
Abstract
Alzheimer’s disease (AD) is a common neurodegenerative disease characterized by amyloid plaque and neurofibrillary tangles (NFT). With the finding that soluble nonfibrillar Aβ levels actually correlate strongly with the severity of the disease, the initial focus on amyloid plaques shifted to the contemporary concept that AD memory failure is caused by soluble Aβ oligomers. The soluble Aβ are known to be more neurotoxicthan fibrillar Aβ species. In this paper, we summarize the essential role of soluble Aβ oligomers in AD and discuss therapeutic strategies that target soluble Aβ oligomers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disease characterized by language breakdown, memory loss, and progressive cognitive impairment. The pathological features of AD are extracellular neuritic plaque composed of misfolded amyloid-beta (Aβ) peptides, intracellular neurofibrillary tangles (NFT) composed of hyperphosphorylated tau, and severe synaptic and neuronal loss. With the finding that soluble nonfibrillar Aβ levels actually correlate strongly with the severity of the disease [1] and synaptic loss [2], researchers’ attentions shifted away from the amyloid plaque, which correlates poorly with cognitive impairment [3, 4], to the soluble Aβ oligomers as possible culprits in the AD pathology. Here, we review the essential role of soluble Aβ oligomers in AD. Given the effects of soluble Aβ oligomers on AD, more therapeutic strategies targeting soluble Aβ oligomers are proposed.
What Are Soluble Aβ Oligomers?
Soluble Aβ oligomers are distinct from fibrillar Aβ aggregates. The term “soluble” refers to any form of Aβ that is soluble in aqueous buffer and remains in solution after high-speed centrifugation. Based on current knowledge, soluble Aβ oligomers include dimers, trimers, Aβ*56 (a putative dodecameric Aβ assembly, 56 kDa), spices immunoreactive to Aβ-derived diffusible ligands (ADDLs) (dodecamers, 35 ~ 60 kDa)/globulomers (dodecamers, 38 ~ 48 kDa) antibodies, and annular protofibrils (APFs) within the spectrum of endogenous assemblies. In both cases of ADDLs and globulomers, there have been some confusion that these “dodecamers” correspond to Aβ*56 [5]. However, several lines of evidence indicate that Aβ*56 and ADDLs/globulomers are different entities [6, 7]. In addition, APFs are pore-like structures that are believed to derive from the circularization of nonfibrillar Aβ assemblies, with a molecular size migrating at 100 ~ 150 kDa.
Production of Soluble Oligomeric Aβ Assemblies
The monomeric Aβ (molecular weight ~4 kDa) is produced in neurons by cleavage of the amyloid precursor protein (APP) (molecular weight ~120 kDa) by β-secretase [(identified to be β-site APP cleaving enzyme 1 (BACE 1))] and γ-secretase. γ-Secretase cleaves the C-terminal of APP at different positions, which leads to C-terminal heterogeneity of the monomeric Aβ and a variety of peptides. Aβ is a heterogeneous mixture of peptides that have different solubilities, stabilities, and biological and toxic properties. To our knowledge, Aβ monomers and trimers are produced and secreted by neurons. Using a large panel of Aβ antibodies, a model for the endogenous production of oligomeric Aβ assemblies was proposed. Aβ oligomers follow a trimer-based expansion in size under physiological conditions. In pathological conditions, monomeric Aβ becomes misfolded into dimers, which can rapidly expand to create dimer-based protofibrils, ultimately contributing to the formation of fibrillar Aβ as amyloid plaques [7].
The mechanism of formation of soluble Aβ oligomers in vivo remains unclear [7] (Fig. 1). The mechanisms of formation might be different for extracellular and intracellular oligomers. The formation of extracellular soluble Aβ oligomers can be induced by GM1 ganglioside, which is formed via a pathway distinct from that of fibril formation [8–10]. Furthermore, nonfibrillar Aβ can be produced in the presence of aB-crystallin [11] and clusterin (also known as ApoJ) [12]. With regard to intracellular soluble Aβ oligomers, Aβ can be localized intracellularly by the uptake of extracellular Aβ or by the cleavage of APP in endosomes generated from the endoplasmic reticulum (ER) or the Golgi apparatus. The uptake of extracellularly produced Aβ oligomers occurs via endocytic pathways or various receptors and transporters such as formyl peptide receptor-like protein 1 (FPRL1) or scavenger receptor for advanced glycation end-products (RAGE). Aβ in the lysosome can leak into the cytosol and form soluble oligomers via the interaction with intracellular proteins such as GM1 [10], prefoldin (PFD) [13] or other molecular chaperones. PFD was reported to prevent further aggregation of Aβ oligomers and stabilize the oligomer structure.

Formation and toxicity mechanisms of intracellular and extracellular Aβ oligomers. Aβ can be localized intracellularly by the uptake of extracellular Aβ or by the cleavage of APP in endosomes generated from the endoplasmic reticulum (ER) or the Golgi apparatus. The uptake of extracellularly produced Aβ oligomers occurs via endocytic pathways or various receptors. These receptor–Aβ complexes are internalized into early endosomes. Although most Aβ in the endosome is degraded, some Aβ in the lysosome can leak into the cytosol and form soluble oligomers via the interaction with intracellular proteins such as GM1, PFD, or other molecular chaperones. Extracellular Aβ is released as a product of proteolytically cleaved, plasma membrane-localized amyloid precursor protein (APP). The formation of extracellular soluble Aβ oligomers can be induced by GM1 ganglioside. Soluble Aβ oligomers may bind with high affinity and specificity to a single receptor type, which then indirectly or directly initiates distinct aberrant downstream signaling cascade leading to different cellular dysfunction. Furthermore, the membrane pore is formed by Aβ oligomers. The pores allow abnormal Ca2+ influx. Also, ER stress that caused by induces Ca2+ release from the ER. The dysregulation of Ca2+ homeostasiscan damage mitochondria and cause oxidative stress
It is important to point out that ADDLs/globulomer are synthetic Aβ molecules and that the origins of the endogenous Aβ species detected by ADDLs/globulomer antibodies are completely unknown. Further studies are needed to determine how these particular Aβ assemblies are produced as well as how and when they form in AD.
The Oligomers Hypothesis
Due to poor correlation between amyloid plaque load and cognitive function, the original amyloid cascade hypothesis of AD has been substituted by the oligomer hypothesis. The oligomer hypothesis attributes dementia to nerve cell death caused by soluble Aβ oligomers, which are the disease-initiating factor and the true culprits of AD [14]. Despite strong evidence supporting the amyloid hypothesis, studies still show that small oligomers can diffuse into synaptic clefts and present much more Aβ surface area to neuronal membranes than large fibrillar plaques [7]. Moreover, synapse loss does not require the presence of amyloid deposits [15] and the antibodies against Aβ can restore memory loss without the removal of amyloid plaques [16]. More eloquent evidence was obtained from the toxicology study associated with clusterin, a protein which prevents amyloid formation and promotes oligomer formation. Surprisingly, clusterin-Aβ solutions seemed to be more cytotoxic than amyloid-containing controls [17]. Therefore, the researchers’ attention has shifted away from amyloid plaque to small soluble oligomers of the Aβ peptides as the initiating toxic agents of AD. It naturally follows that the straightforward therapeutic methods of clearing away fibrils have been changed to focus on blocking the soluble Aβ oligomers.
The Essential Role of Soluble Aβ Oligomers in AD
The Role of Soluble Aβ Oligomers Associated with SPs
Amorphous deposits of soluble Aβ with an α-helix structure can evolve into fibrillar forms with an insoluble β-pleated sheet, and finally form dense amyloid cores placed centrally in the senile plaque (SP) [18]. As the histologically pathologic hallmark of AD, SPs may be a source of soluble oligomers in the untreated AD brains [19, 20]. Consistent with this, a mouse model showed that soluble oligomers are found in high concentration adjacent to dense plaques, with a decline in the concentration of oligomers as the distance from the plaques increases [21]. Moreover, the fact that the volume of plaques keeps stable rather than increases as the disease progresses indicates the conversion of insoluble Aβ in dense plaques back to a soluble form of Aβ and its release into the surrounding brain [22]. Also, the shift of insoluble Aβ in the plaque to soluble Aβ in the extracellular space may be one explanation of the fact that the so-called remnant plaques increase in number but contain a reduced amount of Aβ with the disease duration [23]. These results support the view that there is a dynamic balance between amyloid deposition and soluble Aβ oligomers.
The Role of Soluble Aβ Oligomers Associated with Pathologic Tau
Soluble Aβ oligomers have tight ties to pathologic tau. Recently, the researchers proposed a novel action model in which Aβ is the trigger of the cellular events leading to dementia, with tau acting as a mediator for AD-related synaptic deficits [24]. Soluble oligomers are a stimulus for pathologic tau formation, with the latter leading to neuronal death.
A recent study showed that Aβ trimers and Aβ*56 levels increased with age and were elevated in subjects with total tau (T-tau)/Aβ1–42 ratios greater than a cutoff that distinguished the cognitively intact group from subjects with AD. In the cognitively intact group, T-tau and phospho-tau (p-tau) were found to have a strong correlation with Aβ trimers and Aβ*56 and the strong correlations were found to be attenuated in the AD group. These findings suggest that one or both of the Aβ oligomers is coupled to tau, but this coupling is weakened or broken when AD progesses [25]. However, another study observed that Aβ*56, instead of dimers ortrimmers, correlated with pathological tau proteins [26]. In addition, an in vivo study evidenced that the soluble oligomeric forms of short fragments of Aβ, endogenously identified in AD patient brains, may also directly contribute to the progressive increase in amyloid load and tau pathology [27].
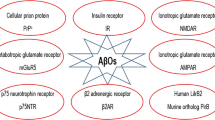
Tau is typically localized to the axons of neurons. The research has demonstrated that soluble Aβ oligomers cause mislocalization of tau to dendrites and that only neurons demonstrating mislocalization display decreased spine density [28]. Furthermore, this mislocalization is tau phosphorylation dependent [29]. Recent findings also indicate that some phosphorylation sites are also necessary for soluble Aβ oligomers to induce tau mislocalization to dendritic spines, impairing synaptic function [30]. Soluble Aβ oligomers are able to cause phosphorylation of tau in vivo [31–33] and in vitro [34]. Using Western blotting and ELISA methods, oligomeric Aβ attaches to tau protein to form a stable complex, which enhances tau phosphorylation by glycogen synthase kinase-3 (GSK3), but the phosphorylation then promotes dissociation of the complex interaction with tau. Therefore, a hypothesis was proposed that in AD, an initial step in the pathogenesis may be the intracellular binding of soluble Aβ to soluble nonphosphorylated tau, thus promoting tau phosphorylation and Aβ nucleation [35]. By some unknown but probably interrelated mechanisms, soluble Aβ oligomers can activate GSK3β and other kinases that are responsible for the hyperphosphorylation of tau [36]. GSK3β activity is regulated by multiple mechanisms; thus, the researchers focused on the major upstream regulatory systems of GSK3β. Cedazo-Mínguez et al. showed that Aβ1–42 effects on GSK3β were biphasic with a strong activation dependent partially on extracellular Ca2+ followed by an inactivation. GSK3β is inactivated by phosphorylation of serine 9 (Ser-9). Protein kinase B (PKB) and protein kinase C (PKC) had been found to mediate this event. Aβ1–42 induced an early and potent activation of PKC-α and a late decrease of PKB activity [36]. Jonathan Brouillette et al. gave evidences that the activation of the caspase-3 could be part of the molecular mechanism by which soluble Aβ1–42 oligomers induce tau hyperphosphorylation in vivo and in vitro [31]. Tokutake et al. also indicated that the mechanism underlying hyperphosphorylation of tau induced by naturally secreted Aβ at nanomolar concentrations was modulated by insulin-dependent Akt-GSK3 signaling pathway because they demonstrated that the phosphorylationof Akt and GSK3 upon insulin stimulation was less activated under that condition [32]. Besides GSK3, exposure of cultured wild-type neurons to Aβ oligomers could cause activation of the nonreceptor tyrosine kinase (nRTK), fyn, the cAMP-regulated protein kinase A (PKA), and calcium-calmodulin kinase II (CaMK II), which respectively phosphorylated tau on Y18, S409, and S416 [37]. Also, the partitioning defective-1 (PAR-1)/microtubule affinity-regulating kinase (MARK) family kinases act as critical mediators of Aβ toxicity, and overexpression of MARK4 leads to tau hyperphosphorylation [38]. Several steps in the pathway between Aβ oligomers and tau mislocalization are currently unknown. One possible mechanism is through a signaling pathway whereby Aβ initiates dysfunction through cell surface receptors on the axons of neurons. A probable receptor for Aβ oligomers is cell prion protein (PrPC) [39] (Fig. 1). Soluble Aβ binds to PrPC at neuronal dendritic spines in vivo and in vitro where it forms a complex with fyn, resulting in the activation of the kinases [40]. Additionally, one recent study has shown a correlation between the mislocalization of tau to dendritic spines and the necessity of calcineurin for soluble Aβ oligomer-induced decreases in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor signaling [30].
