA (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5(6):493–506
Huang TC et al (2011) Resveratrol protects rats from Abeta-induced neurotoxicity by the reduction of iNOS expression and lipid peroxidation. PLoS One 6(12):e29102
Jeon SY et al (2007) Beta-secretase (BACE1)-inhibiting stilbenoids from Smilax Rhizoma. Phytomedicine 14(6):403–8
Marambaud P, Zhao H, Davies P (2005) Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J Biol Chem 280(45):37377–82
Vingtdeux V et al (2010) AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem 285(12):9100–13
Chen J et al (2005) SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem 280(48):40364–74
Ge JF et al (2012) The binding of resveratrol to monomer and fibril amyloid beta. Neurochem Int 61(7):1192–201
Feng Y et al (2009) Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 30(6):986–95
Lu C et al (2013) Design, synthesis, and evaluation of multitarget-directed resveratrol derivatives for the treatment of Alzheimer’s disease. J Med Chem 56(14):5843–5859
Ladiwala AR et al (2011) Polyphenolic glycosides and aglycones utilize opposing pathways to selectively remodel and inactivate toxic oligomers of amyloid beta. Chembiochem 12(11):1749–58
Ansari MA et al (2009) Protective effect of quercetin in primary neurons against Abeta(1-42): relevance to Alzheimer’s disease. J Nutr Biochem 20(4):269–75
Pocernich CB et al (2011) Nutritional approaches to modulate oxidative stress in Alzheimer’s disease. Curr Alzheimer Res 8(5):452–69
Lu J et al (2010) Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J Pathol 222(2):199–212
Shimmyo Y et al (2008) Flavonols and flavones as BACE-1 inhibitors: structure-activity relationship in cell-free, cell-based and in silico studies reveal novel pharmacophore features. Biochim Biophys Acta 1780(5):819–25
Kim H et al (2005) Effects of naturally occurring compounds on fibril formation and oxidative stress of beta-amyloid. J Agric Food Chem 53(22):8537–41
Shimmyo Y et al (2008) Multifunction of myricetin on A beta: neuroprotection via a conformational change of A beta and reduction of A beta via the interference of secretases. J Neurosci Res 86(2):368–77
Akaishi T et al (2008) Structural requirements for the flavonoid fisetin in inhibiting fibril formation of amyloid beta protein. Neurosci Lett 444(3):280–5
Bartolini M et al (2011) Kinetic characterization of amyloid-beta 1-42 aggregation with a multimethodological approach. Anal Biochem 414(2):215–25
Berhanu WM, Masunov AE (2010) Natural polyphenols as inhibitors of amyloid aggregation. Molecular dynamics study of GNNQQNY heptapeptide decamer. Biophys Chem 149(1-2):12–21
Tay WM, da Silva GF, Ming LJ (2013) Metal binding of flavonoids and their distinct inhibition mechanisms toward the oxidation activity of Cu2+-beta-amyloid: not just serving as suicide antioxidants! Inorg Chem 52(2):679–90
DeToma AS et al (2011) Myricetin: a naturally occurring regulator of metal-induced amyloid-beta aggregation and neurotoxicity. ChemBioChem 12(8):1198–201
Fiori J et al (2012) Disclosure of a fundamental clue for the elucidation of the myricetin mechanism of action as amyloid aggregation inhibitor by mass spectrometry. Electrophoresis 33(22):3380–6
Lu JH et al (2011) Baicalein inhibits formation of alpha-synuclein oligomers within living cells and prevents Abeta peptide fibrillation and oligomerisation. ChemBioChem 12(4):615–24
Lemkul JA, Bevan DR (2010) Destabilizing Alzheimer’s Abeta(42) protofibrils with morin: mechanistic insights from molecular dynamics simulations. Biochemistry 49(18):3935–46
Zhao L et al (2013) Apigenin attenuates copper-mediated beta-amyloid neurotoxicity through antioxidation, mitochondrion protection and MAPK signal inactivation in an AD cell model. Brain Res 1492:33–45
Pitozzi V et al (2012) Long-term dietary extra-virgin olive oil rich in polyphenols reverses age-related dysfunctions in motor coordination and contextual memory in mice: role of oxidative stress. Rejuvenation Res 15(6):601–12