The Role of Soluble Aβ Oligomers in the Impairment of Synapse Dysfunction Associated with Memory Loss
Soluble Aβ oligomers can influence the important neurotransmitter glutamate in the synaptic cleft. Soluble Aβ oligomers can also inhibit long-term potentiation (LTP) [41] and facilitate long-term depression (LTD) of excitatory synaptic transmission [42], both of which engage synaptic plasticity mechanisms believed to underlie certain types of learning and memory [43–45]. These findings indicate that soluble Aβ oligomers acutely interfere with normal synaptic functions and contribute significantly to the early memory loss and cognitive dysfunction characteristic of AD.
Soluble Aβ Oligomers Influence Glutamate Concentrations in the Synaptic Cleft
Glutamate is the major fast excitatory neurotransmitter in cortical and hippocampal regions. Consistent with the involvement of the glutamatergic system in learning and memory, disturbances in glutamate neurotransmission have been linked with the pathophysiological processes underlying AD. Glutamate is removed from the extracellular space by transporters in the plasma membrane of astrocytes and neurons. Soluble Aβ oligomers may increase availability/residence of glutamate in the synaptic cleft through inhibition, and even reversal, of uptake mechanisms, or enhancement of release mechanisms; thus, they increase glutamate concentrations in the synaptic cleft.
The possible mechanisms for inhibitory effect on glutamate uptake are associated with oxidative damage induced by soluble Aβ oligomers in AD. Firstly, glutamate transporters are vulnerable to oxidants resulting in reduced uptake function. For example, a major lipid peroxidation product, 4-hydroxynonenal (HNE), which is formed upon incubation of cultured hippocampal neurons with AD, binds to GLT-1, a glial glutamate transporter, and inhibits glutamate uptake [46, 47]. Secondly, because the high-affinity uptake of glutamate by astrocytes depends on Na+ concentration gradients across the astrocytic plasma membrane, oxidative damage from soluble Aβ oligomers impairs Na/K-ATPase activity which precedes loss of calcium homeostasis and neuronal degeneration, and ultimately disrupts glutamate uptake [48]. Different from the possible mechanisms for inhibitory effect on glutamate uptake and less well established, a possible mechanism for the enhancement of release is that soluble Aβ oligomers promote abnormal release of glutamate from hippocampal neurons, accompanied by an increase in postsynaptic activity. This was supported by the result taken from Shi et al. showing that the blockade of glutamate receptors prevents glutamate release prompted by Aβ oligomers [49].
However, soluble Aβ oligomers can also have the opposite effect on glutamate levels. It is reported that Aβ1–42 (20 mM) incubated for 12 ~ 48 h increased glutamate uptake activity in primary cultures of rat cortical astrocytes and neurons [50]. This effect was associated with an increase in cell surface expression of GLT. However, it should be considered a possibility that this increased uptake/expression was a secondary protective mechanism of cells surviving toxicity [51].
Glutamate signal transduction at the postsynaptic terminal is initiated by stimulation of glutamate receptors, especially of the N-methyl-d-aspartate (NMDA) subtype. The binding of glutamate to NMDA receptor initiates an influx of Ca2+, which can cause neuronal intracellular Ca2+ overload due to the prolonged stimulation of the NMDA receptor. The dysfunction of calcium hemostasis results in an activation of a complex cascade of enzymes, messengers, and intracellular reactive oxygen species (ROS) that lead to cell death. Therefore, the different effects of toxic Aβ on glutamate transport mechanisms are likely contributors to excitotoxicity and the neuronal degeneration observed in AD.
Soluble Aβ Oligomers impair LTP
Soluble Aβ oligomers from different sources (cells, transgenic mouse brains, human brains, or synthetic peptides) can rapidly and potently impair glutamatergic synapses and LTP. As noted previously, it was demonstrated in the rat hippocampus that insoluble plaque cores from human brains did not impair LTP unless the cores were first solubilized to release Aβ dimers [52]. Moreover, Aβ oligomers (1 ~ 50 nM) concentration-dependently blocked LTP, and so far, strong effects have been observed at the lowest concentration of Aβ oligomers tested (1 nM) [53]. Remarkably, at subnanomolar doses, the effect on LTP is reversed, and Aβ oligomers markedly enhance LTP.
The precise soluble Aβ oligomer mechanisms underlying synaptic toxicity and memory failure are complex and not fully understood. It is clear that there is no agreement about what mechanism, if any, is most relevant to AD. What can be agreed upon is that soluble Aβ oligomers either directly or indirectly (through the accumulation of extracellular glutamate and excessive stimulation of extrasynaptic receptor) alter extrasynaptic receptor-dependent cascades, thereby leading to LTP impairment. The pathways that have been invoked in different experimental paradigms to explain LTP impairment induced by soluble Aβ oligomers are illustrated in Fig. 2 [60]. In addition, Arancio and colleagues discovered in a detailed analysis that enhancement of LTP occurs over a narrow range of Aβ oligomer concentrations and is associated with presynaptic activation of alpha7 nicotinic acetylcholine receptors (α7-nAchR) [61].

The pathways that have been invoked in different experimental paradigms to explain LTP impairment induced by soluble Aβ oligomers are illustrated in the figure. Soluble Aβ oligomers directly or indirectly alter glutamate-receptor-dependent cascades, thereby leading to LTP impairment. Aberrant activation of calcineurin and NFATc4 leads to the dystrophic changes in neuritis, and calcineurin-dependent dephoshorylation of CaMKII impairs the induction of AMPAR-based LTP in hippocampus. This Aβ oligomer-mediated LTP impairment involves PP1-dependent mechanisms [54]. Abnormal calcineurin hyperactivity can oppose LTP and memory function via dephosphorylative inactivation of CREB. The p38MAPK activation, which was followed by downregulation of CREB protein, was also involved in the Aβ-mediated LTP impairment [55]. Soluble Aβ oligomers also induce LTP impairment via reduced dynamin 1 and PSD-95 protein mediated by the activation of caplain. mGluR5 is involved in the blockage of LTP via activation of c-JNK, cdk5, and p38MAPK [56]. Soluble Aβ oligomers cause the hyperactivation of α7-nAchR, and then downregulation of the ERK2 (MAPK1) cascade leading to an impairment of LTP through decreased phosphorylation of CREB factor [57]. Fyn-dependent signals emanating from the Aβ oligomers: PrPC receptor complex result in the phosphorylation NR2B NMDA receptor subunit, resulting in altered cell surface NR2B expression. LilrB2 may interact with soluble Aβ oligomers, then enhanced cofilin signaling and cause LTP impairment [58]. Aβ-dependent inactivation of the JAK2/STAT3 axis disrupts LTP through cholinergic dysfunction [59]. The caspase-3-mediated pathway might have a role in the Aβ-mediated LTP impairment upstream of GSK3β. The inhibition of the insulin signaling is also related with LTP impairment. mGluR5 metabotropic glutamate receptor 5, ERK2 extracellular signal-regulated kinase 2, MAPK mitogen-activated protein kinase, JNK c-Jun N-terminal kinase, Cdk5 cyclin-dependent kinase 5, GSK3β glycogen synthase kinase-3β, Akt1 serine-threonine protein kinase 1, NMDAR N-methyl-d-aspartate receptor, AMPAR α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate receptor, CaMKII calmodulin kinase II, PKA protein kinase A, PP1 protein phosphatase 1, NFATc4 nuclear factor of activated T-cells, α7-nAChR α7 nicotinic acetylcholine receptor, BDNF brain-derived neurotrophic factor, CREB cAMP-regulatory element binding protein, LilrB2 leukocyte immunoglobulin-like receptor B2, JAK2/STAT3 Janus kinase 2/signal transducer and activator of transcription 3
Soluble Aβ Oligomers Facilitate LTD
Disruption of functional plasticity includes LTD, which is promoted, not inhibited. The mechanisms underlying Aβ-facilitated LTD have not yet been fully elucidated. NMDAR activation can induce either LTP or LTD, depending on the extent of the resultant [Ca2+]i rise in the dendritic spines and the downstream activation of specific intracellular cascades [62]. Activation of synaptic NMDARs and large increases in [Ca2+]i are required for LTP, whereas internalization of synaptic NMDARs, activation of perisynaptic NMDARs, and lower increases in [Ca2+]i are necessary for LTD. As mentioned below, soluble Aβ oligomers may indirectly cause a partial block of NMDARs and shift the activation of NMDAR-dependent signaling cascades toward pathways involved in the induction of LTD, which is consistent with the observation that soluble Aβ oligomers inhibit LTP and enhance LTD. Soluble Aβ oligomers may also enhance LTD by blocking glutamate uptake at synapses, leading to increased glutamate levels at the synaptic cleft, which initially activate synaptic NMDARs followed by desensitization of the receptors [63]. Another consequence of a rise in glutamate levels would be a spillover and activation of perisynaptic NR2B-enriched NMDARs [64]. In light of this, Aβ-induced LTD-like processes may underlie Aβ-induced LTP deficits.
What Are the Receptors by Which Soluble Aβ Oligomers Perturb Synapse Dysfunction Associated with Memory Loss?
Soluble Aβ oligomers may bind with high affinity and specificity to a single receptor type, which then indirectly (interacts with other synaptic proteins) or directly initiates distinct aberrant downstream signaling cascade leading to synaptic malfunction. There are various receptors proposed to act as oligomeric Aβ receptors [65], including AMPAR, NMDAR, metabotropic glutamate receptors (mGluRs), ephrin type B receptor 2 (EphB2), RAGE, a7nAChR, PrPC, insulin receptor (IR), TNF receptor, leukocyte immunoglobulin-like receptor B2 (LilrB2) [58], and so on. We will summarize the main receptors: AMPAR, NMDAR, and PrPC.
AMPAR
Soluble Aβ oligomers can cause synaptic removal of AMPAR [66–68]. The trafficking and activity of AMPAR are modulated by its phosphorylation. Phosphorylation of GluA1 at Ser-845 contributes specifically to the recruitment of new AMPAR into extrasynaptic sites from the synapsis. Soluble Aβ oligomers can induce GluA1 dephosphorylation and reduce receptor surface levels, which are mediated by an increase in calcium influx into neurons through ionotropic glutamate receptors and activation of the calcium-dependent phosphatase calcineurin, which subsequently activate protein phosphatase 1 (PP1). Overactivation of PP1 dephosphorylates CaMKII ultimately causes the dephosphorylation of AMPAR [69]. Correspondingly, AMPAR-mediated synaptic response and ionic currents were inhibited in APP transgenic mice and in cultures treated with Aβ oligomers. Also, the researchers found that the synaptic pool of CaMKII was significantly decreased in cortical neurons from APP transgenic mice, and the density of CaMKII clusters at the synapses was significantly reduced by Aβ oligomer treatment. Furthermore, CaMKII synaptic translocation by Aβ relies on mechanisms involving Ca2+/CaM [70] and F-actin [71] which perhaps alters intracellular calcium signaling and actin.
NMDAR
Synaptic plasticity is disturbed by soluble Aβ oligomers through Aβ-mediated internalization of NMDAR [72]. This internalization of NMDAR requires the activation of α7-nAchR [73]. As mentioned previously, Aβ-induced synaptic depression may result from an initial increase in synaptic activation of NMDARs by glutamate, followed by synaptic NMDAR desensitization, NMDAR internalization, and activation of extrasynaptic NMDARs. Soluble Aβ oligomers were shown to inhibit NMDAR-dependent LTP as a result of overactivation of extrasynaptic NR2B-containing receptors. This effect could be mitigated by reducing extracellular glutamate levels, and the action manner of extrasynaptic NR2B receptor is similar to that of a glutamate reuptake inhibitor [60, 74]. Moreover, aberrant activation of calcineurin and nuclear factor of activated T cells (NFATc4) lead to the dystrophic changes in neurites, and calcineurin-dependent dephoshorylation of CaMKII impairs the induction of AMPAR-based LTP in the hippocampus. The p38 mitogen-activated protein kinase (p38MAPK) activation, which was followed by downregulation of cAMP response element-binding (CREB) protein, was also involved in the Aβ-mediated LTP impairment. Based on the findings about how soluble Aβ oligomers work in synaptic failure and memory impairment, it appears that upon binding with a putative membrane receptor, Aβ activates a molecular cascade that leads to calpain activation and degradation of important proteins involved in synaptic plasticity as well as learning and memory. The postsynaptic density protein 95 (PSD-95) is a postsynaptic scaffolding protein that stabilizes the AMPARs and NMDARs at synapses to establish synaptic plasticity [67]. Reductions in the PSD-95 protein levels were even shown in cultured cortical neurons [67] and in rat hippocampal neurons [75] after exposure to soluble Aβ oligomers which involved NMDAR activity and cyclin-dependent kinase 5 activity. Moreover, dynamin 1, a protein essential for synaptic vesicle recycling and the functioning of the synapse, was degraded by soluble Aβ oligomers due to calpain activation induced by the sustained calcium influx mediated by NMDA receptors in hippocampal neurons [76].
Texidó and coworkers have shown that Aβ oligomers directly interact with and activate NMDARs [77]. Interestingly, NMDARs do not appear to be sufficient for oligomer binding. AβO-attacked and AβO-spared neurons exhibit similar levels of surface NMDARs. This suggests that the process through which Aβ oligomers bind to synapses may involve the NMDAR-promoted assembly of a multiprotein receptor complex that function as a receptor for oligomers. A strong candidate to integrate such a multiprotein receptor complex for Aβ oligomers is PrPC [78].
PrPC
In 2009, PrPC was identified in a genome-wide screen as a high-affinity receptor for Aβ oligomers. The amino-terminal half (aa 95–110) in PrPC is responsible for Aβ oligomer binding PrPC [79]. Of note, Chen et al. even identified a cluster of basic residues at the extreme amino-terminus (aa 23–27) as critical for the interaction [80]. Moreover, PrPC recognizes Aβ oligomers in a size- and conformation-dependent manner.
Synthetic Aβ oligomer-mediated inhibition of LTP at hippocampal synapses in vitro was reported to be dependent on PrPC [81] (Fig. 1). Aβ oligomers, but not monomers or fibrils, potently and selectively bind specific regions of PrPC, especially in the vicinity of amino acids 95–105 [80–82]. Antibodies that bind PrPC within the region of 93–109 or 93–102 prevent the inhibition of hippocampal LTP by synthetic Aβ1–42 oligomers in vitro. Consistent with these reports, the in vivo synaptic plasticity disrupting actions of AD brain extracts containing water-soluble Aβ were dependent on PrPC [83]. Therefore, pre-injection of D13, an antigen-recognizing antibody fragment (Fab) that binds selectively to PrP96–104 C, completely abolished oligomeric Aβ-induced inhibition of LTP, indicating that Aβ oligomers can interact with PrPC to alter LTP. Interestingly, an antibody to the alpha helix of PrPC, which does not overlap with the putative binding site of Aβ oligomers, also prevented the inhibition of LTP by AD brain Aβ oligomers both in vitro and in vivo [82]. The researchers speculated that the antibody to the alpha helix was interfering with the interaction between PrPC. In addition, abnormal cross-linking of PrPC by cell-derived low-n oligomers of Aβ may contribute to synaptic damage in cultured neurons [84]. On the other hand, the property of the interaction between Aβ oligomers and PrPC can be used for treatment. A recent study showed that N1, the main physiological cleavage fragment of PrPC, is necessary and sufficient for binding early oligomeric intermediates during Aβ polymerization. The ability of N1 to bind Aβ oligomers is influenced by positively charged residues in two sites (23–31 and 95–105). These data suggest that N1, or small peptides derived from it, could be a potent inhibitor of Aβ oligomer toxicity and represents an entirely new class of therapeutic agents for AD [85].
What are the main signal transduction responses triggered by the binding of Aβ oligomers to PrPC in postsynaptic density of dendritic spine? Glutamate receptors are central to learning and memory; thus, the potential role of glutamate receptors in the context of PrPC and Aβ oligomer toxicity has stirred researchers’ interest [81, 86–88]. Using Xenopus laevisoocytes that overexpressed distinct ion channels, the researchers observed that neither AMPA (GluR1–4) nor NMDA (NR1 + NR2B or NR1 + NR2D) receptors are directly inhibited by Aβ oligomers in the presence of PrPC, suggesting that any functional connection between PrPC and glutamate receptors may be indirect or require additional molecular components that are not present in Xenopus oocytes [81]. Um et al. noted that in certain cell lines and neuronal cultures, Fyn-dependent signals emanated from the Aβ oligomer: PrPC receptor complex resulted in the phosphorylation of tyrosine at position 1472 of the NR2B NMDA receptor subunit, and this occurred along with altered cell surface NR2B expression and altered calcium signaling [87]. Furthermore, mGluR5 was identified as the linking PrPC to Fyn. Activation of neuronal Fyn requires both mGluR5 and PrPC. Aβ oligomers-PrPC can drive mGluR5-dependent calcium mobilization and eEF2 phosphorylation [89]. Notably, the ∆CR form of PrPC sensitizes neurons to glutamate-induced excitotoxicity, and the binding of Aβ oligomers to PrPC may functionally mimic the ∆CR form of PrPC [90]. Conversely, there are some studies showing contradictory findings that Aβ oligomers impaired LTP independently of PrPC [91]. The explanations for this controversy require further studies.
Soluble Aβ Oligomers, Synaptic Dysfunction, and Memory Loss Are Strongly Interrelated
LTP induction promotes recruitment of AMPARs and growth of dendritic spines, whereas LTD induces spine shrinkage and synaptic loss [62]. Hence, soluble Aβ oligomers impair synaptic function and memory. Moreover, in a study of people with AD, soluble Aβ1–40 correlated best with synaptic degeneration [2]. The amount of soluble Aβ was also a good measure of AD severity [92]. In a mouse model, there was a 60 % loss of excitatory synapses in the immediate vicinity of oligomers and a steep decline in this loss as the concentration of oligomers decreased [21]. Synaptic loss or dysfunction plays an important role in cognitive decline that parallels the accumulation of soluble Aβ oligomers [93]. In a transgenic mouse model, diffuse and soluble deposits of Aβ resulted in significant impairment of spatial memory [94]. The soluble Aβ from human brains even disrupted memory in normal rats [52]. All in all, soluble Aβ oligomers, synaptic dysfunction, and memory loss are strongly interrelated.
The Role of Soluble Aβ Oligomers in Oxidative Stress
Oxidative stress plays a significant role in the pathogenesis of AD; the AD brain is closely linked to extensive oxidative stress [95]. In the ATP generation, up to 2 % of electrons are incompletely reduced to yield superoxide (O2 −•) instead of H2O because oxidative phosphorylation (OxPhos) is not totally efficient. O2 −• can also combine with nitric oxide (NO) to yield peroxynitrite, a reactive nitrogen species (RNS). The redox balance is highly regulated in the healthy human brain. The molecules involved in antioxidant defense mechanisms such as glutathione and superoxide dismutase (SOD) keep the redox balance in the brain. Of note, there are also relatively high-concentration redox-active metal ions such as iron (Fe) and copper (Cu) which facilitate the conversion of H2O2 to other potent ROS [65, 96]. In AD brains, the levels of the antioxidants are significantly reduced, and the levels of lipid peroxidation (e.g., 4-hydroxynonenal (HNE)) and products of oxidative damage to DNA (e.g., 8-hydroxy-2-deoxyguanosine), and of lipid peroxidation (e.g., HNE) are elevated. Lipid peroxidation is correlated with brain degeneration [97]. Soluble Aβ oligomers are able to induce oxidative stress in AD [98, 99], and the possible mechanisms are reviewed as follows. Accordingly, some antioxidants are being considered as possible candidates for the treatment of AD [100–103].
Aβ was demonstrated to be a pro-oxidant in in vitro [104]. The ability of Aβ in aqueous solution to generate free radicals has even been shown in cell-free media. The capacity of Aβ to enhance generation of reactive oxygen species in isolated cells of neural origin has also been reported [105–107]. The mechanism by which such free radicals are produced by Aβ acting on neural tissues may be mediated by the generation of NO [108]. Moreover, as a pro-oxidant, free radical transfer reactions via oxidation of the sulfur atom of methionine residue 35 in Aβ1–42 or shorter fragments are critical to the free radical toxicity of Aβ [109]. Considering that transition metals including Fe and Cu in excess may be involved in chemical reactions that form ROS, the researchers reported that Aβ, in combination with the redox-active metals Fe and Cu, facilitated their reduction and the consequent pro-oxidant activity of these Aβ metal ion complexes [110]. In contrast, an in vitro study showed that Zn2+ quenched Aβ-Cu2+ complexes, which generated an antioxidant function [111].
Aβ can also induce oxidative stress through other indirect mechanisms. It has been reported that soluble Aβ induces mitochondrial dysfunction and oxidative damage. In APP transgenic mice, hydrogen peroxide levels were found to be significantly increased and directly correlated with levels of soluble Aβ, and cytochrome-c oxidase (also known as COX IV) activity was found to be decreased [112]. Aβ seems to aim at COX IV in the oxidative phosphorylation system (OXPHOS) and even the deregulation of COX IV was Aβ-dependent [113]. Aβ produced by APP processing in mitochondria or uptaken out of mitochondria can dysregulate the COX IV function and then injure the electron transport chain (ETC). Injured ETC produces ROS pathologically [114]. Aside from directly acting as a mitochondrial toxin, Aβ can damage mitochondria indirectly via excessive intraneuronal Ca2+ levels. Aβ oligomers are postulated to disrupt NMDA receptor activity, causing an excessive Ca2+ influx intoadjacent neurons, which makes ETC inefficient and increases ROS production. Moreover, neuronal nitric oxide synthetase (nNOS), which can produce NO, is also activated by NMDAR-mediated Ca2+ influx [115].
In addition, intracellular cysteine is rate-limiting for the synthesis of the antioxidant glutathione (GSH), and soluble Aβ oligomers could inhibit the excitatory amino acid transporter 3 (EAAT3)-mediated cysteine uptake, causing a decrease in redox regulation as well as intracellular cysteine and GSH levels [99]. Furthermore, aging makes synapses more vulnerable to soluble Aβ oligomers’ toxicity. Age is also related to decline in mitochondrial activity, reduced antioxidant contents, and increased oxidative stress markers in resting and depolarized synaptic terminals [116].
The Role of Soluble Aβ Oligomers in Neuroinflammation in AD
The brain inflammatory process has a fundamental role in the pathogenesis of AD. Aβ deposition activates a potentially pathological innate immune response in AD. Inflammation is a response to eliminate the initial cause of cell injury as the necrotic cells and tissues resulting from the original insult. If tissue health is not restored within a short time frame, inflammation becomes chronic and continues to damage surrounding tissues. Aβ plaques and tangles stimulate a chronic inflammatory reaction to clear this debris [117]. Actually, soluble Aβ oligomers are demonstrated to be the most effective pro-inflammatory mediator [118].