Farr SA et al (2012) Extra virgin olive oil improves learning and memory in SAMP8 mice. J Alzheimers Dis 28(1):81–92
Berr C et al (2009) Olive oil and cognition: results from the three-city study. Dement Geriatr Cogn Disord 28(4):357–64
Bazoti FN et al (2006) Noncovalent interaction between amyloid-beta-peptide (1-40) and oleuropein studied by electrospray ionization mass spectrometry. J Am Soc Mass Spectrom 17(4):568–75
Bazoti FN et al (2008) Localization of the noncovalent binding site between amyloid-beta-peptide and oleuropein using electrospray ionization FT-ICR mass spectrometry. J Am Soc Mass Spectrom 19(8):1078–85
Galanakis PA et al (2011) Study of the interaction between the amyloid beta peptide (1-40) and antioxidant compounds by nuclear magnetic resonance spectroscopy. Biopolymers 96(3):316–27
Diomede L et al (2013) Oleuropein aglycone protects transgenic C. elegans strains expressing Abeta42 by reducing plaque load and motor deficit. PLoS One 8(3):e58893
Grossi C et al (2013) The polyphenol oleuropein aglycone protects TgCRND8 mice against ass plaque pathology. PLoS One 8(8):e71702
Chen YP et al (2013) Syntheses and evaluation of novel isoliquiritigenin derivatives as potential dual inhibitors for amyloid-beta aggregation and 5-lipoxygenase. Eur J Med Chem 66:22–31
Tarozzi A et al (2010) Neuroprotective effects of cyanidin 3-O-glucopyranoside on amyloid beta (25-35) oligomer-induced toxicity. Neurosci Lett 473(2):72–6
Ono K et al (2004) Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer’s beta-amyloid fibrils in vitro. Biochim Biophys Acta 1690(3):193–202
Li G, Pomes R (2013) Binding mechanism of inositol stereoisomers to monomers and aggregates of Abeta(16-22). J Phys Chem B 117(22):6603–13
Mohamed T et al (2013) Tau-derived-hexapeptide 306VQIVYK311 aggregation inhibitors: nitrocatechol moiety as a pharmacophore in drug design. ACS Chem Neurosci 4(12):1559–70
Landau M et al (2011) Towards a pharmacophore for amyloid. PLoS Biol 9(6):e1001080
Jiang L et al (2013) Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. Elife 2:e00857
Eisenberg DS, et al (2013) Pharmacophores for amyloid fibers involved in Alzheimer’s disease. World Patent WO2013010176A2
Miller Y, Ma B, Nussinov R (2010) Polymorphism in Alzheimer Abeta amyloid organization reflects conformational selection in a rugged energy landscape. Chem Rev 110(8):4820–38
Sato M et al (2013) Site-specific inhibitory mechanism for amyloid beta42 aggregation by catechol-type flavonoids targeting the Lys residues. J Biol Chem 288(32):23212–24
Rao PPN et al (2015) Curcumin binding to beta amyloid: a computational study. Chem Biol Drug Des. doi:10.1111/cbdd.12552
Lipinski CA et al (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46(1-3):3–26
Cirrito JR et al (2003) In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci 23(26):8844–53
Ferri P et al (2015) Enhancement of flavonoid ability to cross the blood-brain barrier of rats by co-administration with alpha-tocopherol. Food Funct 6(2):394–400
Shu XH et al (2015) Diffusion efficiency and bioavailability of resveratrol administered to rat brain by different routes: therapeutic implications. Neurotherapeutics 12(2):491–501
Sinha S et al (2011) Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc 133(42):16958–69
Acknowledgments
This work is supported by the National Natural Science Foundation of China (grant numbers 81270423 and 81471296) for Y.-J.W. and NSERC-Discovery RGPIN 03830-2014, Mitacs-Canada, and Early Researcher Award from the Ministry of Research and Innovation, Government of Ontario, Canada, for P.P.N.R.
Conflict of interests
The authors declare no financial or other conflicts of interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Bu, XL., Rao, P.P.N. & Wang, YJ. Anti-amyloid Aggregation Activity of Natural Compounds: Implications for Alzheimer’s Drug Discovery. Mol Neurobiol 53, 3565–3575 (2016). https://doi.org/10.1007/s12035-015-9301-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9301-4