Aβ fibrils are not the only species responsible for microglial activation, as soluble Aβ oligomers are also able to trigger microglial activation. Aβ oligomers as abnormal, misfolded proteins present themselves to microglial cells, which are armed with a vast array of pattern recognition receptors (PRR) that can specifically recognize Aβ, including Toll-like receptors 2 (TLR2) and 4, formyl peptide receptors (FPRL1) and receptor for RAGE [119]. Upon interaction with PRR, Aβ oligomers can activate microglia [120]. Soluble Aβ oligomers have been shown to stimulate microglial secretion of pro-inflammatory cytokines and free radicals, which may all cause neurotoxicity in AD. A nuclear factor-kappaB (NFκB)-dependent pathway is required for soluble Aβ oligomers to produce cytokine [121]. Compared to fibrillar Aβ, soluble Aβ oligomers exert more potent effects on the expressions of inflammatory factors TLR4 and TNF-α and activation of NF-κB signaling, indicating that soluble Aβ oligomers are more neurotoxic than fibrillar Aβ [122]. Additionally, soluble Aβ oligomers also induce the activation of astrocytes, which are responsible for the reaction activating inflammatory mediators [123–125]. Recently, a polyphenol compound rutin that could inhibit Aβ aggregation was shown to significantly decrease oligomeric Aβ level, downregulate microgliosis and astrocytosis, decrease interleukin (IL)-1 and IL-6 levels in the brain, and attenuated memory deficits in AD transgenic mice [103].
In turn, inflammatory mediators and stress conditions enhance APP production and its amyloidogenic processing to generate Aβ [126]. Astrocytes are known to be important for Aβ clearance and degradation. Under certain conditions related to chronic stress, however, the role of astrocytes may not be beneficial. A report suggests that activated astrocytes can also be a source for Aβ, because they overexpress β-secretase of APP (BACE1) in response to chronic stress [127]. Thus, Aβ induces expression of pro-inflammatory cytokines in glia cells in a vicious cycle. In particular, activated glial cells in AD brains may produce excessive superoxide through NADPH oxidase [128].
The Role of Soluble Aβ Oligomers in Cholinergic Dysfunction
Acholinergic hypothesis of AD was proposed nearly 30 years ago [129], and there is general consensus that dysregulated cholinergic signaling is an early hallmark of AD [130, 131]. Soluble Aβ oligomers have been found to be involved in the cholinergic dysfunction.
Soluble Aβ oligomers can induce a major reduction in choline acetyltransferase (ChAT) activity in the absence of changes in neuronal viability or in total ChAT expression, whereas other elements of the cholinergic synapse remain unaffected. This ChAT inhibition was determined to be clearly dependent on glutamate receptors, particularly of the NMDA subtype. Oxidative damage to the enzyme instigated by Aβ oligomers though NMDA receptors can possibly explain this effect. Moreover, antioxidant polyunsaturated fatty acids can fully block ChAT inhibition [132].
There is considerable evidence to support the hypothesis that the interaction of soluble Aβ oligomers with different subtypes of nAchRs has a significant role in AD pathogenesis (Fig. 1). Moreover, soluble Aβ oligomers are reported to create a variety of effects on nAchRs, indicating that there are complex mechanisms involved in the Aβ modulation of nAchR function. Some studied were able to confirm the high-affinity binding of Aβ to α7-nAchRs [133–135], which might result in Aβ internalization [136]. Picomolar concentrations of Aβ have been observed to act as an agonist at α7-nAchRs [137, 138] and mediate activation of the ERK2-mitogen-activated protein kinase (MAPK) signaling cascade [137, 139]. However, nanomolar concentrations of Aβ cause functional antagonism of both human and rathomomeric α7-nAchRs, which might be due to inhibition of presynaptic nAchRs, resulting in changes in presynaptic Ca2+ levels [140–142]. Soluble Aβ42 is also capable of modulating postsynaptic transmission by activating presynaptic α4β2 nAchRs in septohippocampal neurons of the basal forebrain, an important pathway for learning and memory [143]. In addition, selective high-affinity interactions between pathologically relevant concentrations of soluble Aβ oligomers and α7β2-nAchRs, a novel subtype, could contribute to deficits in cholinergic signaling that could occur early in the etiopathogenesis of AD [144]. These different findings might depend on a number of important factors, such as the nAchR subtype being activated, and these receptors could serve as potentially useful therapeutic targets in this condition. However, the precise nature of such interplay requires further study.
The Role of Soluble Aβ Oligomers in ER Stress in AD
The endoplasmic reticulum (ER) serves many crucial cellular functions. However, when misfolded or unfolded proteins accumulate in the ER, the stress of ER will be induced. ER stress is characterized by the overexpression of ER molecular chaperones and unfolded protein response (UPR) in order to promote normal protein folding and degrade the abnormal. When activation of the UPR is severe or prolonged enough, the final cellular outcome is pathologic apoptotic cell death. Perturbations in the function of ER are emerging as relevant factors of AD [145].
Because Aβ is synthesized and accumulates in the ER, soluble Aβ1–42 oligomers can directly induce ER stress and mediate apoptosis in cortical neurons in culture, as reflected by that Aβ1–42 oligomers increased GRP78 levels and activated caspase-12, which are two ER stress markers. In addition, ER Ca2+ release which occurs during oligomer-induced GSK3β activation and tau phosphorylation could cause AD [146] (Fig. 1).
The Role of Soluble Aβ Oligomers in Calcium Dyshemostasis
The neurotoxic effects of AD-associated soluble Aβ oligomers are considered a result of the dysregulation of Ca2+ homeostasis (Fig. 1). A mounting body of evidence shows that the fluxes of Ca2+ across the plasma membrane and its release from intracellular stores are induced by cell exposure to soluble Aβ oligomers [147–150]. A number of mechanisms by which Aβ elicits its effects on intracellular Ca2+ homeostasis have been put forward. Firstly, extracellular Aβ oligomers directly disrupt the integrity of the plasma membrane through three different ways; the oligomers destabilize the plasma membrane’s structure [151, 152], induce a generalized increase in membrane permeability, and insert themselves into the membrane to form cation-conducting pores [153, 154]. Secondly, the dysregulation of Ca2+ homeostasis induced by Aβ is also mediated by plasma membrane receptors, including NMDA receptors coupled with Ca2+ influx, to alter neuronal excitability. This, in turn, influences the extent of Ca2+ influx and induces dysregulation of ER Ca2+ homeostasis [155]. Thirdly, in addition to the effects of the extracellular Aβ, intracellular Aβ has been shown to target the ER and the mitochondria, inducing a stress response. ER stress induces Ca2+ release from the ER and causes excessive entry of Ca2+ into mitochondria [156]. Lately, a new study found that inositol 1,4,5-trisphosphate (InsP3) signaling contributed to soluble oligomeric Aβ42-stimulated Ca2+ release from ER stores. However, when using DT40 cells deficient in InsP3R receptors that were permeabilized to allow direct access of Aβ42 to the ER, the same Ca2+ mobilizing effect of Aβ42 was also observed, revealing an additional direct effect of Aβ42 upon the ER, and a mechanism for induction of toxicity by intracellular Aβ42 [157]. Globally, calcium serves as the central pieces of multiple parallel signaling pathways for Aβ-mediated neurodegeneration and synaptic deficits [24]. Moreover, calcium chelator BAPTA could prevent inhibition of signaling by Aβ, whereas Aβ-induced inhibition of signaling was not prevented by application of MK-801 or nimodipine (antagonists of the NMDA receptor and L-type voltage-sensitive calcium channel, respectively). This suggests that soluble Aβ ligomers may induce influx by either channel, or additional channels, or that the neurons contained sufficient calcium to mediate the impact of Aβ [158].
The Soluble Aβ Oligomers Inhibit IIS
AD is also referred to as “type 3 diabetes” because epidemiological, clinical, and experimental evidence suggest a link between AD and insulin resistance [159–163]. The concentrations of insulin and insulin-like growth factors (IGFs), as well as their receptor-mediated signaling, are markedly reduced [164, 165]. Insulin/IGF-1-like signaling (IIS) are the major regulators of longevity and protein homoeostasis with direct relevance to AD [166–168].
Extracellular soluble Aβ oligomers were shown to inhibit insulin signaling by way of decreasing the expressions of cell surface IRs [169, 170]. Felice et al. suggest that extracellular ADDLs bind at specific synaptic sites on neuron dendrites [171], causing oxidative stress and decreasing the abundance of cell surface IRs through internalization and thus leading to a concomitant decrease in insulin signaling. However, ADDLs do not bind directly to the IRs and do not compete with insulin for receptor binding. It is possible that ADDLs bind with low affinity to protein complexes containing NMDA receptors, leading to internalization through an unknown mechanism. In addition, soluble Aβ oligomers also significantly decreased the expressions of insulin receptor substrate (IRS) [170].
Intracellular soluble Aβ oligomers also interfere with the IIS. The activation of IIS mediated by the IR/IGF-1R mainly occurs via activation of phosphoinositide 3-kinase (PI3K) and thereby the serine threonine kinase Akt (also known as PKB) [172] (Fig. 1). Akt in turn phosphorylates a plethora of target protein, including GSK3β, the mammalian target of rapamycin (mTOR), p70/S6 kinase, and inactivates the forkhead box O (FOXO) forkhead transcription factors. The primary negative regulator of this pathway is the lipid and protein phosphatase PTEN (phosphatase and tensinhomologue deleted on chromosome 10). In the nervous system, the downstream protein networks were targeted by Akt regulate survival, migration, neuronal polarity, inflammatory responses/stress resistance, protein translation, synaptic plasticity, learning and memory, autophagy, cell cycle, protein transport/trafficking, metabolism, myelination, and so on. The IIS pathway becomes overactive with age in people at risk of developing AD [173], and some findings lead to the hypothesis that an excessive and inappropriate hyperactivation of Akt in AD neurons contribute to overactivate the IIS pathway [174, 175]. Also, soluble Aβ oligomers were shown to cause the dysfunction of Akt. This initiates the increased production of Aβ and a decreased ability to clear Aβ. These excessive soluble Aβ oligomers in turn induce a progressive sustained activation of the PI3K/Akt/mTOR [176–178] pathway in neurons, which serves to turn off both FOXO-induced stress resistance machinery and mTOR-driven autophagy pathways, and to increase cap-independent protein translation. In a parallel manner, the sustained activation of IIS causes feedback inhibition and desensitization of IGF-1R and IR through inactivation of the key adaptor IRS proteins and disables normal activation of IIS by insulin, IGF-1, and other growth factors [171, 174, 179]. In brief, soluble Aβ oligomers may derail and/or compete for the IGF-1R/IR signaling system in AD neurons through inappropriately increased activation of the IIS pathway and feedback shut-off of normal IIS activation. The mechanism by which Aβ causes sustained activation of the PI3K/Akt pathway is unknown but may occur via Aβ-induced inactivation of PTEN [180] or other major brakes on the IIS pathway.
All in all, soluble Aβ oligomers inhibit IIS. A number of studies have shown toxic action induced by soluble Aβ oligomers through this pathway. Soluble Aβ oligomers showed significant effects leading to increased gene expression and protein amount of GSK3β as well as to decreased levels of gene and protein expression of INSR [181]. Moreover, the researchers reported that natural and synthetic Aβ oligomers, acting through growth factor receptors, inhibit the prosurvival signaling PI3K/Akt/GSK3, and that synthetic Aβ oligomers blocked the effect mediated by different neurotrophins including insulin and IGF-1 [182]. Heras-Sandoval et al. also found similar results showing that pretreatment with Aβ antagonized insulin’s effect on the hippocampal synaptosomes, but not vice versa [179]. In addition, the mechanism underlying the Aβ-induced tau hyperphosphorylation was found to be mediated by the impaired insulin signal transduction because the researchers demonstrated that the phosphorylation of Akt and GSK3 upon insulin stimulation is less activated under this condition [32]. Also, soluble Aβ oligomers inhibited EAAT3-mediated cysteine uptake through IGF-1 signaling, causing a decrease in intracellular GSH levels. These metabolic effects of soluble Aβ oligomers coincided with changes in the expression of redox and methylation pathway genes which may contribute to the pathology of AD [99]. Specifically, controlled on/off signals through the PI3K/Akt/GSK3β/mTOR axis regulate key elements of LTD/LTP [183–186]; thus, the IIS mechanism is closely related to cognitive function.
Soluble Aβ Oligomers Disrupt Axonal Transport in AD
Soluble Aβ oligomers were also demonstrated to disrupt axonal transport (AT). Perfusion of soluble Aβ oligomers into isolated giant squid axoplasm inhibited bidirectional transports of vesicles by the activation of endogenous CK2 and further phosphorylation of motor protein kinesin light chains (KLCs) [187], leading to the release of kinesin from its cargoes. In cultured hippocampal neurons, soluble Aβ oligomers negatively impacted AT by a mechanism that was initiated by NMDARs and mediated by GSK3β without the changes in microtubule stability [188]. Phosphorylation of KLCs by GSK3β [189] promotes the detachment of kinesin from cargo membranes. However, Aβ oligomers induced early and selective diminutions in velocities of synaptic cargoes but had no effect on mitochondrial motility, contrary to previous reports. Soluble Aβ oligomers may lead to decreased cargo-motor engagement. One possible explanation for a lack of inhibition of mitochondrial transport is that such Aβ-induced transport defects are most pronounced for synaptic (and possibly other) cargoes that have a greater rate of cargo attachment/detachment, but more continuously engaged cargoes such as mitochondria are relatively less affected [190]. Additionally, a recent study showed that extracellular Aβ can bind to a so far unknown receptor causing localized Ca2+ elevation, activation of several kinases including MARK, CDK5, and JNK (but not GSK3β), and tau phosphorylation, leading to traffic defects and mitochondria depletion [28].
Tau protein, a typical microtubule-associated protein (MAP), can bind to microtubules that act as the track in AT and plays a key role in the stabilization and spacing of microtubules. Hyperphosphorylation can lower tau’s affinity for the microtubules, which most likely leads to destabilization and disruption of AT. Therefore, soluble Aβ oligomers can cause rapid dissociation of tau from microtubules and the collapse of axonal structure, leading first to defects in AT and ultimately to the death of neurons [191].
Development of Therapeutic Strategies Which Target Soluble Aβ Oligomers
It is hypothesized that early soluble oligomeric forms of Aβ may precede plaque formation and are responsible for neuronal death and the development of AD. Thus, removal of such oligomeric species of Aβ would be beneficial for the disease process. Naturally, researchers have two research directions: preventing the production of soluble Aβ oligomers and eliminating soluble Aβ oligomers.
The first possible action to take is to inhibit the accumulation of Aβ obtained by APP proteolysis. Compounds capable of interfering with the proteases that generate the Aβ peptide from APP have been actively researched. However, the results of those studies are not satisfying. For example, the β-secretase was found to be particularly difficult to achieve, and only a few compounds have reached clinical testing so far [192]. Among inhibitors of γ-secretase, the clinical development of LY450139 (semagacestat) has been halted [193]. Aside from weak points found in the promising drugs, other possible reasons for the failure are that some treatments that initially reduced the concentration of soluble oligomers encouraged release of soluble Aβ from plaques to restore the equilibrium between Aβ in the plaques and the extracellular environment.
Preventing the formation of cytotoxic oligomers should also be an important goal for treating AD. Soluble Aβ oligomers form during the early aggregation process. Monomers are the building block for Aβ assemblies, hence natural removal of the Aβ monomer by the action of degradative enzymes in the brain, or stabilizing the Aβ monomer through binding with small molecules should prevent oligomerization. Some molecules capable of disrupting preformed oligomers have been identified, but there is still a long way to go before they reach the clinical trial stage [194–197]. Oxidative stress is a key factor of the oligomerization induced by Aβ overproduction in vivo, thus the antioxidants that stabilize Aβ monomer(s) can effectively prevent any further aggregation [100, 101, 198].
On the other hand, strategies to eliminate excessive brain Aβ by immunization have become more important in therapy. The most innovative of the pharmacological approaches was the stimulation of Aβ clearance from the brain of AD patients via the administration of Aβ antigens (active vaccination) or anti-Aβ antibodies (passive vaccination). The mechanism by which anti-Aβ antibodies stimulate Aβ clearance from the brains includes direct disassembly of Aβ deposits [199], inhibition of Aβ aggregation [199], and activation of microglia by eliciting Fc-mediated phagocytosis [200]. Unfortunately, the first active vaccine (AN1792, consisting of preaggregate Aβ and an immune adjuvant, QS-21) was abandoned because it caused meningoencephalitis in approximately 6 % of treated patients. Later, several other active and passive anti-Aβ immunization preparations were developed with the aim of abolishing or reducing the inflammatory adverse events observed with AN1792. However, vasogenic edema or amyloid-related imaging abnormalities (ARIA) were observed to be a complication in some passive immunization trials [201, 202]. Anti-Aβ monoclonal antibodies (bapineuzumab and solanezumab) are now being developed. Major safety challenges of this immune therapeutic approach are brain microhemorrhage, off-target cross-reactivity, and loss of the antibody to a peripheral sink phenomenon [203]. The clinical results of the initial studies with bapineuzumab were equivocal in terms of cognitive benefit. Solanezumab is the closest competitor of the humanized anti-Aβ monoclonal antibody bapineuzumaba and is able to directly interact with the mid-region of the Aβ peptide. It has been shown to be able to neutralize soluble Aβ species. Solanezumab may change the equilibrium of Aβ levels between brain interstitial fluids and blood, thereby accelerating Aβ efflux [16, 204]; therefore, this antibody drives an efflux of Aβ from the brain to the blood plasma, providing a peripheral sink for Aβ clearance. Nevertheless, there are some problems which can lead to unsuccessful treatment. For instance, directly removing soluble oligomers from extracellular space (ECS) could encourage release of soluble oligomers from their reservoir in SPs. Plaque-removing trials have also failed [205], suggesting the possibility that during plaque removal, soluble Aβ may have been released from the affected plaques [206]. Hence, eliminating soluble Aβ oligomers before the formation of SPs may be a more effective treatment.
Dysfunction of the autophagy-lysosome system also contributes to Aβ accumulation and insoluble aggregates. Induction of autophagy might enhance the clearance of both soluble and aggregated forms of Aβ. The mTOR pathway plays a central role in controlling protein homeostasis and negatively regulates autophagy, therefore, inhibition of mTOR by rapamycin rescues Aβ pathology [207]. Recently, a study showed that somatostatin receptor subtype-4 agonist NNC 26-9100 mitigated the effect of soluble Aβ42 oligomers via a metalloproteinase-dependent mechanism [208].
According to the role of soluble Aβ oligomers in AD pathology, a number of targets were exploited toward the development of effective therapeutics for AD, including acetylcholinesterase inhibitors (AChEI), various receptor antagonists, nonsteroidal anti-inflammatory drugs (NSAID), antioxidants, insulin sensitizers, estrogen [209], and so on. Throughout the soluble Aβ oligomers’ induced signal pathways, some molecules such as GSK act in overlapping, synergistic, and circulating ways. Therapeutic interventions can be aimed at the important molecules in the Aβ-induced pathways.
Concluding Remarks
Soluble Aβ oligomers are implicated in the early memory impairment seen in Alzheimer’s disease before the onset of discernable neurodegeneration. In this review, we summarized ten essential roles of soluble Aβ oligomers in AD and their possible acting pathways. Although Aβ oligomers have various acting pathways, all pathways ultimately converge to the neuron loss and death that underpin the dementia of AD. Soluble Aβ oligomers are more toxic than fibrillar Aβ aggregates. However, more studies should be done regarding the detailed mechanisms in order to make a better foundation for effective therapies. More research, for example, is required to investigate which mechanisms takes place under which condition and which mechanisms play a pivotal role. Many research groups have demonstrated that soluble Aβ oligomers can mediate various types of neuronal damage when applied to rodent neurons in culture, yet what happens when this situation is recapitulated in vivo? Due to the different roles of soluble Aβ oligomers, any singular kind of therapy might mitigate parts of the defects in AD, but cannot cover all defects as a cohesive whole; thus, the combined therapies are a new trend. All in all, we sincerely hope researchers will devote more effort to study the precise mechanism of soluble Aβ oligomers in AD for the sake of new treatments that will surely contribute to the protection of AD patients.
References
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46(6):860–866
Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol 155(3):853–862
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30(4):572–580. doi:10.1002/ana.410300410
Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 16(3):285–298, discussion 298-304
Wilcox KC, Lacor PN, Pitt J, Klein WL (2011) Abeta oligomer-induced synapse degeneration in Alzheimer’s disease. Cell Mol Neurobiol 31(6):939–948. doi:10.1007/s10571-011-9691-4
Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL (2003) Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A 100(18):10417–10422. doi:10.1073/pnas.1834302100
Glabe CG (2008) Structural classification of toxic amyloid oligomers. J Biol Chem 283(44):29639–29643. doi:10.1074/jbc.R800016200
Yamamoto N, Matsubara E, Maeda S, Minagawa H, Takashima A, Maruyama W, Michikawa M, Yanagisawa K (2007) A ganglioside-induced toxic soluble Abeta assembly. Its enhanced formation from Abeta bearing the Arctic mutation. J Biol Chem 282(4):2646–2655. doi:10.1074/jbc.M606202200
Yanagisawa K (2007) Role of gangliosides in Alzheimer’s disease. Biochim Biophys Acta 1768(8):1943–1951. doi:10.1016/j.bbamem.2007.01.018
Yuyama K, Yamamoto N, Yanagisawa K (2008) Accelerated release of exosome-associated GM1 ganglioside (GM1) by endocytic pathway abnormality: another putative pathway for GM1-induced amyloid fibril formation. J Neurochem 105(1):217–224. doi:10.1111/j.1471-4159.2007.05128.x
Stege GJ, Renkawek K, Overkamp PS, Verschuure P, van Rijk AF, Reijnen-Aalbers A, Boelens WC, Bosman GJ, de Jong WW (1999) The molecular chaperone alphaB-crystallin enhances amyloid beta neurotoxicity. Biochem Biophys Res Commun 262(1):152–156. doi:10.1006/bbrc.1999.1167
Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TE, Rozovsky I, Stine WB, Snyder SW, Holzman TF et al (1995) Clusterin (apoJ) alters the aggregation of amyloid beta-peptide (A beta 1-42) and forms slowly sedimenting A beta complexes that cause oxidative stress. Exp Neurol 136(1):22–31
Sakono M, Zako T, Ueda H, Yohda M, Maeda M (2008) Formation of highly toxic soluble amyloid beta oligomers by the molecular chaperone prefoldin. FEBS J 275(23):5982–5993. doi:10.1111/j.1742-4658.2008.06727.x
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356. doi:10.1126/science.1072994
Mucke HA (2000) Apoptosis modulators: targeting cellular suicide. IDrugs 3(5):506–511
Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci 5(5):452–457. doi:10.1038/nn842
Oda T, Pasinetti GM, Osterburg HH, Anderson C, Johnson SA, Finch CE (1994) Purification and characterization of brain clusterin. Biochem Biophys Res Commun 204(3):1131–1136. doi:10.1006/bbrc.1994.2580
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Cruz L, Urbanc B, Buldyrev SV, Christie R, Gomez-Isla T, Havlin S, McNamara M, Stanley HE, Hyman BT (1997) Aggregation and disaggregation of senile plaques in Alzheimer disease. Proc Natl Acad Sci U S A 94(14):7612–7616
Hyman BT, Marzloff K, Arriagada PV (1993) The lack of accumulation of senile plaques or amyloid burden in Alzheimer’s disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol 52(6):594–600
Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106(10):4012–4017. doi:10.1073/pnas.0811698106
Serrano-Pozo A, Mielke ML, Muzitansky A, Gomez-Isla T, Growdon JH, Bacskai BJ, Betensky RA, Frosch MP, Hyman BT (2012) Stable size distribution of amyloid plaques over the course of Alzheimer disease. J Neuropathol Exp Neurol 71(8):694–701. doi:10.1097/NEN.0b013e31825e77de
Oide T, Kinoshita T, Arima K (2006) Regression stage senile plaques in the natural course of Alzheimer's disease. Neuropathol Appl Neurobiol 32(5):539–556. doi:10.1111/j.1365-2990.2006.00767.x
Liao D, Miller EC, Teravskis PJ (2014) Tau acts as a mediator for Alzheimer’s disease-related synaptic deficits. Eur J Neurosci 39(7):1202–1213. doi:10.1111/ejn.12504
Handoko M, Grant M, Kuskowski M, Zahs KR, Wallin A, Blennow K, Ashe KH (2013) Correlation of specific amyloid-beta oligomers with tau in cerebrospinal fluid from cognitively normal older adults. JAMA Neurol 70(5):594–599. doi:10.1001/jamaneurol.2013.48
Lesne SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH (2013) Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain 136(Pt 5):1383–1398. doi:10.1093/brain/awt062
Zussy C, Brureau A, Keller E, Marchal S, Blayo C, Delair B, Ixart G, Maurice T, Givalois L (2013) Alzheimer’s disease related markers, cellular toxicity and behavioral deficits induced six weeks after oligomeric amyloid-beta peptide injection in rats. PLoS One 8(1):e53117. doi:10.1371/journal.pone.0053117
Zempel H, Thies E, Mandelkow E, Mandelkow EM (2010) Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci Off J Soc Neurosci 30(36):11938–11950. doi:10.1523/JNEUROSCI. 2357-10.2010
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68(6):1067–1081. doi:10.1016/j.neuron.2010.11.030
Miller EC, Teravskis PJ, Dummer BW, Zhao X, Huganir RL, Liao D (2014) Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur J Neurosci 39(7):1214–1224. doi:10.1111/ejn.12507
Brouillette J, Caillierez R, Zommer N, Alves-Pires C, Benilova I, Blum D, De Strooper B, Buee L (2012) Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-beta1-42 oligomers are revealed in vivo by using a novel animal model. J Neurosci Off J Soc Neurosci 32(23):7852–7861. doi:10.1523/JNEUROSCI. 5901-11.2012
Tokutake T, Kasuga K, Yajima R, Sekine Y, Tezuka T, Nishizawa M, Ikeuchi T (2012) Hyperphosphorylation of Tau induced by naturally secreted amyloid-beta at nanomolar concentrations is modulated by insulin-dependent Akt-GSK3beta signaling pathway. J Biol Chem 287(42):35222–35233. doi:10.1074/jbc.M112.348300
Chabrier MA, Blurton-Jones M, Agazaryan AA, Nerhus JL, Martinez-Coria H, LaFerla FM (2012) Soluble abeta promotes wild-type tau pathology in vivo. J Neurosci Off J Soc Neurosci 32(48):17345–17350. doi:10.1523/JNEUROSCI. 0172-12.2012
Selenica ML, Brownlow M, Jimenez JP, Lee DC, Pena G, Dickey CA, Gordon MN, Morgan D (2013) Amyloid oligomers exacerbate tau pathology in a mouse model of tauopathy. Neurodegener Dis 11(4):165–181. doi:10.1159/000337230
Guo JP, Arai T, Miklossy J, McGeer PL (2006) Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc Natl Acad Sci U S A 103(6):1953–1958. doi:10.1073/pnas.0509386103
Cedazo-Minguez A, Popescu BO, Blanco-Millan JM, Akterin S, Pei JJ, Winblad B, Cowburn RF (2003) Apolipoprotein E and beta-amyloid (1-42) regulation of glycogen synthase kinase-3beta. J Neurochem 87(5):1152–1164
Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, Roberson ED, Bloom GS (2013) Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J Cell Sci 126(Pt 5):1278–1286. doi:10.1242/jcs.1125880
Yu W, Polepalli J, Wagh D, Rajadas J, Malenka R, Lu B (2012) A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Abeta on synapses and dendritic spines. Hum Mol Genet 21(6):1384–1390. doi:10.1093/hmg/ddr576
Larson ME, Lesne SE (2012) Soluble Abeta oligomer production and toxicity. J Neurochem 120(Suppl 1):125–139. doi:10.1111/j.1471-4159.2011.07478.x
Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesne SE (2012) The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J Neurosci Off J Soc Neurosci 32(47):16857–16871
Cullen WK, Suh YH, Anwyl R, Rowan MJ (1997) Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport 8(15):3213–3217
Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ (2001) Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci Off J Soc Neurosci 21(4):1327–1333
Lynch MA (2004) Long-term potentiation and memory. Physiol Rev 84(1):87–136. doi:10.1152/physrev.00014.2003
Neves G, Cooke SF, Bliss TV (2008) Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci 9(1):65–75. doi:10.1038/nrn2303
Collingridge GL, Peineau S, Howland JG, Wang YT (2010) Long-term depression in the CNS. Nat Rev Neurosci 11(7):459–473. doi:10.1038/nrn2867
Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP (1997) 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience 80(3):685–696
Blanc EM, Keller JN, Fernandez S, Mattson MP (1998) 4-hydroxynonenal, a lipid peroxidation product, impairs glutamate transport in cortical astrocytes. Glia 22(2):149–160
Mark RJ, Hensley K, Butterfield DA, Mattson MP (1995) Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosc Off J Soc Neurosci 15(9):6239–6249
Shi C, Wu F, Xu J (2010) H2O2 and PAF mediate Abeta1-42-induced Ca2+ dyshomeostasis that is blocked by EGb761. Neurochem Int 56(8):893–905. doi:10.1016/j.neuint.2010.03.016
Ikegaya Y, Matsuura S, Ueno S, Baba A, Yamada MK, Nishiyama N, Matsuki N (2002) Beta-amyloid enhances glial glutamate uptake activity and attenuates synaptic efficacy. J Biol Chem 277(35):32180–32186. doi:10.1074/jbc.M203764200
Fernandez-Tome P, Brera B, Arevalo MA, de Ceballos ML (2004) Beta-amyloid25-35 inhibits glutamate uptake in cultured neurons and astrocytes: modulation of uptake as a survival mechanism. Neurobiol Dis 15(3):580–589. doi:10.1016/j.nbd.2003.12.006
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14(8):837–842. doi:10.1038/nm1782
Rammes G, Hasenjager A, Sroka-Saidi K, Deussing JM, Parsons CG (2011) Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of beta-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology 60(6):982–990. doi:10.1016/j.neuropharm.2011.01.051
Knobloch M, Farinelli M, Konietzko U, Nitsch RM, Mansuy IM (2007) Abeta oligomer-mediated long-term potentiation impairment involves protein phosphatase 1-dependent mechanisms. J Neurosci Off J Soc Neurosci 27(29):7648–7653. doi:10.1523/JNEUROSCI. 0395-07.2007
Reese LC, Zhang W, Dineley KT, Kayed R, Taglialatela G (2008) Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell 7(6):824–835. doi:10.1111/j.1474-9726.2008.00434.x
Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R (2004) Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci Off J Soc Neurosci 24(13):3370–3378. doi:10.1523/JNEUROSCI. 1633-03.2004
Chen L, Wang H, Zhang Z, Li Z, He D, Sokabe M, Chen L (2010) DMXB (GTS-21) ameliorates the cognitive deficits in beta amyloid(25-35(-) ) injected mice through preventing the dysfunction of alpha7 nicotinic receptor. J Neurosci Res 88(8):1784–1794. doi:10.1002/jnr.22345
Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT, Shatz CJ (2013) Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341(6152):1399–1404. doi:10.1126/science.1242077
Chiba T, Yamada M, Sasabe J, Terashita K, Shimoda M, Matsuoka M, Aiso S (2009) Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol Psychiatry 14(2):206–222. doi:10.1038/mp.2008.105
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci Off J Soc Neurosci 31(18):6627–6638. doi:10.1523/JNEUROSCI. 0203-11.2011
Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O (2008) Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci Off J Soc Neurosci 28(53):14537–14545. doi:10.1523/JNEUROSCI. 2692-08.2008
Kullmann DM, Lamsa KP (2007) Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci 8(9):687–699. doi:10.1038/nrn2207
Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62(6):788–801. doi:10.1016/j.neuron.2009.05.012
Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304(5673):1021–1024. doi:10.1126/science.1096615
Benilova I, Karran E, De Strooper B (2012) The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 15(3):349–357. doi:10.1038/nn.3028
Gu Z, Liu W, Yan Z (2009) {beta}-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem 284(16):10639–10649. doi:10.1074/jbc.M806508200
Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, Morabito M, Almeida OF (2005) Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci Off J Soc Neurosci 25(48):11061–11070. doi:10.1523/JNEUROSCI. 3034-05.2005
Rui Y, Gu J, Yu K, Hartzell HC, Zheng JQ (2010) Inhibition of AMPA receptor trafficking at hippocampal synapses by beta-amyloid oligomers: the mitochondrial contribution. Mol Brain 3:10. doi:10.1186/1756-6606-3-10
Minano-Molina AJ, Espana J, Martin E, Barneda-Zahonero B, Fado R, Sole M, Trullas R, Saura CA, Rodriguez-Alvarez J (2011) Soluble oligomers of amyloid-beta peptide disrupt membrane trafficking of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. J Biol Chem 286(31):27311–27321. doi:10.1074/jbc.M111.227504
Barria A, Derkach V, Soderling T (1997) Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem 272(52):32727–32730
Ouyang Y, Wong M, Capani F, Rensing N, Lee CS, Liu Q, Neusch C, Martone ME, Wu JY, Yamada K, Ellisman MH, Choi DW (2005) Transient decrease in F-actin may be necessary for translocation of proteins into dendritic spines. Eur J Neurosci 22(12):2995–3005. doi:10.1111/j.1460-9568.2005.04521.x
Decker H, Jurgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, Epstein AL, De Felice FG, Jerusalinsky D, Ferreira ST (2010) N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-beta peptide oligomers. J Neurochem 115(6):1520–1529. doi:10.1111/j.1471-4159.2010.07058.x
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8(8):1051–1058. doi:10.1038/nn1503
Ronicke R, Mikhaylova M, Ronicke S, Meinhardt J, Schroder UH, Fandrich M, Reiser G, Kreutz MR, Reymann KG (2011) Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol Aging 32(12):2219–2228. doi:10.1016/j.neurobiolaging.2010.01.011
Dinamarca MC, Colombres M, Cerpa W, Bonansco C, Inestrosa NC (2008) Beta-amyloid oligomers affect the structure and function of the postsynaptic region: role of the Wnt signaling pathway. Neurodegener Dis 5(3–4):149–152. doi:10.1159/000113687
Kelly BL, Ferreira A (2006) beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem 281(38):28079–28089. doi:10.1074/jbc.M605081200
Texido L, Martin-Satue M, Alberdi E, Solsona C, Matute C (2011) Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium 49(3):184–190. doi:10.1016/j.ceca.2011.02.001
Paula-Lima AC, Brito-Moreira J, Ferreira ST (2013) Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer’s disease. J Neurochem 126(2):191–202. doi:10.1111/jnc.12304
Lauren J (2014) Cellular prion protein as a therapeutic target in Alzheimer’s disease. J Alzheimers Dis 38(2):227–244. doi:10.3233/JAD-130950
Chen S, Yadav SP, Surewicz WK (2010) Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem 285(34):26377–26383. doi:10.1074/jbc.M110.145516
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457(7233):1128–1132. doi:10.1038/nature07761
Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J (2011) Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun 2:336. doi:10.1038/ncomms1341
Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ (2011) Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci Off J Soc Neurosci 31(20):7259–7263. doi:10.1523/JNEUROSCI. 6500-10.2011
Bate C, Williams A (2011) Amyloid-beta-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J Biol Chem 286(44):37955–37963. doi:10.1074/jbc.M111.248724
Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, Borsello T, Gobbi M, Harris DA (2013) An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J Biol Chem 288(11):7857–7866. doi:10.1074/jbc.M112.423954
Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Muller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J (2011) The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J 30(10):2057–2070. doi:10.1038/emboj.2011.86
Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM (2012) Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15(9):1227–1235. doi:10.1038/nn.3178
You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW (2012) Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A 109(5):1737–1742. doi:10.1073/pnas.1110789109
Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79(5):887–902. doi:10.1016/j.neuron.2013.06.036
Biasini E, Unterberger U, Solomon IH, Massignan T, Senatore A, Bian H, Voigtlaender T, Bowman FP, Bonetto V, Chiesa R, Luebke J, Toselli P, Harris DA (2013) A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J Neurosci Off J Soc Neurosci 33(6):2408–2418. doi:10.1523/JNEUROSCI. 3406-12.2013
Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G (2010) Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A 107(5):2295–2300. doi:10.1073/pnas.0911829107
Niedowicz DM, Beckett TL, Matveev S, Weidner AM, Baig I, Kryscio RJ, Mendiondo MS, LeVine H 3rd, Keller JN, Murphy MP (2012) Pittsburgh compound B and the postmortem diagnosis of Alzheimer disease. Ann Neurol 72(4):564–570. doi:10.1002/ana.23633
Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298(5594):789–791. doi:10.1126/science.1074069
Koistinaho M, Ort M, Cimadevilla JM, Vondrous R, Cordell B, Koistinaho J, Bures J, Higgins LS (2001) Specific spatial learning deficits become severe with age in beta -amyloid precursor protein transgenic mice that harbor diffuse beta-amyloid deposits but do not form plaques. Proc Natl Acad Sci U S A 98(25):14675–14680. doi:10.1073/pnas.261562998
Chen Z, Zhong C (2014) Oxidative stress in Alzheimer’s disease. Neurosci Bull 30(2):271–281. doi:10.1007/s12264-013-1423-y
Bondy SC, Guo-Ross SX, Truong AT (1998) Promotion of transition metal-induced reactive oxygen species formation by beta-amyloid. Brain Res 799(1):91–96
Butterfield DA, Lauderback CM (2002) Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med 32(11):1050–1060
Cenini G, Dowling AL, Beckett TL, Barone E, Mancuso C, Murphy MP, Levine H 3rd, Lott IT, Schmitt FA, Butterfield DA, Head E (2012) Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta 1822(2):130–138. doi:10.1016/j.bbadis.2011.10.001
Hodgson N, Trivedi M, Muratore C, Li S, Deth R (2013) Soluble oligomers of amyloid-beta cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J Alzheimers Dis 36(1):197–209. doi:10.3233/JAD-130101
Murakami K, Murata N, Noda Y, Tahara S, Kaneko T, Kinoshita N, Hatsuta H, Murayama S, Barnham KJ, Irie K, Shirasawa T, Shimizu T (2011) SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J Biol Chem 286(52):44557–44568. doi:10.1074/jbc.M111.279208
Murakami K, Murata N, Ozawa Y, Kinoshita N, Irie K, Shirasawa T, Shimizu T (2011) Vitamin C restores behavioral deficits and amyloid-beta oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J Alzheimers Dis 26(1):7–18. doi:10.3233/JAD-2011-101971
Cimini A, Gentile R, D’Angelo B, Benedetti E, Cristiano L, Avantaggiati ML, Giordano A, Ferri C, Desideri G (2013) Cocoa powder triggers neuroprotective and preventive effects in a human Alzheimer's disease model by modulating BDNF signaling pathway. J Cell Biochem 114(10):2209–2220. doi:10.1002/jcb.24548
Xu PX, Wang SW, Yu XL, Su YJ, Wang T, Zhou WW, Zhang H, Wang YJ, Liu RT (2014) Rutin improves spatial memory in Alzheimer’s disease transgenic mice by reducing Abeta oligomer level and attenuating oxidative stress and neuroinflammation. Behav Brain Res 264:173–180. doi:10.1016/j.bbr.2014.02.002
Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA (1994) A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A 91(8):3270–3274
Harris ME, Carney JM, Cole PS, Hensley K, Howard BJ, Martin L, Bummer P, Wang Y, Pedigo NW Jr, Butterfield DA (1995) beta-Amyloid peptide-derived, oxygen-dependent free radicals inhibit glutamate uptake in cultured astrocytes: implications for Alzheimer’s disease. Neuroreport 6(14):1875–1879
Manelli AM, Puttfarcken PS (1995) beta-Amyloid-induced toxicity in rat hippocampal cells: in vitro evidence for the involvement of free radicals. Brain Res Bull 38(6):569–576
Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, Kimura H (1995) Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci U S A 92(6):1989–1993
Ii M, Sunamoto M, Ohnishi K, Ichimori Y (1996) beta-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res 720(1–2):93–100. doi:10.1016/0006-8993(96)00156-4
Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang J, Sultana R, Bredesen DE (2010) In vivo oxidative stress in brain of Alzheimer disease transgenic mice: requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med 48(1):136–144. doi:10.1016/j.freeradbiomed.2009.10.035
Greenough MA, Camakaris J, Bush AI (2013) Metal dyshomeostasis and oxidative stress in Alzheimer's disease. Neurochem Int 62(5):540–555. doi:10.1016/j.neuint.2012.08.014
Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI (2000) Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem 275(26):19439–19442. doi:10.1074/jbc.C000165200
Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH (2006) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15(9):1437–1449. doi:10.1093/hmg/ddl066
Rhein V, Baysang G, Rao S, Meier F, Bonert A, Muller-Spahn F, Eckert A (2009) Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell Mol Neurobiol 29(6–7):1063–1071. doi:10.1007/s10571-009-9398-y
Young KJ, Bennett JP (2010) The mitochondrial secret(ase) of Alzheimer’s disease. J Alzheimers Dis 20(Suppl 2):S381–S400. doi:10.3233/JAD-2010-100360
Garthwaite J, Boulton CL (1995) Nitric oxide signaling in the central nervous system. Annu Rev Physiol 57:683–706. doi:10.1146/annurev.ph.57.030195.003343
Quiroz-Baez R, Flores-Dominguez D, Arias C (2013) Synaptic aging is associated with mitochondrial dysfunction, reduced antioxidant contents and increased vulnerability to amyloid-beta toxicity. CurrAlzheimer Res 10(3):324–331
Town T, Nikolic V, Tan J (2005) The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation 2:24. doi:10.1186/1742-2094-2-24
Paranjape GS, Gouwens LK, Osborn DC, Nichols MR (2012) Isolated amyloid-beta(1-42) protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem Neurosci 3(4):302–311. doi:10.1021/cn2001238
Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2009) Inflammation in Alzheimer's disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 87(3):181–194
Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J (2008) Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci Off J Soc Neurosci 28(45):11650–11661. doi:10.1523/JNEUROSCI. 3024-08.2008
Dewapriya P, Li YX, Himaya SW, Pangestuti R, Kim SK (2013) Neoechinulin A suppresses amyloid-beta oligomer-induced microglia activation and thereby protects PC-12 cells from inflammation-mediated toxicity. Neurotoxicology 35:30–40. doi:10.1016/j.neuro.2012.12.004
He Y, Zheng MM, Ma Y, Han XJ, Ma XQ, Qu CQ, Du YF (2012) Soluble oligomers and fibrillar species of amyloid beta-peptide differentially affect cognitive functions and hippocampal inflammatory response. Biochem Biophys Res Commun 429(3–4):125–130. doi:10.1016/j.bbrc.2012.10.129
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21(3):383–421
DaRocha-Souto B, Scotton TC, Coma M, Serrano-Pozo A, Hashimoto T, Sereno L, Rodriguez M, Sanchez B, Hyman BT, Gomez-Isla T (2011) Brain oligomeric beta-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J Neuropathol Exp Neurol 70(5):360–376. doi:10.1097/NEN.0b013e318217a118
White JA, Manelli AM, Holmberg KH, Van Eldik LJ, Ladu MJ (2005) Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiol Dis 18(3):459–465. doi:10.1016/j.nbd.2004.12.013
Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, Smith MA, Martins RN (2003) Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res Brain Res Rev 43(1):1–16
Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR (2005) Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J Neurochem 92(2):226–234. doi:10.1111/j.1471-4159.2004.02857.x
Choi DY, Lee YJ, Hong JT, Lee HJ (2012) Antioxidant properties of natural polyphenols and their therapeutic potentials for Alzheimer’s disease. Brain Res Bull 87(2–3):144–153. doi:10.1016/j.brainresbull.2011.11.014
Bartus RT, Dean RL 3rd, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217(4558):408–414
Contestabile A (2011) The history of the cholinergic hypothesis. Behav Brain Res 221(2):334–340. doi:10.1016/j.bbr.2009.12.044
Schliebs R, Arendt T (2011) The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221(2):555–563. doi:10.1016/j.bbr.2010.11.058
Nunes-Tavares N, Santos LE, Stutz B, Brito-Moreira J, Klein WL, Ferreira ST, de Mello FG (2012) Inhibition of choline acetyltransferase as a mechanism for cholinergic dysfunction induced by amyloid-beta peptide oligomers. J Biol Chem 287(23):19377–19385. doi:10.1074/jbc.M111.321448
Wang HY, Lee DH, Davis CB, Shank RP (2000) Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem 75(3):1155–1161
Liu Q, Kawai H, Berg DK (2001) beta-Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A 98(8):4734–4739. doi:10.1073/pnas.081553598
Buckingham SD, Jones AK, Brown LA, Sattelle DB (2009) Nicotinic acetylcholine receptor signalling: roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol Rev 61(1):39–61. doi:10.1124/pr.108.000562
Nagele RG, D’Andrea MR, Anderson WJ, Wang HY (2002) Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110(2):199–211
Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD (2001) Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci Off J Soc Neurosci 21(12):4125–4133
Fodero LR, Mok SS, Losic D, Martin LL, Aguilar MI, Barrow CJ, Livett BG, Small DH (2004) Alpha7-nicotinic acetylcholine receptors mediate an Abeta(1-42)-induced increase in the level of acetylcholinesterase in primary cortical neurones. J Neurochem 88(5):1186–1193
Dineley KT, Bell KA, Bui D, Sweatt JD (2002) beta-Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem 277(28):25056–25061. doi:10.1074/jbc.M200066200
Lee DH, Wang HY (2003) Differential physiologic responses of alpha7 nicotinic acetylcholine receptors to beta-amyloid1-40 and beta-amyloid1-42. J Neurobiol 55(1):25–30. doi:10.1002/neu.10203
Pettit DL, Shao Z, Yakel JL (2001) beta-Amyloid(1-42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci Off J Soc Neurosci 21(1):RC120
Tozaki H, Matsumoto A, Kanno T, Nagai K, Nagata T, Yamamoto S, Nishizaki T (2002) The inhibitory and facilitatory actions of amyloid-beta peptides on nicotinic ACh receptors and AMPA receptors. Biochem Biophys Res Commun 294(1):42–45. doi:10.1016/S0006-291X(02)00429-1
Chin JH, Ma L, MacTavish D, Jhamandas JH (2007) Amyloid beta protein modulates glutamate-mediated neurotransmission in the rat basal forebrain: involvement of presynaptic neuronal nicotinic acetylcholine and metabotropic glutamate receptors. J Neurosci Off J Soc Neurosci 27(35):9262–9269. doi:10.1523/JNEUROSCI. 1843-07.2007
Liu Q, Huang Y, Xue F, Simard A, DeChon J, Li G, Zhang J, Lucero L, Wang M, Sierks M, Hu G, Chang Y, Lukas RJ, Wu J (2009) A novel nicotinic acetylcholine receptor subtype in basal forebrain cholinergic neurons with high sensitivity to amyloid peptides. J Neurosci Off J Soc Neurosci 29(4):918–929. doi:10.1523/JNEUROSCI. 3952-08.2009
Li JQ, Yu JT, Jiang T, Tan L (2014) Endoplasmic reticulum dysfunction in Alzheimer’s disease. Mol Neurobiol. doi:10.1007/s12035-014-8695-8
Resende R, Ferreiro E, Pereira C, Oliveira CR (2008) ER stress is involved in Abeta-induced GSK-3beta activation and tau phosphorylation. J Neurosci Res 86(9):2091–2099. doi:10.1002/jnr.21648
Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31(9):454–463. doi:10.1016/j.tins.2008.06.005
Green KN, LaFerla FM (2008) Linking calcium to Abeta and Alzheimer’s disease. Neuron 59(2):190–194. doi:10.1016/j.neuron.2008.07.013
Berridge MJ (2011) Calcium signalling and Alzheimer’s disease. Neurochem Res 36(7):1149–1156. doi:10.1007/s11064-010-0371-4
Demuro A, Parker I, Stutzmann GE (2010) Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 285(17):12463–12468. doi:10.1074/jbc.R109.080895
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300(5618):486–489. doi:10.1126/science.1079469
Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416(6880):507–511. doi:10.1038/416507a
Simakova O, Arispe NJ (2006) Early and late cytotoxic effects of external application of the Alzheimer’s Abeta result from the initial formation and function of Abeta ion channels. Biochemistry 45(18):5907–5915. doi:10.1021/bi060148g
Demuro A, Smith M, Parker I (2011) Single-channel Ca(2+) imaging implicates Abeta1-42 amyloid pores in Alzheimer’s disease pathology. J Cell Biol 195(3):515–524. doi:10.1083/jcb.201104133
Resende R, Ferreiro E, Pereira C, Resende de Oliveira C (2008) Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 155(3):725–737. doi:10.1016/j.neuroscience.2008.06.036
Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H (2011) Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res 89(7):1031–1042. doi:10.1002/jnr.22640
Jensen LE, Bultynck G, Luyten T, Amijee H, Bootman MD, Roderick HL (2013) Alzheimer's disease-associated peptide Abeta42 mobilizes ER Ca(2+) via InsP3R-dependent and -independent mechanisms. Front Mol Neurosci 6:36. doi:10.3389/fnmol.2013.00036
Lee S, Zemianek J, Shea TB (2013) Rapid, reversible impairment of synaptic signaling in cultured cortical neurons by exogenously-applied amyloid-beta. J Alzheimers Dis 35(2):395–402. doi:10.3233/JAD-122452
Schnaider Beeri M, Goldbourt U, Silverman JM, Noy S, Schmeidler J, Ravona-Springer R, Sverdlick A, Davidson M (2004) Diabetes mellitus in midlife and the risk of dementia three decades later. Neurology 63(10):1902–1907
Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, Butler PC (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53(2):474–481
Allen KV, Frier BM, Strachan MW (2004) The relationship between type 2 diabetes and cognitive dysfunction: longitudinal studies and their methodological limitations. Eur J Pharmacol 490(1–3):169–175. doi:10.1016/j.ejphar.2004.02.054
Cukierman T, Gerstein HC, Williamson JD (2005) Cognitive decline and dementia in diabetes–systematic overview of prospective observational studies. Diabetologia 48(12):2460–2469. doi:10.1007/s00125-005-0023-4
Craft S (2006) Insulin resistance syndrome and Alzheimer disease: pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis Assoc Disord 20(4):298–301. doi:10.1097/01.wad.0000213866.86934.7e
Hoyer S (2003) Memory function and brain glucose metabolism. Pharmacopsychiatry 36(Suppl 1):S62–S67. doi:10.1055/s-2003-40452
de la Monte SM, Wands JR (2005) Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis 7(1):45–61
Kenyon CJ (2010) The genetics of ageing. Nature 464(7288):504–512. doi:10.1038/nature08980
Panowski SH, Dillin A (2009) Signals of youth: endocrine regulation of aging in Caenorhabditis elegans. Trends Endocrinol Metab 20(6):259–264. doi:10.1016/j.tem.2009.03.006
Douglas PM, Dillin A (2010) Protein homeostasis and aging in neurodegeneration. J Cell Biol 190(5):719–729. doi:10.1083/jcb.201005144
Townsend M, Mehta T, Selkoe DJ (2007) Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem 282(46):33305–33312. doi:10.1074/jbc.M610390200
Han X, Ma Y, Liu X, Wang L, Qi S, Zhang Q, Du Y (2012) Changes in insulin-signaling transduction pathway underlie learning/memory deficits in an Alzheimer’s disease rat model. J Neural Transm 119(11):1407–1416. doi:10.1007/s00702-012-0803-1
De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL (2009) Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A 106(6):1971–1976. doi:10.1073/pnas.0809158106
Franke TF (2008) PI3K/Akt: getting it right matters. Oncogene 27(50):6473–6488. doi:10.1038/onc.2008.313
Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C (2005) Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J Neurochem 93(1):105–117. doi:10.1111/j.1471-4159.2004.02949.x
Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL (2008) Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J 22(1):246–260. doi:10.1096/fj.06-7703com
Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT (2009) The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener 4:14. doi:10.1186/1750-1326-4-14
Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magri A, Oddo S (2011) Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem 286(11):8924–8932. doi:10.1074/jbc.M110.180638
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S (2010) Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 285(17):13107–13120. doi:10.1074/jbc.M110.100420
Majumder S, Caccamo A, Medina DX, Benavides AD, Javors MA, Kraig E, Strong R, Richardson A, Oddo S (2012) Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging cell 11(2):326–335. doi:10.1111/j.1474-9726.2011.00791.x
Heras-Sandoval D, Ferrera P, Arias C (2012) Amyloid-beta protein modulates insulin signaling in presynaptic terminals. Neurochem Res 37(9):1879–1885. doi:10.1007/s11064-012-0800-7
Kwak YD, Ma T, Diao S, Zhang X, Chen Y, Hsu J, Lipton SA, Masliah E, Xu H, Liao FF (2010) NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener 5:49. doi:10.1186/1750-1326-5-49
Bartl J, Meyer A, Brendler S, Riederer P, Grunblatt E (2013) Different effects of soluble and aggregated amyloid beta42 on gene/protein expression and enzyme activity involved in insulin and APP pathways. J Neural Transm 120(1):113–120. doi:10.1007/s00702-012-0852-5
Jimenez S, Torres M, Vizuete M, Sanchez-Varo R, Sanchez-Mejias E, Trujillo-Estrada L, Carmona-Cuenca I, Caballero C, Ruano D, Gutierrez A, Vitorica J (2011) Age-dependent accumulation of soluble amyloid beta (Abeta) oligomers reverses the neuroprotective effect of soluble amyloid precursor protein-alpha (sAPP(alpha)) by modulating phosphatidylinositol 3-kinase (PI3K)/Akt-GSK-3beta pathway in Alzheimer mouse model. J Biol Chem 286(21):18414–18425. doi:10.1074/jbc.M110.209718
Ehninger D, de Vries PJ, Silva AJ (2009) From mTOR to cognition: molecular and cellular mechanisms of cognitive impairments in tuberous sclerosis. J Intellect Disabil Res 53(10):838–851. doi:10.1111/j.1365-2788.2009.01208.x
Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL (2007) LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53(5):703–717. doi:10.1016/j.neuron.2007.01.029
Hou L, Klann E (2004) Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci Off J Soc Neurosci 24(28):6352–6361. doi:10.1523/JNEUROSCI. 0995-04.2004
Hoeffer CA, Klann E (2010) mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 33(2):67–75. doi:10.1016/j.tins.2009.11.003
Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S (2009) Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A 106(14):5907–5912. doi:10.1073/pnas.0901229106
Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA (2010) Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci Off J Soc Neurosci 30(27):9166–9171. doi:10.1523/JNEUROSCI. 1074-10.2010
Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST (2002) Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J 21(3):281–293. doi:10.1093/emboj/21.3.281
Tang Y, Scott DA, Das U, Edland SD, Radomski K, Koo EH, Roy S (2012) Early and selective impairments in axonal transport kinetics of synaptic cargoes induced by soluble amyloid beta-protein oligomers. Traffic 13(5):681–693. doi:10.1111/j.1600-0854.2012.01340.x
Wang ZX, Tan L, Yu JT (2014) Axonal transport defects in Alzheimer’s disease. Mol Neurobiol. doi:10.1007/s12035-014-8810-x
Panza F, Solfrizzi V, Frisardi V, Capurso C, D'Introno A, Colacicco AM, Vendemiale G, Capurso A, et al (2009) Disease-modifying approach to the treatment of Alzheimer’s disease: from alpha-secretase activators to gamma-secretase inhibitors and modulators. Drugs Aging 26(7):537–555. doi:10.2165/11315770-000000000-00000
Imbimbo BP, Panza F, Frisardi V, Solfrizzi V, D'Onofrio G, Logroscino G, Seripa D, Pilotto A (2011) Therapeutic intervention for Alzheimer’s disease with gamma-secretase inhibitors: still a viable option? Expert Opin Investig Drugs 20(3):325–341. doi:10.1517/13543784.2011.550572
Ono K, Yamada M (2011) Low-n oligomers as therapeutic targets of Alzheimer’s disease. J Neurochem 117(1):19–28. doi:10.1111/j.1471-4159.2011.07187.x
Belluti F, Rampa A, Gobbi S, Bisi A (2013) Small-molecule inhibitors/modulators of amyloid-beta peptide aggregation and toxicity for the treatment of Alzheimer’s disease: a patent review (2010–2012). Expert Opin Ther Patents 23(5):581–596. doi:10.1517/13543776.2013.772983
Zhang Y, Zhen Y, Dong Y, Xu Z, Yue Y, Golde TE, Tanzi RE, Moir RD, Xie Z (2011) Anesthetic propofol attenuates the isoflurane-induced caspase-3 activation and Abeta oligomerization. PLoS One 6(11):e27019. doi:10.1371/journal.pone.0027019
O'Hare E, Scopes DI, Kim EM, Palmer P, Jones M, Whyment AD, Spanswick D, Amijee H, Nerou E, McMahon B, Treherne JM, Jeggo R (2013) Orally bioavailable small molecule drug protects memory in Alzheimer’s disease models. Neurobiol Aging 34(4):1116–1125. doi:10.1016/j.neurobiolaging.2012.10.016
Parachikova A, Green KN, Hendrix C, LaFerla FM (2010) Formulation of a medical food cocktail for Alzheimer’s disease: beneficial effects on cognition and neuropathology in a mouse model of the disease. PLoS One 5(11):e14015. doi:10.1371/journal.pone.0014015
Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT (2001) Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 7(3):369–372. doi:10.1038/85525
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T (2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6(8):916–919. doi:10.1038/78682
Wisniewski T, Konietzko U (2008) Amyloid-beta immunisation for Alzheimer’s disease. Lancet Neurol 7(9):805–811. doi:10.1016/S1474-4422(08)70170-4
Panza F, Frisardi V, Solfrizzi V, Imbimbo BP, Logroscino G, Santamato A, Greco A, Seripa D, Pilotto A (2012) Immunotherapy for Alzheimer’s disease: from anti-beta-amyloid to tau-based immunization strategies. Immunotherapy 4(2):213–238. doi:10.2217/imt.11.170
Wisniewski T, Goni F (2014) Immunotherapy for Alzheimer’s disease. Biochem Pharmacol 88(4):499–507. doi:10.1016/j.bcp.2013.12.020
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM (2001) Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 98(15):8850–8855. doi:10.1073/pnas.151261398
Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM, Team ANS (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64(9):1553–1562. doi:10.1212/01.WNL.0000159740.16984.3C
Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C (2006) Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol 65(11):1040–1048. doi:10.1097/01.jnen.0000240466.10758.ce
Cai Z, Zhao B, Li K, Zhang L, Li C, Quazi SH, Tan Y (2012) Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer's disease? J Neurosci Res 90(6):1105–1118. doi:10.1002/jnr.23011
Sandoval KE, Farr SA, Banks WA, Crider AM, Morley JE, Witt KA (2013) Somatostatin receptor subtype-4 agonist NNC 26-9100 mitigates the effect of soluble Abeta(42) oligomers via a metalloproteinase-dependent mechanism. Brain Res 1520:145–156. doi:10.1016/j.brainres.2013.05.006
Logan SM, Sarkar SN, Zhang Z, Simpkins JW (2011) Estrogen-induced signaling attenuates soluble Abeta peptide-mediated dysfunction of pathways in synaptic plasticity. Brain Res 1383:1–12. doi:10.1016/j.brainres.2011.01.038
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81471309, 81371406, 81171209), the Shandong Provincial Collaborative Innovation Center for Neurodegenerative Disorders, Shandong Provincial Outstanding Medical Academic Professional Program, Qingdao Key Health Discipline Development Fund, and Qingdao Outstanding Health Professional Development Fund.
Conflict of Interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wang, ZX., Tan, L., Liu, J. et al. The Essential Role of Soluble Aβ Oligomers in Alzheimer’s Disease. Mol Neurobiol 53, 1905–1924 (2016). https://doi.org/10.1007/s12035-015-9143-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9143-0